
Abstract
Casticin, a flavonoid isolated from Vitex species, has been found to have anti-tumor property in multiple human cancers. The present study aimed to investigate the effect of casticin on the proliferation and apoptosis of esophageal cancer (EC) cells, and further illustrate the underlying mechanisms. In in vitro studies, human EC cell lines TE-1 and ECA-109 were treated with various concentrations of casticin (low-, middle-, and high-dose groups). The results showed that casticin dose-dependently inhibited the proliferation and clonogenicity of EC cells and induced cell cycle arrest in sub-G1 and G2 phases. Furthermore, casticin markedly enhanced EC cell apoptosis as detected by flow cytometry and Hoechst 33342 staining. The level of anti-apoptotic Bcl-2 protein was decreased, while the levels of pro-apoptotic Bax, cleaved-caspase-3, cleaved-caspase-9, and cleaved-PARP were conversely increased in casticin-treated TE-1 and ECA-109 cells. Moreover, casticin decreased the mitochondrial membrane potential and increased the release of mitochondrial cytochrome C into cytoplasm. In addition, the JNK signaling pathway was involved in casticin-medicated anti-proliferation and pro-apoptosis. Cells pretreated with SP600125, a JNK pathway inhibitor, partially abolished the effect of casticin. Finally, the anti-tumor property of casticin was confirmed in in vivo xenograft models. Overall, we provided both in vitro and in vivo evidences that casticin inhibited the proliferation and induced apoptosis of EC cells, and the anti-tumor action of casticin was mediated, in part, by the mitochondrial-dependent apoptosis and the activation of JNK signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Esophageal cancer (EC) is the eighth most common malignant tumor and the sixth leading cause of cancer mortality worldwide (Napier et al. 2014). As reported, it affects more than 450,000 people worldwide and the incidence of EC varies widely by region. China has a very high incidence of EC, accounting for 100 cases per 100,000 population annually (Pennathur et al. 2013). Currently, the treatment for EC patients includes surgery, chemotherapy, radiation, or combination of these methods, but it still shows a low overall 5-year survival rate. Therefore, there is an urgent need for searching effective treatments to improve the survival rate of EC.
Casticin (3′, 5-dihydroxy-3, 4′, 6, 7-tetramethoxyflavone), also known as vitexicarpin, is a flavonoid isolated from Vitex species (Hu et al. 2007; Rasul et al. 2014). Previous studies have demonstrated that casticin possesses multiple pharmacological properties (Rasul et al. 2014), such as anti-inflammatory (Lee et al. 2015), anti-malarial (Elford et al. 1987), anti-oxidation (Choudhary et al. 2009; Hajdu et al. 2007), and neuroprotective effects (de Sampaio e Spohr et al. 2010). In addition, recent studies have shown that casticin also has an inhibitory effect on numerous cancer cells, and its inhibitory impact is associated with regulation of cell apoptosis, cell cycle, and cell proliferation (Chan et al. 2018). For example, Song et al. reported that casticin induced G0/G1 phase arrest and apoptosis in gallbladder cancer cells (Song et al. 2017). Casticin induced apoptosis in colon cancer cells by activating apoptosis signal-regulating kinase 1 (ASK1)-c-Jun N-terminal kinase (JNK)-Bim signaling pathway (Qu et al. 2014). Casticin was also able to induce breast cancer cell apoptosis by inhibiting forkhead box protein M1 expression (Liu et al. 2014). These findings indicated the pleiotropic roles of casticin in cancer. At present, casticin has not been reported to have the anti-cancer effect on esophageal cancer cells.
In this study, we performed both in vitro and in vivo experiments to evaluate the potential role of casticin in the proliferation and apoptosis of esophageal cancer cells, and further illustrated the molecular mechanisms underlying its anti-tumor functions.
Materials and method
Cell culture and treatment
Human esophageal cancer cell lines TE-1 and ECA-109 were purchased from ZhongQiaoXinZhou Biotechnology Co., Ltd. (Shanghai, China). The cells were cultured in RPMI-1640 (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA) and maintained at 37 °C in a humidified atmosphere containing 5% CO2. Casticin (purity ≥ 98.0%) was purchased from Chengdu Biopurify Phytochemicals Ltd. (Chengdu, China) and dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, USA). The concentrations of casticin (low, L: 10 μM, medium, M: 20 μM, and high, H: 30 μM) were chosen according to the previous preliminary experiments.
MTT assay
Cell viability was tested by MTT assay. In brief, TE-1 or ECA-109 cells (5 × 103/well) were seeded in 96-well plates; then, casticin was added to the cells at different concentrations (0, 1, 2, 4, 10, 20, 40 μM) and incubated for 24 h, 48 h, or 72 h. For inhibition of the JNK signaling pathway, cells were pretreated with the JNK pathway inhibitor SP600125 (2.5 μM) for 30 min, then incubated with casticin for 48 h. After that, MTT (0.5 mg/ml, KeyGEN, Nanjing, China) was added into cells and incubated for additional 4 h. Formazan crystals were dissolved in DMSO and the absorbance was taken at 570 nm under a microplate reader (BioTek, VT, USA).
Colony formation assay
For the colony formation assay, cells were seeded into 35-mm dishes (300 cells/dish), and then treated with different concentrations of casticin for 2 weeks. Colonies were fixed with 4% paraformaldehyde (Aladdin, Shanghai, China) and then stained with Wright-Giemsa solution (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The colonies consisting of at least 50 cells were counted under a microscope (AE31, Motic, Xiamen, China). The colony formation rate = number of clones/number of seeded cells × 100%.
Flow cytometry analysis
Cells were treated with different concentrations of casticin for 48 h. For cell cycle analysis, the cells with/without casticin treatment were fixed in 70% pre-cooled ethanol at 4 °C for 2 h, followed by incubation with staining buffer (containing propidium iodide and RNase A) for 30 min at 37 °C in the dark. Then, the cells were analyzed using flow cytometer (C6, BD, Franklin Lakes, NJ, USA).
For apoptosis analysis, cells with different treatments were fixed and detected using Annexin V-FITC Apoptosis Detection Kit (Beyotime) according to the manufacturer’s instructions. Apoptotic cells were assessed by flow cytometry (C6, BD), and the apoptosis rate was calculated.
Hoechst 33342 staining
After treatment with various concentrations of casticin for 48 h, the cells were fixed with 4% paraformaldehyde for 20 min and stained with Hoechst 33324 reagent (Beyotime) for 5 min. At last, the cells were observed under a fluorescence microscope (IX53, Olympus, Tokyo, Japan) at 400× magnification.
Mitochondria membrane potential measurement
The MMP of cells was measured using the lipophilic cationic dye JC-1 (Beyotime) according to the manufacturer’s instructions. Briefly, after 48 h of casticin treatment, the cells were washed with PBS and incubated in 0.5-ml medium containing JC-1 staining probe for 20 min at 37 °C. The proportion of mitochondrial depolarization is measured by the relative proportion of red (aggregates) and green (monomer) fluorescence. The fluorescence intensity was detected using a microplate reader (Infinite M200PRO, TECAN, Männedorf, Switzerland).
Western blot
Total protein from cells or tissues was extracted with RIPA lysis buffer (Beyotime) containing 1% (v/v) phenylmethanesulfonyl fluoride (PMSF, Beyotime). Endochylema and mitochondria were separated by Cell Mitochondria Isolation Kit (Beyotime). Protein concentrations were determined with a BCA assay kit (Beyotime). Equal amounts of proteins from each group were separated by sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membrane (Millipore, Boston, MA, USA). After blocking with 5% skim milk or 1% BSA, the membrane was incubated with primary antibody followed by incubation with corresponding secondary antibody. All antibodies were purchased commercially and specific information was listed as follows: anti-CyclinA (1: 1500, Abcam, Cambridge, UK), anti-CyclinB1, anti-CyclinD1, anti-p-JNK, and anti-JNK (1: 500, Santa Cruz Biotechnology, Dallas, USA), anti-Bcl-2 and anti-cleaved caspase 3 (1: 500, Abcam). Anti-Bax, anti-cleaved-caspase-9, and anti-cleaved-PARP (1: 1000, Cell signaling Technology, Beverly, MA, USA), anti-Cytochrome C (1: 500, Proteintech, Wuhan, China), anti-β-actin, and anti-COX IV (1: 500; Bioss, Beijing, China), and horseradish peroxidase-labeled secondary antibody (1: 5000; Beyotime). Finally, the targeted protein bands were visualized using an enhanced chemiluminescence kit (Beyotime) and analyzed by Gel-Pro-Analyzer (Media Cybernetics, USA). β-Actin or COX IV was served as internal reference.
Xenograft tumor study
Eighteen male nude mice (4 weeks old, weighing 18~20 g) were obtained from HFK Bioscience Co., LTD (Beijing, China) and housed in sterile environment under standard conditions (22 ± 1 °C; 50 ± 5% humidity; 12 h light/12 h dark). Mice were randomly divided into three groups: control, casticin (2 mg/kg), and casticin (10 mg/kg). To establish xenograft models, ECA-109 cells (1 × 106 cells/mouse) were subcutaneously injected into the armpit of nude mice. Then, the tumor sizes were monitored every 3 days, and the tumor volumes were calculated following the formula: xenograft volume = major axis × minor axis2/2. When the tumor volume reached to 100 mm3, mice in the casticin treatment groups were intraperitoneally injected with different concentrations of casticin (dissolved in olive oil), and control mice received the same volume of olive oil, 5 times/7 days, for continuous 14 days. At the end of drug treatment, all animals were sacrificed and the tumor tissues were removed, weighted, and prepared for subsequent experiments.
Hematoxylin and eosin staining
H&E staining was performed to evaluate histologic alterations. Briefly, xenograft tumor tissues were fixed in 10% formalin (Sinopharm Chemical Reagent Beijing Co., Ltd) overnight, then embedded in paraffin, and sliced into 5-μm sections. The slices were then stained with hematoxylin and eosin, and the pathologic changes in xenograft tissues were examined under a light microscope (DP73; Olympus Corp, Tokyo, Japan) at 200× magnification.
Terminal deoxynucleoitidyl transferase dUTP nick end labeling staining
The paraffin-embedded tissue sections were prepared as above mentioned. Cellular apoptosis in xenograft tissues was evaluated using an In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. TUNEL-positive cells were quantified under a light microscope (DP73; Olympus Corp, Tokyo, Japan) at a magnification of 400×.
Statistical analysis
All experimental data are presented as the mean ± standard deviation (SD) and analyzed by GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA). Statistical difference was evaluated using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. A p value less than 0.05 was considered statistically significant.
Results
Casticin decreased the viability of EC cells
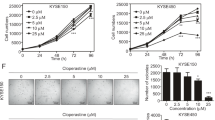
To verify whether casticin affected the proliferation of EC cells, TE-1 and ECA-109 cells were treated with various concentrations of casticin for 24, 48 and 72 h. Cell viability was measured by MTT assay (Fig. 1a), and the results indicated that casticin significantly decreased the cell viability of TE-1 and ECA-109 in a time- and dose-dependent manner. Based on MTT results, the concentrations of casticin at 10, 20, and 30 μM were used in TE-1 cells and ECA-109 cells. Colony formation assay further confirmed that casticin treatment observably suppressed the colony-forming ability of both TE-1 and ECA-109 cells (Fig. 1b, p < 0.05 or p < 0.01 vs. untreated cells), indicating an anti-proliferative property of casticin in EC cells.

Casticin decreases the viability EC cells. a The effect of casticin on EC cell proliferation was measured by MTT assay. According to the results, concentrations of casticin (low, L: 10 μM, medium, M: 20 μM, and high, H: 30 μM) were selected for subsequent experiments. b Colony formation assay was performed in TE-1 and ECA-109 cells, and the colony formation rate was calculated. Casticin reduced cell colony-forming ability in a dose-dependent manner. c Cell cycle distribution in TE-1 and ECA-109 cells with/without casticin treatment was detected by flow cytometry. d Expression levels of CyclinA, CyclinB1, and CyclinD1 were detected by Western blot. β-actin was served as the internal control. All results were presented as mean ± SD. *p < 0.05, **p < 0.01 compared with TE-1 or ECA-109 group, respectively
In addition, the effect of casticin on the cell cycle progression was determined by flow cytometry. Following casticin treatment, the ratios of cells in sub-G1 and G2 phases were strikingly increased, while the ratios of cells in G1 and S phases were significantly decreased (Fig. 1c, p < 0.05 or p < 0.01). Cell cycle regulatory proteins were further detected by Western blot assay. The levels of CyclinA, CyclinB1, and CyclinD1 were significantly reduced by casticin treatment in both TE-1 and ECA-109 cells (Fig. 1d, p < 0.05 or p < 0.01). Together, these findings suggested that casticin dose-dependently inhibited the proliferation and induced cell cycle arrest in EC cells.
Casticin enhanced the apoptosis of EC cells
Cell apoptosis was detected using Annexin V/PI staining and analyzed by flow cytometry. As demonstrated in Fig. 2a, treatment with casticin significantly increased the percentage of apoptotic cells in EC cells (TE-1: 4.62 ± 0.44, casticin-L: 20.48 ± 1.42, casticin-M: 36.69 ± 1.94, casticin-H: 49.31 ± 3.90; ECA-109: 2.52 ± 0.46, casticin-L: 25.52 ± 3.33, casticin-M: 40.09 ± 5.07, casticin-H: 49.86 ± 2.20). Moreover, cell apoptosis was confirmed by Hoechst 33342 staining (Fig. 2b). There were more shrunken and fractured nuclei in cells with casticin treatment, and the pro-apoptotic effect of casticin was dose-dependent.

Casticin promotes apoptosis of EC cells. a Apoptosis of TE-1 and ECA-109 cells with/without casticin treatment was analyzed by flow cytometry, and the percentage of apoptotic cells was calculated. All results were presented as mean ± SD. *p < 0.05, **p < 0.01 compared with TE-1 or ECA-109 group, respectively. b Effects of casticin on apoptosis in TE-1 and ECA-109 cells were confirmed by Hoechst 33342 staining. Scale bar = 50 μm
Involvement of mitochondrial-dependent apoptosis signaling pathway in casticin-treated EC cells
As mitochondrial dysfunction plays key roles in regulating cell death, here further determined whether casticin was able to induce mitochondrial dysfunction in EC cells. The loss of MMP and mitochondrial cytochrome C (Cyto-C) release were used to evaluate mitochondrial dysfunction. The results showed that casticin treatment resulted in significant reduction in MMP in both TE-1 and ECA-109 cells (Fig. 3a). The levels of Cyto-C in cytosol and mitochondria were determined by Western blot. As shown in Fig. 3b, administration of casticin led to significant increase in the level of cytosolic Cyto-C, but decrease in mitochondrial Cyto-C level suggested that casticin induced mitochondrial dysfunction in EC cells. We further determined the levels of mitochondrial-apoptosis-related proteins. As presented in Fig. 3c, the level of anti-apoptotic Bcl-2 was down-regulated, while the levels of pro-apoptotic Bax, cleaved-caspase-3, cleaved-caspase-9, and cleaved-PARP were conversely up-regulated in casticin-treated cells. Together, these results indicated that casticin induced cell apoptosis through affecting mitochondrial events in EC cells.

Involvement of mitochondrial-dependent apoptosis signaling pathway in casticin-treated EC cells. a Mitochondrial membrane potential (MMP) was measured by JC-1 staining. Casticin dose-dependently decreased the MMP in TE-1 and ECA-109 cells compared with un-treated cells. b Expression levels of cytochrome C (Cyto-C) in the cytoplasm and mitochondria were determined by Western blot. Relative cyto-C expression in the cytoplasm was normalized to β-actin and relative Cyto-C in mitochondria was normalized to COX IV. c Expression levels of apoptotic apoptosis-related proteins Bcl-2, Bax, cleaved-caspase 3, 9, and PARP were determined by Western blot. β-actin was served as the internal control. All data were presented as mean ± SD. *p < 0.05, **p < 0.01 compared with TE-1 or ECA-109 group, respectively
Casticin activated the JNK signaling pathway in EC cells
In order to determine the signaling pathway through which casticin regulated cell proliferation and apoptosis, we further performed Western blot to detect JNK pathway-related proteins. The results showed that treatment with casticin significantly increased the phosphorylation of JNK (p-JNK) in both TE-1 and ECA-109 cells (Fig. 4a, p < 0.01), indicating the activation of the JNK pathway by casticin. Furthermore, when cells pretreated with the specific JNK inhibitor SP600125, the anti-proliferative action of casticin was partially inhibited (Fig. 4b, p < 0.05 vs. SP600125-treated cells). Similarly, SP600125 also partially abolished the pro-apoptotic effect of casticin in TE-1 and ECA-109 cells (Fig. 4c, p < 0.01 vs. SP600125-treated cells). The protein levels of p-JNK and JNK after treatment with SP600125 were verified by Western blot (Fig. 4d). Together, these results indicated that casticin activated the JNK signaling pathway in EC cells, which was essential for the regulation of proliferation and apoptosis by casticin.

Casticin activates the JNK signaling pathway in EC cells. a Western blot was performed to determine the expression levels of p-JNK and JNK in TE-1 and ECA-109 cells with/without casticin treatment. β-actin was served as the internal control. b The proliferation (b) and apoptosis (c) of EC cells treated with casticin or combined combination with specific JNK inhibitor SP600125 were measured by MTT assay and flow cytometry. d The protein levels of p-JNK and JNK were detected by Western blot. All results were presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared with TE-1 or ECA-109 group, respectively. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with casticin group
Casticin suppresses the growth of xenograft tumor in vivo
We further performed in vivo experiments to confirm the anti-cancer effect of casticin in EC. Animals were injected with ECA-109 cells to initiate tumor xenograft and then were treated with casticin. Tumor volume and weight were measured to assess tumor growth and results are shown in Fig. 5a. Nude mice without treatment exhibited larger tumor volume and weight, while casticin treatment, especially at 10 mg/kg, significantly reduced the tumor volume and weight as compared with the control group (p < 0.05 or p < 0.01). The final weights of tumors in control, casticin 2 mg, and casticin 10 mg groups were 510.83 ± 94.95 mg, 369.83 ± 70.04 mg, and 193.50 ± 33.73 mg, respectively. Together, these results indicated that casticin inhibited EC cell growth both in vitro and in vivo. Histologic changes of xenograft tumors were further detected by H&E and TUNEL staining (Fig. 5b). Results demonstrated that the tumor cells in control group arranged more closely, and no obvious apoptosis and necrosis areas were observed, whereas casticin treatment significantly affected cellular morphology and increased the number of apoptotic cells in xenograft tumor tissues. The pro-apoptotic effect of casticin was more effective at 10-mg/kg dose. Apoptosis-related proteins were further detected by Western blot. As shown in Fig. 5c, casticin treatment markedly increased the levels of Bax, cleaved caspase-3 and cleaved PARP, and decreased Bcl-2 expression, which were in line with TUNEL results and confirmed the pro-apoptotic property of casticin. Moreover, we found that casticin activated the JNK signaling pathway, as evidenced by increased p-JNK level in casticin-treated groups (Fig. 5d, p < 0.01 vs. control group). Consistent with this, as assessed by immunohistochemical staining, the expression of Bcl-2 was decreased, while the expression of Bax and p-JNK was enhanced in casticin-treated tumor tissues (Fig. 5e).

Casticin suppresses the growth of xenograft tumor in vivo. a Animals were injected with ECA-109 cells to initiate tumor xenograft and then were treated with casticin. The tumor volume was recorded every 3 days, and the weight was measured to assess tumor growth. b Histological changes in tumor tissues were analyzed by H&E staining. Scale bar = 100 μm. Cell apoptosis was observed with TUNEL staining. Scale bar = 50 μm. c Expression levels of Bcl-2, Bax, cleaved caspase 3, and PARP, d p-JNK and JNK in xenograft tissues were determined by Western blot. β-actin was served as the internal control. e The expression of Bcl-2, Bax, and p-JNK in xenograft tissues was determined by immunohistochemical staining. All results were presented as mean ± SD. *p < 0.05, **p < 0.01 compared with control group
Discussion
Increasing evidence has indicated that casticin plays an important role in regulating tumor cell proliferation and apoptosis (Dai et al. 2018). However, its functional roles in EC have not been clarified. Herein, we investigated whether casticin exerted anti-cancer effects on EC. From our in vitro studies, casticin was shown to suppress cell proliferation, induce cell cycle arrest, and promote the apoptosis of TE-1 and ECA-109 cells. In vivo, casticin also repressed tumor growth as well as promoted apoptosis in xenograft tumor tissues. Furthermore, the antitumor effect of casticin was associated with mitochondrial apoptosis and activation of JNK signaling pathway. In summary, we provided both in vitro and in vivo evidence that casticin has an antitumor effect on EC, and this molecule may be a potential anticancer agent for EC in future.
Various mechanisms have been suggested to the anti-cancer function of casticin. In the present study, we mainly focused on its anti-proliferative and pro-apoptotic properties in EC. TE-1 and ECA-109 cells were treated with different concentrations of casticin. Our data showed that casticin dose-dependently inhibited the cell viability and clonogenicity and arrested the cell cycle at G2 phase. Meanwhile, the levels of cell cycle regulatory proteins including CyclinA, CyclinB1, and CyclinD1 were significantly decreased after casticin treatment. Furthermore, Annexin V/PI and Hoechst staining revealed that casticin induced apoptosis of EC cells. Our in vitro studies confirmed the anti-tumor effect of casticin in EC cells. Previous studies have demonstrated that casticin plays a role in modulating cell cycle progression. Shang et al. reported casticin induced cell cycle arrest at G2/M phase and decreased cell numbers in G0/G1 phase in colon cancer cells (Shang et al. 2017). The study of Enyu L et al. also indicated that casticin could induce glioma cell cycle arrest in G2/M phase (Liu et al. 2013). Our results were consistent with these findings. Moreover, the pro-apoptotic effect of casticin has been demonstrated in many cancer cells, such as gastric cancer cells (Zhou et al. 2013), colon cancer cells (Shang et al. 2017), bladder cancer cells (Chung and Kim 2016), and leukemia cells (Shen et al. 2009). Consistent with these data, here we also demonstrated that casticin could induce EC cell apoptosis in a dose-dependent manner, and the molecular mechanisms were subsequently investigated.
At present, it is believed that apoptosis occurs mainly through the following two pathways, the death receptor pathway and the mitochondrial pathway (Ghobrial et al. 2005). The former pathway initiates apoptosis through recruiting and activating caspase-2 precursors via a linker protein like Fas-associated death domain (FADD) (Hengartner 2000). The latter mitochondrial-dependent apoptosis can be affected by many factors, such as the Bcl-2 family proteins, MMP change, and the formation of mitochondrial membrane-transmigration, and the release of apoptosis-related factors such as Cyto-C (Schapira 2012). Cyto-C is released from the mitochondria, forming an apoptotic complex (caspase-9/apoptotic protease activating factor (Apaf-1), Cyto-C, and ATP/dATP, etc.) in the cytoplasm and following activating caspase-9. Both of these pathways eventually induce the activation of caspase-3 and the proteolysis of the caspase substrate, leading to apoptosis (Hajnoczky and Hoek 2007; Joza et al. 2001). The anti-apoptotic proteins like Bcl-2 and Bcl-xL reside in the outer mitochondrial wall and inhibit Cyto-C release. While the pro-apoptotic protein like Bax promotes the release of Cyto-C, the Bcl-2 family participates in the regulation of mitochondrial function thereby regulating cell apoptosis (Leibowitz and Yu 2010). To further assess the mechanisms of casticin-induced apoptosis in EC cells, we detected the mitochondrial-apoptotic pathway. The results showed that the MMP markedly decreased and cytoplasmic Cyto-C level significantly up-regulated in casticin-treated EC cells, indicating mitochondrial dysfunction by casticin treatment. In addition, casticin treatment decreased Bcl-2 expression and up-regulated Bax, cleaved caspase 9, cleaved caspase-3, and cleaved-PARP levels, which implied casticin induced EC cell apoptosis via the mitochondrial apoptotic pathway. Consistently, Chen et al. reported that casticin inducted apoptosis in cervical cancer cells through reactive oxygen species-mediated mitochondrial signaling pathways (Chen et al. 2011).
JNK, as one of the major members of the MAPK family, is involved in various cellular processes like cell survival, growth, differentiation, and cell death (Ji et al. 2009). For cell apoptosis, the JNK pathway can affect the dynamics of mitochondria, thus promotes Cyto-C release, activates caspase-3 protein, and finally induces apoptosis. Previous studies have found that casticin can induce apoptosis through the activation of JNK pathway (Qu et al. 2014; Zeng et al. 2012). For instance, Zeng et al. (2012) showed that casticin sustained JNK activation thereby modulating casticin-induced apoptosis in cervical cancer cells. Chung et al. demonstrated that activation of JNK/p38 MAPK by casticin was able to affect the activation of the caspase cascade and damage of the MMP (Chung and Kim 2016). In this study, our results showed that casticin was able to activate the JNK pathway in a dose-dependent manner. When cells pretreated with JNK inhibitor SP600125, the anti-proliferative and pro-apoptotic effects of casticin were partially reversed. These findings suggested that the antitumor effect of casticin was mediated partially through the JNK signaling pathway.
At last, the antitumor effect of casticin was evaluated by in vivo studies. In xenograft tumor models, casticin treatment significantly inhibited tumor growth and induced cell apoptosis. Furthermore, the expression of Bcl-2 was decreased, and Bax, cleaved-caspase-3, and cleaved-PARP levels were increased by casticin treatment. Moreover, the JNK signaling pathway was activated by casticin treatment. Together, both in vitro and in vivo evidences suggested the antitumor effect of casticin on EC through the JNK pathway.
In conclusion, our results demonstrate that casticin inhibits proliferation and induces apoptosis of EC cells both in vitro and in vivo. The antitumor effect of casticin was mediated by the mitochondrial-apoptotic pathway and JNK signaling pathway. Our studies indicate that casticin may be a potential effective agent for the treatment of esophageal cancer.
References
Chan EWC, Wong SK, Chan HT (2018) Casticin from Vitex species: a short review on its anticancer and anti-inflammatory properties. J Integr Med 16:147–152. https://doi.org/10.1016/j.joim.2018.03.001
Chen D, Cao J, Tian L, Liu F, Sheng X (2011) Induction of apoptosis by casticin in cervical cancer cells through reactive oxygen species-mediated mitochondrial signaling pathways. Oncol Rep 26:1287–1294. https://doi.org/10.3892/or.2011.1367
Choudhary MI, Jalil S, Nawaz SA, Khan KM, Tareen RB (2009) Antiinflammatory and lipoxygenase inhibitory compounds from Vitex agnus-castus. Phytother Res 23:1336–1339. https://doi.org/10.1002/ptr.2639
Chung YH, Kim D (2016) RIP kinase-mediated ROS production triggers XAF1 expression through activation of TAp73 in casticin-treated bladder cancer cells. Oncol Rep 36:1135–1142. https://doi.org/10.3892/or.2016.4895
Dai Y, Cheng R, Gao J, Li Y, Lou C (2018) Casticin inhibits PDGF-induced proliferation and migration of airway smooth muscle cells. Eur J Pharmacol 830:39–46. https://doi.org/10.1016/j.ejphar.2018.04.016
de Sampaio e Spohr TC et al (2010) Effects of the flavonoid casticin from Brazilian Croton betulaster in cerebral cortical progenitors in vitro: direct and indirect action through astrocytes. J Neurosci Res 88:530–541. https://doi.org/10.1002/jnr.22218
Elford BC, Roberts MF, Phillipson JD, Wilson RJ (1987) Potentiation of the antimalarial activity of qinghaosu by methoxylated flavones. Trans R Soc Trop Med Hyg 81:434–436
Ghobrial IM, Witzig TE, Adjei AA (2005) Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 55:178–194
Hajdu Z et al (2007) Diterpenoids and flavonoids from the fruits of Vitex agnus-castus and antioxidant activity of the fruit extracts and their constituents. Phytother Res 21:391–394. https://doi.org/10.1002/ptr.2021
Hajnoczky G, Hoek JB (2007) Cell signaling. Mitochondrial longevity pathways. Science 315:607–609. https://doi.org/10.1126/science.1138825
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776. https://doi.org/10.1038/35037710
Hu Y, Xin HL, Zhang QY, Zheng HC, Rahman K, Qin LP (2007) Anti-nociceptive and anti-hyperprolactinemia activities of Fructus Viticis and its effective fractions and chemical constituents. Phytomedicine 14:668–674. https://doi.org/10.1016/j.phymed.2007.01.008
Ji RR, Gereau RW, Malcangio M, Strichartz GR (2009) MAP kinase and pain. Brain Res Rev 60:135–148. https://doi.org/10.1016/j.brainresrev.2008.12.011
Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CYJ, Sasaki T, Elia AJ, Cheng HYM, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zúñiga-Pflücker JC, Kroemer G, Penninger JM (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410:549–554. https://doi.org/10.1038/35069004
Lee H, Jung KH, Park S, Choi W, Bae H (2015) Casticin, an active compound isolated from Vitex Fructus, ameliorates the cigarette smoke-induced acute lung inflammatory response in a murine model. Int Immunopharmacol 28:1097–1101. https://doi.org/10.1016/j.intimp.2015.07.041
Leibowitz B, Yu J (2010) Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol Ther 9:417–422
Liu E, Kuang Y, He W, Xing X, Gu J (2013) Casticin induces human glioma cell death through apoptosis and mitotic arrest. Cell Physiol Biochem 31:805–814. https://doi.org/10.1159/000350098
Liu LP, Cao XC, Liu F, Quan MF, Sheng XF, Ren KQ (2014) Casticin induces breast cancer cell apoptosis by inhibiting the expression of forkhead box protein M1. Oncol Lett 7:1711–1717. https://doi.org/10.3892/ol.2014.1911
Napier KJ, Scheerer M, Misra S (2014) Esophageal cancer: a review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol 6:112–120. https://doi.org/10.4251/wjgo.v6.i5.112
Pennathur A, Gibson MK, Jobe BA, Luketich JD (2013) Oesophageal carcinoma. Lancet 381:400–412. https://doi.org/10.1016/S0140-6736(12)60643-6
Qu L, Liu FX, Cao XC, Xiao Q, Yang X, Ren KQ (2014) Activation of the apoptosis signal-regulating kinase 1/c-Jun N-terminal kinase pathway is involved in the casticin-induced apoptosis of colon cancer cells. Exp Ther Med 8:1494–1500. https://doi.org/10.3892/etm.2014.1934
Rasul A, Zhao BJ, Liu J, Liu B, Sun JX, Li J, Li XM (2014) Molecular mechanisms of casticin action: an update on its antitumor functions. Asian Pac J Cancer Prev 15:9049–9058
Schapira AH (2012) Mitochondrial diseases. Lancet 379:1825–1834. https://doi.org/10.1016/S0140-6736(11)61305-6
Shang HS, Liu JY, Lu HF, Chiang HS, Lin CH, Chen A, Lin YF, Chung JG (2017) Casticin induced apoptotic cell death and altered associated gene expression in human colon cancer Colo 205 cells. Environ Toxicol 32:2041–2052. https://doi.org/10.1002/tox.22381
Shen JK, Du HP, Yang M, Wang YG, Jin J (2009) Casticin induces leukemic cell death through apoptosis and mitotic catastrophe. Ann Hematol 88:743–752. https://doi.org/10.1007/s00277-008-0677-3
Song XL, Zhang YJ, Wang XF, Zhang WJ, Wang Z, Zhang F, Zhang YJ, Lu JH, Mei JW, Hu YP, Chen L, Li HF, Ye YY, Liu YB, Gu J (2017) Casticin induces apoptosis and G0/G1 cell cycle arrest in gallbladder cancer cells. Cancer Cell Int 17:9. https://doi.org/10.1186/s12935-016-0377-3
Zeng F, Tian L, Liu F, Cao J, Quan M, Sheng X (2012) Induction of apoptosis by casticin in cervical cancer cells: reactive oxygen species-dependent sustained activation of Jun N-terminal kinase. Acta Biochim Biophys Sin 44:442–449. https://doi.org/10.1093/abbs/gms013
Zhou Y, Tian L, Long L, Quan M, Liu F, Cao J (2013) Casticin potentiates TRAIL-induced apoptosis of gastric cancer cells through endoplasmic reticulum stress. PLoS One 8:e58855. https://doi.org/10.1371/journal.pone.0058855
Funding
This study was supported by a grant from the Science and Technology Development Project of Shaanxi Province (No. S2015YFSF0111).
Author information
Authors and Affiliations
Contributions
ZQ conceived and designed study; ZQ, YC, and SLiu performed experiments; ZQ, ZM, SLi, and WZ analyzed data. ZQ and YC wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All the animal experiments were conducted in strict accordance with the Guidelines for the care and use of laborary animals and were approved by the Experimental Animal Ethics of the Second Affiliated Hospital of Xi’an Jiaotong University.
Rights and permissions
About this article

Cite this article
Qiao, Z., Cheng, Y., Liu, S. et al. Casticin inhibits esophageal cancer cell proliferation and promotes apoptosis by regulating mitochondrial apoptotic and JNK signaling pathways. Naunyn-Schmiedeberg's Arch Pharmacol 392, 177–187 (2019). https://doi.org/10.1007/s00210-018-1574-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-018-1574-5