
Abstract
Nitrate and nitrite have emerged as an important novel source of nitric oxide (NO). We have previously demonstrated that sodium nitrite is an antihypertensive compound that exerts antioxidant effects in experimental hypertension. These unpredicted antioxidant effects of nitrite raised the question whether the beneficial effects found were caused by its conversion to NO or simply due to reversal of endothelial dysfunction as a consequence of its antioxidant effects. Here, we evaluated the antihypertensive effects of a daily dose of sodium nitrite for 4 weeks in l-NAME-induced hypertension in rats. We studied the effects of nitrite on markers of NO bioavailability, vascular oxidative stress, and expression of xanthine oxidoreductase. Moreover, we tested if xanthine oxidoreductase inhibition could attenuate the acute hypotensive effects of sodium nitrite in l-NAME hypertensive rats. We found that a single pharmacological dose of sodium nitrite exerts antihypertensive effects in l-NAME-induced hypertension. While the beneficial antihypertensive properties of nitrite were associated with increased levels of NO metabolites, hypertension increased vascular xanthine oxidoreductase expression by approximately 40 %, with minor increases in vascular superoxide production. The inhibition of xanthine oxidoreductase by oxypurinol attenuated the acute hypotensive effects of nitrite. Taken together, our results show that nitrite exerts antihypertensive effects in l-NAME hypertensive rats and provide evidence that xanthine oxidoreductase plays an important role in this antihypertensive effect.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nitrate and nitrite have emerged as an important novel source of nitric oxide (NO) (Lundberg and Weitzberg 2013). After initial misconceptions regarding the lack of biological effects (Moncada and Higgs 1993; Lauer et al. 2001) and association with some types of cancer (McKnight et al. 1999), there is now a strong evidence that the nitrate/nitrite-derived NO is part of an alternative pathway for NO formation, which is complementary to the classical NO formation mediated by NO synthases (Lundberg et al. 2008, 2009).
Two main mechanisms have been described for NO formation from nitrite. The first involves enzymatic reduction of nitrite anion to NO by several enzymes and heme-containing metalloproteins including (but not limited to) deoxyhemoglobin and deoxymyoglobin (Cosby et al. 2003; Rassaf et al. 2007), cytochromes P450 (Kozlov et al. 2003), enzymes of the mitochondrial respiratory chain (Kozlov et al. 1999), guanylate cyclase (Alzawahra et al. 2008), and xanthine oxidoreductase (Tripatara et al. 2007; Webb et al. 2004). The second mechanism involves the enterosalivary circulation of nitrate and nitrite. The diet is an important source of nitrate/nitrite that once ingested, enters to the enterosalivary circulation (Lundberg et al. 2004). While part of nitrite is promptly reduced to NO and other N-oxides products at the low pH of stomach, most of ingested nitrate/nitrite is reabsorbed and concentrated at the salivary glands that secrete saliva containing nitrate that is reduced by commensal bacteria of the mouth to nitrite (McKnight et al. 1997). When the saliva is swallowed, the nitrite is reduced again to NO at low gastric pH, completing the enterosalivary cycle (Lundberg et al. 2004).
Although these two possible mechanisms explain NO production from nitrite, the relevance of each pathway and the role of nitrite in hypertensive disorders are still poorly understood (Montenegro et al. 2011). For example, the plasma levels of nitrite are lower in experimental and clinical hypertensive disorders such as hypertension and preeclampsia as compared to normotensive controls (Gomes et al. 2013; Montenegro et al. 2010, 2011; Sandrim et al. 2010). This difference in plasma nitrite levels is ascribed mostly to endothelial dysfunction or to increased production to reactive oxygen species associated with hypertensive diseases, which decrease the NO bioavailability (Cai and Harrison 2000). In this regard, we have previously demonstrated that oral treatment with sodium nitrite restores plasma nitrite levels and exerts antihypertensive and antioxidant effects in experimental renovascular hypertension (Montenegro et al. 2011, 2012). The unexpected antioxidant effects induced by nitrite in this model of hypertension raises the question whether the beneficial effects of nitrite were due to the activation of nitrate-nitrite-NO pathway fed by exogenously administrated nitrite or by reversal of endothelial dysfunction as a consequence of antioxidant effects of nitrite (Montenegro et al. 2011).
In the present study, we decided to evaluate the antihypertensive effects of sodium nitrite in an experimental model of hypertension caused by the NO synthase inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) because the antioxidant effects of nitrite cannot revert l-NAME-induced NO synthases inhibition, which causes endothelial dysfunction. Therefore, the goal of the current study was to examine whether restoring nitrite levels by exogenous nitrite administration could attenuate the hypertension induced by l-NAME. We have also studied the effects of sodium nitrite treatment on NO metabolites levels, vascular reactive oxygen species production, and xanthine oxidoreductase expression. Finally, we tested the effects of xanthine oxidoreductase inhibition on the acute hypotensive effects exerted by sodium nitrite.
Methods
Animals and treatments
This study complied with guidelines of the Faculty of Medicine of Ribeirao Preto, University of Sao Paulo, and the animals were handled according to the guiding principles published by the National Institutes of Health Guide for the care and use of laboratory animals. Male Wistar rats (180–200 g) obtained from the colony at the University of Sao Paulo (Ribeirao Preto Campus, Brazil) were maintained on a 12-h light/dark cycle at room temperature (22–25 °C) with free access to standard rat chow and water.
The rats were randomly divided into four experimental groups (n = 10/group): control + vehicle (animals who received only tap water and daily gavage of vehicle), control + nitrite (animals who received tap water and daily gavage of nitrite), l-NAME + vehicle (animals who received Nω-nitro-l-arginine methyl ester (l-NAME; 1 g/l) in their drinking water and daily gavage of vehicle), and l-NAME + nitrite (animals who received l-NAME (1 g/l) in their drinking water and daily gavage of nitrite). After 2 weeks of treatment with tap water (control groups) or l-NAME (l-NAME hypertensive groups), the animals received a daily gavage of vehicle or sodium nitrite (15 mg/kg) for four additional weeks. This dose of sodium nitrite was chosen with basis on our previous studies showing antihypertensive effects of nitrite (Montenegro et al. 2011, 2012).
Body weight and tail systolic blood pressure (SBP) were assessed weekly by tail-cuff plethysmography. Aiming to minimize the effects of stress induced by this method on blood pressure measurement, the animals were trained for a week before the protocol started.
Arterial blood samples were collected in tubes containing heparin and immediately centrifuged at 1,000×g for 10 min. Plasma aliquots were stored at −70 °C until analyzed.
Measurement of plasma nitrite concentrations
Plasma aliquots were analyzed in duplicate for their nitrite content using an ozone-based reductive chemiluminescence assay as previously described (Feelisch et al. 2002; Montenegro et al. 2011). Briefly, to assess nitrite content in plasma, 200 μl of plasma samples were injected into a solution of acidified triiodide, purged with nitrogen in-line with a gas-phase chemiluminescence NO analyzer (Sievers Model 280 NO analyzer, Boulder, CO, USA). Approximately 8 ml of triiodide solution (2 g potassium iodide and 1.3 g iodine dissolved in 40 ml water with 140 ml acetic acid) was placed in the purge vessel into which plasma samples were injected. The triiodide solution reduces nitrites to NO gas, which is detected by the NO analyzer. The data were analyzed using the software Origin Lab 8.5.
Measurement of plasma nitrate concentrations
The plasma nitrate concentrations were determined in duplicate by using the Griess reaction as we described previously (Montenegro et al. 2009; Sandrim et al. 2010). Briefly, 40 μl of plasma were incubated with the same volume of nitrate reductase buffer (0.1 M potassium phosphate, pH 7.5, containing 1 mM beta nicotinamide adenine dinucleotide phosphate, and 2 units of nitrate reductase/ml) in individual wells of a 96-well plate. Samples were allowed to incubate overnight at 37 °C in the dark. Eighty microliters of freshly prepared Griess reagent (1 % sulphanilamide, 0.1 % naphthylethylenediamine dihydrochloride in 5 % phosphoric acid) were added to each well and the plate was incubated for an additional 15 min at room temperature. A standard nitrate curve was obtained by incubating sodium nitrate (0.2–200 μM) with the same reductase buffer. The absorbance was measured at 540 nm, using a microplate reader.
Measurement of vascular reactive oxygen species production
Reactive oxygen species (ROS) production in the aortas were measured by dihydroethidium (DHE), a ROS-sensitive fluorescent dye. The aortic cryosections (5-μm thick) were incubated at room temperature with DHE (10 μmol/l) for 30 min. Sections were examined by fluorescence microscopy (Leica Imaging Systems Ltd., Cambridge, England), and the image was captured at ×400. Red fluorescence from 20 fields around the vessel was evaluated using image software ImageJ (http://rsbweb.nih.gov/ij/), as described before (Montenegro et al. 2010).
Western blotting analysis for xanthine oxidoreductase
We evaluated the vascular expression of xanthine oxidoreductase in aortic samples from all animals at the end of treatments. Western blotting analysis was performed as we described previously (Montenegro et al. 2011) with primary antibodies (1:500) directed against xanthine oxidoreductase (code sc-20991, Santa Cruz Inc., Santa Cruz, CA, USA), and the expression was corrected by β-actin expression (dilution 1:10,000, code MAB1501, Millipore, Temecula, CA, USA).
Assessment of mean arterial pressure in unanesthetized freely moving rats
One day before mean arterial pressure (MAP) assessment, the animals were anesthetized with tribromoethanol (250 mg/kg, i.p.) and both the femoral artery and vein were cannulated (3-cm segment of a PE-10 tube connected to 14 cm of a PE-50 tubing; Clay Adams, Parsippany, NJ, USA). The catheter were then tunneled subcutaneously and exteriorized through the back of the neck. After surgery, the nonsteroidal anti-inflammatory flunixin meglumine (2.5 mg/kg, s.c., Banamine®, Schering Plough, Brazil) was administered for postoperative analgesia. Following 12 h of fasting, the arterial cannula was connected to a pressure transducer and the MAP was recorded in freely moving rats using a data acquisition system (MP150CE; Biopac Systems Inc. CA, USA) connected to a computer (Acknowledge 3.2, for Windows). Before collecting the data, we allowed at least 15 min of stabilization.
Effects of xanthine oxidoreductase inhibition on the acute hypotensive responses to intravenous administration of sodium nitrite
We studied the role of xanthine oxidoreductase in the hypotensive effects of intravenously administered sodium nitrite in l-NAME hypertensive rats by examining the hypotensive responses to this anion in the presence or in the absence of oxypurinol (a xanthine oxidoreductase inhibitor). The rats received a single gavage of l-NAME (100 mg/kg) to increase the MAP by approximately 40 mmHg, as described previously (Amaral et al. 2013; Pinheiro et al. 2012). MAP data recording started 30 min after l-NAME treatment and was stable for at least 10 min. Thereafter, the rats received infusions of vehicle (NaOH 0.15 mol/l; 25 μl/min) or oxypurinol (25 mg/kg; 25 μl/min) for 10 min, and 15 min later the rats received cumulative doses of sodium nitrite (1, 3, 10, 30, and 100 μmol/kg) or saline intravenously. Nitrite dose were injected every 15 min, and the maximum changes in MAP were evaluated.
Drugs and solutions
All drugs and reagents were purchased from Sigma Chemical Co. (St Louis, MO, USA), and solutions were prepared immediately before use.
Statistical analysis
The results are expressed as means ± SEM. The comparisons between groups were assessed by two-way analysis of variance using Bonferroni correction or one-way analysis of variance followed by Dunnett’s multiple comparison tests. A probability value <0.05 was considered significant.
Results
Sodium nitrite exerts antihypertensive effect in l-NAME hypertension
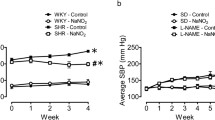
Baseline systolic blood pressure (SBP) and body weight were similar in the four experimental groups (Fig. 1a, b). We found no significant changes in SBP in the control + vehicle and control + nitrite groups throughout the experimental protocol. The l-NAME treatment induced a sustained increase in SBP after the first and second week of treatment (Fig. 1a). While the SBP increased in the l-NAME + vehicle group and peaked at week 4, a daily gavage with sodium nitrite (15 mg/kg) significantly decreased SBP in the l-NAME + nitrite group (Fig. 1a; P < 0.05). We found no significant differences in body weight between the control groups (Fig. 1b, P > 0.05). However, l-NAME-treated animals gained less weight than the control groups (Fig. 1b, P < 0.05).

Systolic blood pressure measured by noninvasive tail-cuff method (mmHg; panel a) and Bodyweight changes (g; panel b) throughout the study. Data are shown as mean ± SEM (n = 10–12 per group). *P < 0.01 for l-NAME + vehicle group versus control + nitrite 15 mg/kg group. #P < 0.01 for l-NAME + nitrite 15 mg/kg group versus l-NAME + vehicle group
Effects of sodium nitrite treatment on plasma nitrite, nitrate, and NOx (nitrate + nitrite) levels
We evaluated NO formation by measuring plasma nitrite and plasma nitrate concentrations using reductive chemiluminescence and Griess reagent, respectively. We found significant lower nitrite plasma levels in l-NAME + vehicle group compared with those found in the control + vehicle group (P < 0.05; Fig. 2a). Treatment with sodium nitrite enhanced the plasma nitrite concentrations in both control and l-NAME nitrite-treated groups (P < 0.05; Fig. 2a). No changes were found in plasma nitrate or NOx concentrations in the l-NAME + vehicle group when compared to the control + vehicle group, but the treatment with sodium nitrite enhanced both plasma nitrate and NOx concentrations in both the control and l-NAME nitrite-treated groups (P < 0.05; Fig. 2b, c, respectively).

Plasma nitrite, plasma nitrate, and plasma NOx (nitrate + nitrite) levels at end of treatments. Panel a shows plasma nitrite concentration (nmol/l) assessed by reductive chemiluminescence. Panel b shows plasma nitrate concentrations assessed by Griess reagent. Panel c shows plasma NOx (nitrate + nitrite) concentrations. Data are shown as mean ± SEM (n = 8 per group). *P < 0.05 versus control + vehicle group
Effects of sodium nitrite treatment on vascular ROS production
We assessed ROS production by using the sensitive fluorescent dye dihydroethidine (DHE) in aortic slices from the animals. While DHE oxidation was significantly increased in the l-NAME + vehicle group when compared to the control + vehicle group (P < 0.05, Fig. 3a, b), nitrite treatment exerted no significant effects on DHE oxidation, suggesting that the treatment with nitrite does not affect l-NAME-induced oxidative stress (P > 0.05, Fig. 3a, b).

Effects of nitrite treatment on in situ vascular O2 •− production measured by dihydroethidium (DHE) fluorescence. Panel a shows the fluorescence intensity in each experimental group at the end of period of treatment. Panel b shows representative photomicrographs (×400) of aortic arteries incubated with the O2 •−-sensitive dye DHE, which produces red fluorescence when oxidized. Data are shown as mean ± SEM (n = 5 per group). *P < 0.05 versus control + vehicle group
Effects of sodium nitrite on aortic xanthine oxidoreductase expression
The analysis of xanthine oxidoreductase expression in the aortas showed significant increases in xanthine oxidoreductase expression in the l-NAME hypertensive rats when compared to the control + vehicle group (P < 0.05; Fig. 4a, b). The treatment with nitrite exerted no significant effects on xanthine oxidoreductase expression (P > 0.05; Fig. 4a, b).

l-NAME-induced hypertension increases aortic expression of xanthine oxidoreductase (XOR). Panel a shows a graph with the densitometric analysis for xanthine oxidoreductase normalized by β-actin content. Panel b shows a representative western blotting gel showing the aortic expression of xanthine oxidoreductase at the end of treatment. Data are shown as mean ± SEM (n = 6–7 per group) *P < 0.05 versus control + vehicle group
Xanthine oxidoreductase contributes for acute hypotensive effects of sodium nitrite
Because we found increased xanthine oxidoreductase expression in aortic tissues from l-NAME hypertensive rats, we decided to evaluate if oxypurinol (a xanthine oxidoreductase inhibitor) could attenuate the hypotensive effects of sodium nitrite. The infusion of vehicle or oxypurinol (25 mg/kg) alone for 10 min induced no significant changes in MAP (MAP before and after infusions: 142 ± 6 and 142 ± 7 mmHg, respectively, for vehicle-treated, and 138 ± 5 and 140 ± 6 mmHg, respectively, for oxypurinol-treated; both P > 0.05). Sodium nitrite infused 15 min after vehicle induced significant, dose-dependent hypotension in unanesthetized freely moving rats (P < 0.05; Fig. 5). The hypotensive responses to sodium nitrite were significantly attenuated when sodium nitrite was infused 15 min after oxypurinol (P < 0.05; Fig. 5).

Changes in mean arterial pressure (MAP) measured in unanesthetized free-moving hypertensive rats in response to increasing doses of sodium nitrite. The rats received l-NAME (100 mg/kg, by gavage) to increase MAP by approximately 40 mmHg, followed 30 min later by vehicle or oxypurinol (25 mg/kg; i.v.). Then 15 min later, the changes in MAP in response to intravenous injections of vehicle (saline) or increasing doses of sodium nitrite (1, 3, 10, 30, and 100 μmol/kg) were recorded. Data are shown as mean ± SEM (n = 4–6 per group). *P < 0.001 for oxypurinol versus vehicle at each and every dose of sodium nitrite
Discussion
In this study, we demonstrated that (1) a single pharmacological dose of sodium nitrite exerts antihypertensive effects in l-NAME hypertensive rats. (2) This antihypertensive effect was associated with increased concentrations of NO metabolites, and minor changes in vascular xanthine oxidoreductase expression and superoxide production. (3) The inhibition of xanthine oxidoreductase by oxypurinol attenuated the acute hypotensive effects of nitrite. Together, our data suggest that nitrite exerts antihypertensive effects in l-NAME hypertensive rats and provide evidence that xanthine oxidoreductase plays an important role in this effect.
Nitrite has been described as an important alternative to NO formation in vivo, and as a result, this molecule became a potential candidate in the management of hypertension. Indeed, we have demonstrated that treatment with sodium nitrite restores NO metabolites in renovascular hypertension and exerts important antihypertensive and antioxidant effects (Montenegro et al. 2011, 2012). Carlström et al. reported similar findings using nitrate instead of nitrite in a salt-induced hypertension model (Carlström et al. 2011). In the present study, we expanded these previous findings and evaluated if nitrite treatment would revert the hypertension induced by l-NAME. We found significant antihypertensive effects after the first week of treatment with a single oral dose of nitrite, and this effect improved with time. Nitrite treatment was started 2 weeks after hypertension was induced, so that we would make sure that the animals were really hypertensive before the nitrite treatment was started. These findings are consistent with a previous report by Tsuchiya et al. showing that supplementation of drinking water with nitrite attenuated the increases in blood pressure caused by l-NAME (Tsuchiya et al. 2005). These beneficial antihypertensive effects were associated with increased concentrations of markers of NO bioavailability. While no changes were found in plasma NOx (nitrite + nitrate) in the l-NAME hypertensive rats, plasma nitrite levels were significantly lower than the controls, consistent with endothelial dysfunction induced by NO synthase inhibition by l-NAME. As expected, the nitrite treatment induced a significant increase in both plasma NOx and plasma nitrite levels.
The NO deprivation caused by l-NAME treatment increased the in situ production of reactive oxygen species in aortas, consistent with the alterations found in hypertension models. However, these alterations were not reverted by nitrite treatment, suggesting that nitrite exerted minor antioxidant effects (if any) in l-NAME-induced hypertension. This finding contrasts with those reported in the renovascular hypertension model (two kidney, one clip), which showed reversible oxidative stress after nitrite treatment (Montenegro et al. 2012). Indeed, the antioxidant effects of nitrite involve NADPH oxidase inhibition, which is upregulated in the renovascular hypertension model (Montenegro et al. 2011).
We found increased xanthine oxidoreductase expression in the aortas from l-NAME hypertensive rats, which were not reverted by nitrite treatment. This particular finding prompted us to believe that xanthine oxidoreductase could be using nitrite as a substrate for NO production, rather than simply being a source or reactive oxygen species. Indeed, previous studies reported that xanthine oxidoreductase uses nitrite as a substrate under ischemia/reperfusion conditions (Tripatara et al. 2007; Webb et al. 2004, 2008). Moreover, Gosh et al. have recently reported that the beneficial effects of nitrite are associated with elevated erythrocytic xanthine oxidoreductase expression and xanthine oxidoreductase-dependent nitrite reductase activity (Ghosh et al. 2013). Therefore, we decided to examine whether oxypurinol, a xanthine oxidoreductase inhibitor, would attenuate the hypotensive effects elicited by intravenous administration of sodium nitrite in l-NAME hypertensive rats. While sodium nitrite induced dose-dependent hypotensive responses in control animals, pretreatment with oxypurinol significantly attenuated the hypotensive responses to nitrite, suggesting that xanthine oxidoreductase plays an important role in the hypotensive responses to sodium nitrite. Our findings agree with previous studies showing that xanthine oxidoreductase is responsible for the metabolism of nitrite to NO within the lung, and therefore explains the protective effect of nitrite against the development of pulmonary arterial hypertension (Zuckerbraun et al. 2010).
Although acute affects do not necessarily correspond to chronic effects, our findings corroborate previous studies showing increased vascular expression of xanthine oxidoreductase in hypertension (Ghosh et al. 2013). Together, these results support the idea that increased xanthine oxidoreductase activity may be a compensatory mechanism to enhance the responses to nitrite and increase NO bioactivity in hypertension, rather than only promoting deleterious effects.
Some details of this study deserve some comments. First, we found that treatment with the NO synthase inhibitor l-NAME decreased plasma nitrite concentrations by more than 60 %, whereas no significant decreases (<10 %) were found in plasma nitrate concentrations. These results are consistent with the idea that nitrate concentration does not reflect NO synthase activity and depends on a variety of factors. Conversely, nitrite concentrations, rather than nitrate concentrations, reflect endogenous NO formation in vivo (Kleinbongard et al. 2003; Metzger et al. 2006). Second, the antihypertensive effects of sodium nitrite in l-NAME-induced hypertension were shown in previous studies (Amin et al. 2012; Kanematsu et al. 2008). However, sodium nitrite treatment started before the animals really became hypertensive in those studies, whereas we started sodium nitrite after the animals were clearly hypertensive, and therefore our results show a therapeutic effect of sodium nitrite. Third, we used oxypurinol, the major active product of allopurinol, to inhibit xanthine oxidoreductase in the present study in order to test how the acute inhibition of xanthine oxidoreductase affects the responses to sodium nitrite. In parallel with allopurinol, this drug may inhibit other enzymes involved in purine and pyrimidine metabolism (Takano et al. 2005) and therefore exerts nonspecific biological effects that have not been examined in the present study. However, we used oxypurinol only to examine acute effects, and therefore nonspecific effects may be less relevant under the conditions using oxypurinol. Moreover, a number of previous studies have used oxypurinol or allopurinol to examine the role of xanthine oxidoreductase in the formation of NO from nitrite (Zuckerbraun et al. 2010; Ghosh et al. 2013).
In conclusion, our results show that nitrite exerts antihypertensive effects in l-NAME hypertensive rats and provide evidence that xanthine oxidoreductase participates in this beneficial antihypertensive effects.
References
Alzawahra WF, Talukder MA, Liu X, Samouilov A, Zweier JL (2008) Heme proteins mediate the conversion of nitrite to nitric oxide in the vascular wall. Am J Physiol Heart Circ Physiol 295(2):H499–H508
Amaral JH, Montenegro MF, Pinheiro LC, Ferreira GC, Barroso RP, Costa-Filho AJ, Tanus-Santos JE (2013) Tempol enhances the antihypertensive effects of sodium nitrite by mechanisms facilitating nitrite-derived gastric nitric oxide formation. Free Radic Biol Med 65C:446–455
Amin A, Choi SK, Osman-Elazeik Y, Badr El-Din NK, Kevil CG, Navar LG, Kadowitz P, Trebak M, Matrougui K (2012) Sodium nitrite therapy rescues ischemia-induced neovascularization and blood flow recovery in hypertension. Pflugers Arch 464(6):583–592
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87(10):840–844
Carlström M, Persson AEG, Larsson E, Hezel M, Scheffer PG, Teerlink T, Weitzberg E, Lundberg JO (2011) Dietary nitrate attenuates oxidative stress, prevents cardiac and renal injuries, and reduces blood pressure in salt-induced hypertension. Cardiovasc Res 89:574–585
Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO 3rd, Gladwin MT (2003) Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9(12):1498–1505
Feelisch M, Rassaf T, Mnaimneh S, Singh N, Bryan NS, Jourd'Heuil D, Kelm M (2002) Concomitant s-, n-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of no in vivo. Faseb J 16(13):1775–1785
Ghosh SM, Kapil V, Fuentes-Calvo I, Bubb KJ, Pearl V, Milsom AB, Khambata R, Maleki-Toyserkani S, Yousuf M, Benjamin N, Webb AJ, Caulfield MJ, Hobbs AJ, Ahluwalia A (2013) Enhanced vasodilator activity of nitrite in hypertension: critical role for erythrocytic xanthine oxidoreductase and translational potential. Hypertension 61(5):1091–1102
Gomes HF, Palei AC, Machado JS, da Silva LM, Montenegro MF, Jordao AA, Duarte G, Tanus-Santos JE, Cavalli RC, Sandrim VC (2013) Assessment of oxidative status markers and no bioavailability in hypertensive disorders of pregnancy. J Hum Hypertens 27(6):345–348
Kanematsu Y, Yamaguchi K, Ohnishi H, Motobayashi Y, Ishizawa K, Izawa Y, Kawazoe K, Kondo S, Kagami S, Tomita S, Tsuchiya K, Tamaki T (2008) Dietary doses of nitrite restore circulating nitric oxide level and improve renal injury in l-NAME-induced hypertensive rats. Am J Physiol Renal Physiol 295(5):F1457–F1462
Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, Scheeren T, Godecke A, Schrader J, Schulz R, Heusch G, Schaub GA, Bryan NS, Feelisch M, Kelm M (2003) Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med 35(7):790–796
Kozlov AV, Staniek K, Nohl H (1999) Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett 454(1–2):127–130
Kozlov AV, Dietrich B, Nohl H (2003) Various intracellular compartments cooperate in the release of nitric oxide from glycerol trinitrate in liver. Br J Pharmacol 139(5):989–997
Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M (2001) Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA 98(22):12814–12819
Lundberg JO, Weitzberg E (2013) Biology of nitrogen oxides in the gastrointestinal tract. Gut 62(4):616–629
Lundberg JO, Weitzberg E, Cole JA, Benjamin N (2004) Nitrate, bacteria and human health. Nat Rev Microbiol 2(7):593–602
Lundberg JO, Weitzberg E, Gladwin MT (2008) The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7(2):156–167
Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR Jr, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E (2009) Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol 5(12):865–869
McKnight GM, Smith LM, Drummond RS, Duncan CW, Golden M, Benjamin N (1997) Chemical synthesis of nitric oxide in the stomach from dietary nitrate in humans. Gut 40(2):211–214
McKnight GM, Duncan CW, Leifert C, Golden MH (1999) Dietary nitrate in man: friend or foe? Br J Nutr 81(5):349–358
Metzger IF, Sertorio JT, Tanus-Santos JE (2006) Relationship between systemic nitric oxide metabolites and cyclic GMP in healthy male volunteers. Acta Physiol (Oxf) 188(2):123–127
Moncada S, Higgs A (1993) The l-arginine-nitric oxide pathway. N Engl J Med 329(27):2002–2012
Montenegro MF, Pessa LR, Gomes VA, Desta Z, Flockhart DA, Tanus-Santos JE (2009) Assessment of vascular effects of tamoxifen and its metabolites on the rat perfused hindquarters vascular bed. Basic Clin Pharmacol Toxicol 104(5):400–407
Montenegro MF, Neto-Neves EM, Dias-Junior CA, Ceron CS, Castro MM, Gomes VA, Kanashiro A, Tanus-Santos JE (2010) Quercetin restores plasma nitrite and nitroso species levels in renovascular hypertension. Naunyn Schmiedebergs Arch Pharmacol 382(4):293–301
Montenegro MF, Amaral JH, Pinheiro LC, Sakamoto EK, Ferreira GC, Reis RI, Marcal DM, Pereira RP, Tanus-Santos JE (2011) Sodium nitrite downregulates vascular NADPH oxidase and exerts antihypertensive effects in hypertension. Free Radic Biol Med 51(1):144–152
Montenegro MF, Pinheiro LC, Amaral JH, Marcal DM, Palei AC, Costa-Filho AJ, Tanus-Santos JE (2012) Antihypertensive and antioxidant effects of a single daily dose of sodium nitrite in a model of renovascular hypertension. Naunyn Schmiedebergs Arch Pharmacol 385(5):509–517
Pinheiro LC, Montenegro MF, Amaral JH, Ferreira GC, Oliveira AM, Tanus-Santos JE (2012) Increase in gastric pH reduces hypotensive effect of oral sodium nitrite in rats. Free Radic Biol Med 53(4):701–709
Rassaf T, Flogel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J (2007) Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res 100(12):1749–1754
Sandrim VC, Montenegro MF, Palei AC, Metzger IF, Sertorio JT, Cavalli RC, Tanus-Santos JE (2010) Increased circulating cell-free hemoglobin levels reduce nitric oxide bioavailability in preeclampsia. Free Radic Biol Med 49(3):493–500
Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA (2005) Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci 76(16):1835–1847
Tripatara P, Patel NS, Webb A, Rathod K, Lecomte FM, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C (2007) Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J Am Soc Nephrol 18(2):570–580
Tsuchiya K, Kanematsu Y, Yoshizumi M, Ohnishi H, Kirima K, Izawa Y, Shikishima M, Ishida T, Kondo S, Kagami S, Takiguchi Y, Tamaki T (2005) Nitrite is an alternative source of no in vivo. Am J Physiol Heart Circ Physiol 288(5):H2163–H2170
Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A (2004) Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA 101(37):13683–13688
Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ, Lecomte FM, Perrett D, Raimondo C, Khoshbin E, Ahmed Z, Uppal R, Benjamin N, Hobbs AJ, Ahluwalia A (2008) Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res 103(9):957–964
Zuckerbraun BS, Shiva S, Ifedigbo E, Mathier MA, Mollen KP, Rao J, Bauer PM, Choi JJ, Curtis E, Choi AM, Gladwin MT (2010) Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation 121(1):98–109
Acknowledgments
This study was funded by Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP-Brazil) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil).
Conflicts of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Montenegro, M.F., Pinheiro, L.C., Amaral, J.H. et al. Vascular xanthine oxidoreductase contributes to the antihypertensive effects of sodium nitrite in l-NAME hypertension. Naunyn-Schmiedeberg's Arch Pharmacol 387, 591–598 (2014). https://doi.org/10.1007/s00210-014-0970-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-014-0970-8