
Abstract
Xanthomonas campestris pv. campestris is a bacterial pathogen and the causal agent of black rot in crucifers. In this study, a clpX mutant was obtained by EZ-Tn5 transposon mutagenesis of the X. campestris pv. campestris. The clpX gene was annotated to encode ClpX, the ATP-binding subunit of ATP-dependent Clp protease. The clpX mutant exhibited reduced bacterial attachment, extracellular enzyme production and virulence. Mutation of clpX also resulted in increased sensitivity to a myriad of stresses, including heat, puromycin, and sodium dodecyl sulfate. These altered phenotypes of the clpX mutant could be restored to wild-type levels by in trans expression of the intact clpX gene. Proteomic analysis revealed that the expression of 211 proteins differed not less than twofold between the wild-type and mutant strains. Cluster of orthologous group analysis revealed that these proteins are mainly involved in metabolism, cell wall biogenesis, chaperone, and signal transduction. The reverse transcription quantitative real-time polymerase chain reaction analysis demonstrated that the expression of genes encoding attachment-related proteins, extracellular enzymes, and virulence-associated proteins was reduced after clpX mutation. The results in this study contribute to the functional understanding of the role of clpX in Xanthomonas for the first time, and extend new insights into the function of clpX in bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phytopathogenic bacteria of the Xanthomonas genus cause severe diseases in approximately 400 host plants, including a wide variety of economically important crops (Ryan et al. 2011). Xanthomonas campestris, the most dominant species within the Xanthomonas genus, has at least 141 pathovars capable of infecting a wide range of plants with agronomical interest (He and Zhang 2008). X. campestris pv. campestris, the causal agent of black rot disease, infects cruciferous plants and is responsible for major yield and quality losses in Brassica crops, such as broccoli, cabbage, cauliflower, and kale (He and Zhang 2008; Vicente and Holub 2013).
X. campestris pv. campestris is known to produce a range of factors such as exopolysaccharides and extracellular enzymes (cellulase, mannanase, pectinase, and protease), which collectively have an important role in pathogenesis (Chan and Goodwin 1999; Dow et al. 2003). The exopolysaccharides can cause tissue necrosis and leaf wilting by obstructing the water flow in xylem vessels (He and Zhang 2008; Buttner and Bonas 2010), and the extracellular enzymes can degrade plant cell components and play a role in facilitating plant tissue maceration (Chan and Goodwin 1999). In addition, experimental evidences indicate that biofilm formation and bacterial adhesion are associated with the virulence of X. campestris pv. campestris; as shown for genes encoding extracellular mannanase (manA), isocitrate dehydrogenase (icd2), major outer-membrane protein (mopB), tail-specific protease (prc), and UTP-glucose-1-phosphate uridylyltransferase (galU), mutation in these genes has an impact in biofilm formation or bacterial attachment and leads to an attenuation of bacterial virulence (Dow et al. 2003; Chen et al. 2010; Liao et al. 2014, 2016; Chiang et al. 2017).
To identify novel genes related to X. campestris pv. campestris biofilm formation, a previously constructed mutant library of X. campestris pv. campestris strain 17 (hereinafter, Xcc17) (Liao et al. 2016) was screened in a biofilm formation assay. Here, one mutant designated as J67 exhibiting a reduced ability in bacterial attachment was characterized. The mutant strain J67 was found to have a transposon inserted in the locus_tag AAW18_RS04855 in the recently completed genome sequence of Xcc17 (GenBank accession no. NZ_CP011946) (Liu et al. 2015), which was annotated to encode ClpX, the ATP-binding subunit of ATP-dependent Clp protease.
The ATP-dependent Clp protease is widely conserved within the bacterial domain and has a multitude of functions in bacteria, such as protein quality control, stress tolerance, and virulence factor expression (Malik and Brotz-Oesterhelt 2017). Clp protease consists of two functional units with separate activities: ATPase-active chaperone rings and a proteolytic core (Kress et al. 2009; Schmidt et al. 2009). The ATPase component is responsible for substrate recognition, unfolding, and translocation into the proteolytic component (Gottesman 2003; Kress et al. 2009; Schmidt et al. 2009). The proteolytic component possesses multiple active sites of serine or threonine-type, which enable protein hydrolysis (Kress et al. 2009). The existence of two different proteolytic cores (ClpP and ClpQ) and multiple different chaperone rings (ClpA, ClpC, ClpE, ClpX, and ClpY) results in an inventory of several ATP-dependent Clp proteases (Kress et al. 2009). In Escherichia coli, various ATP-dependent Clp proteases exist, including ClpAP, ClpXP, and ClpYQ (also called HslUV) (Porankiewicz et al. 1999; Gottesman 2003; Kress et al. 2009; Schmidt et al. 2009). The ClpP is classified as a serine-type protease and associates with either ClpA or ClpX, whereas the ClpQ (HslV) has a threonine active site and associates with ClpY (HslU) (Porankiewicz et al. 1999). An adaptor protein, such as ClpS, may also be associated with ClpA and influences the ClpAP complex (Erbse et al. 2006). ClpXP is the most ubiquitous of Clp proteases as it is found in almost all bacteria (Kress et al. 2009). ClpYQ exists alongside ClpAP in most proteobacteria and is also found in certain Gram-positive bacteria (Kress et al. 2009). The ClpP protease core can also interact with ClpC and ClpE, which is found in Gram-positive bacteria (Kress et al. 2009). In the fully sequenced genomes of several X. campestris pv. campestris strains, such as ATCC33913 (da Silva et al. 2002), 8004 (Qian et al. 2005) as well as Xcc17 (Liu et al. 2015), several putative Clp protein encoding genes (such as clpA, clpX, clpY, clpP, and clpQ) have been annotated, whereas none of them have been studied. Here, the X. campestris pv. campestris clpX gene was characterized.
Materials and methods
Bacterial strains, media and growth conditions
Escherichiacoli strain DH5α ECOS™ 101 (Yeastern Biotech, Taiwan) was used as cloning host. X. campestris pv. campestris strain Xcc17 is a virulent wild-type strain isolated from the leaves of the infected plants in Taiwan (Yang and Tseng 1988). J67, a derivative of Xcc17, was a clpX mutant constructed in this study. E. coli strains were cultured in Luria–Bertani (LB) medium (Sambrook et al. 1989) at 37 °C. X. campestris pv. campestris strains were grown in LB medium or XOLN medium (Fu and Tseng 1990) at 28 °C. Glucose or glycerol were supplemented (2%) as necessary. Liquid cultures were grown with shaking at 180 rpm. Solid media contained 1.5% agar. Ampicillin (50 μg/mL), kanamycin (50 μg/mL), and tetracycline (15 μg/mL) were supplemented as necessary.
Recombinant DNA techniques
Bacterial genomic and plasmid DNAs were extracted by the Wizard® Genomic DNA Purification Kit (Promega) and the Gene-Spin™ Miniprep Purification Kit (Protech), respectively. Polymerase chain reaction (PCR) was done as previously described (Hsiao et al. 2005). Restriction endonuclease digestion, DNA ligation, agarose gel electrophoresis, and transformation were performed by standard protocols as described previously (Sambrook et al. 1989).
Generation of transposon mutants and determination of transposon insertion site
X. campestris pv. campestris transposon mutants were generated using EZ-Tn5™ < R6Kγori/KAN-2 > Tnp Transposome Kit (Epicentre) as described by Liao et al. (2016). The transposon insertion site was determined by “rescue cloning”, as described by Liu et al. (2018).
Construction of plasmid for complementation
To construct the plasmid for complementation of the clpX mutant, the fragment encompassing the upstream 52-bp region, the entire coding region and 6 bp downstream of the clpX stop codon in Xcc17 were amplified by PCR with primers clpX-CF (5′-AAGCTTCTCGAAATCGAACCCCCGTGC-3′, HindIII restriction site underlined) and clpX-CR (5′-TCTAGAGGCCTCAGTCGCCAGACGC-3′, XbaI restriction site underlined). The product was cloned into cloning vector yT&A (Yeastern, Taiwan). After sequence confirmation, the fragment containing clpX (1345-bp) was excised with HindIII and XbaI, then ligated into broad-host-range vector pRK415 (Keen et al. 1988) to generate pRKclpX. For complementation, pRKclpX was transferred into the clpX mutant J67 by electroporation (2.5 kV, 25 μF, 200 Ω), yielding the complementary strain J67c. For comparison, Xcc17 and J67 carrying the empty vector pRK415 were generated in parallel and were designated as Xcc17v and J67v, respectively.
Bacterial attachment analysis, extracellular enzyme production assay, and virulence test
Bacterial attachment was analyzed by examining the ability of cells to adhere to the 96-well polystyrene microtiter plates (Nunc) and was quantified by staining of the attached cells with crystal violet, as the previously described method (Liao et al. 2016). Quantitative analysis of adhesion was also examined on glass surface as described previously (Ryan et al. 2007) with some modifications. Briefly, bacteria from overnight cultures were diluted to an OD550 of 0.35 in XOLN medium containing 2% glucose. Then, 1.0 mL of cultures prepared as aforementioned was added in borosilicate glass tubes (10 cm × 1.3 cm, Kimble) and incubated at 28 °C for 24 h without shaking. After incubation, the medium and unattached cells were removed carefully. Then, surface-attached bacteria were washed with 1.5 mL distilled water (three times) and stained with 1.5 mL crystal violet (0.1%) for 5 min at room temperature. The free dye was removed and the tube was washed three times with distilled water. The dye that was incorporated into the attached cells was solubilized with 2.0 mL ethanol (95%) and quantified by spectrophotometry at 595 nm. Bacterial adhesion to leaves surface was conducted as described previously (Hsiao et al. 2012). The experiments were carried out at least three times.
Extracellular enzyme production was evaluated by inoculating bacteria strains on substrate-supplementary agar plates and was detected by monitoring the appearance of hydrolytic zones around bacterial colonies according to previously described methods (Hsiao et al. 2011) with minor modifications. Briefly, 3 μL of diluted overnight culture (OD550 = 1) was deposited onto the surface of XOLN plates containing the appropriate substrates. The supplemented substrates were carboxymethyl cellulose (0.5%, for cellulase), locus bean gum (0.2%, for mannanase), sodium polypectate (0.2%, for pectinase), and skim milk (1%, for protease). Activity of extracellular enzymes was assayed after incubation at 28 °C for 2 days as described previously (Hsiao et al. 2011). Each experiment was done at least three times.
Bacterial virulence was assayed using the scissors-clipping method as described (Liao et al. 2016). The lesion length was measured and the disease symptom was photographed 14 days after inoculation. Three independent experiments, each with six replicates, were done.
Stress tolerance assay
For temperature tolerance assay, bacteria were grown overnight and diluted with fresh LB medium to a starting density of OD550 = 0.5 (5 × 108 cells/mL), and then followed a tenfold dilution. Five microliters of resulting cultures (5 × 108 to 5 × 105 cells/mL) were spotted on LB plates and incubated at either 28 °C or 37 °C for 3 days. To evaluate the tolerance of bacteria to H2O2, polymyxin B, puromycin, and sodium dodecyl sulfate (SDS), disk diffusion assays were performed. Bacterial strains were grown in LB medium until the OD550 of the cultures reached about 0.6 (mid-exponential phase). The mid-exponential-phase cultures were collected and suspended in fresh LB broth to yield an OD550 = 1. One milliliter of resulting cultures was spread on LB plate and a disc (6 mm) containing 10 μl of H2O2 (3%), polymyxin B (5 mg/mL), puromycin (5 mg/mL), and SDS (5%) was placed on top. The plates were incubated at 28 °C for 3 days and the growth-inhibition zones surrounding the discs were measured. All assays were performed at least three times.
RNA extraction, reverse transcription (RT), and quantitative real-time PCR (qPCR)
Bacteria were cultivated in XOLN–glycerol medium to mid-exponential phase (OD550 = 0.6) and the cells were collected for RNA extraction and RT-qPCR analysis as previously described by Liao et al. (2019). Briefly, total RNA was extracted from cells of Xcc17 and J67 using the RNeasy Mini Kit (Qiagen), subjected to DNase I treatment using the RNase-Free DNase Set (Qiagen), and reverse-transcribed to cDNA using the iScript™ cDNA Synthesis Kit (BIO-RAD). Subsequent qPCR was then performed in a CFX96 Real Time PCR system (BIO-RAD) using the iQ™ SYBR® Green Supermix (BIO-RAD). Each assay was carried out in triplicate with 16S rRNA gene as a reference gene. Primer sequences of the tested target genes are listed in Table S1. The fold change for transcript was calculated by the 2−ΔΔCt method.
Cell extract preparation and gel electrophoresis
X. campestris pv. campestris strains were cultivated in XOLN–glycerol medium. After reaching an OD550 of approximately 0.6 (mid-exponential phase), the cells were harvested by centrifugation at 10,000g for 5 min at 4 °C. The cell pellets were washed with 10 mM Tris–HCl (pH 7.5) three times. After washing, proteins from the pelleted cells were prepared using ExtractPRO™ Protein Extraction Reagent (Visual Protein, Taiwan), according to the manufacturer’s instructions. The protein concentration was estimated using the Bradford method. Equal amounts of protein (15 μg) were loaded and resolved on 12.5% SDS-PAGE. After electrophoresis, the gel was stained with the VisPRO 5 min Protein Stain Kit (Visual Protein). After staining, the gel was washed with distilled water and stored at 4 °C until processing for in-gel digestion.
In-gel digestion and peptide preparation
In-gel digestion was performed according to a method described previously (Liao et al. 2019). Briefly, the gel was incubated with sequencing-grade trypsin (6.25 ng/μL) overnight at 25 ºC after reduction with dithiothreitol (10 mM) for 1 h at 56 ºC and alkylation with iodoacetamide (25 mM) in the dark for 45 min at room temperature. The tryptic peptides were then extracted and concentrated as the previously described method (Liao et al. 2019).
Mass spectrometric analysis, mass data searching, protein identification and quantification
Tryptic peptides obtained as described above were separated by nano liquid chromatography (nLC) using the Ultimate 3000 nLC system (Dionex) and then analyzed by tandem mass spectrometry (MS/MS) through the LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific) as described previously (Liao et al. 2019). BioWorks 3.3 (Thermo Fisher Scientific) was used to interpret the acquired MS/MS spectra. Database search was carried out against the X. campestris pv. campestris strain 8004 database from UniProt (4252 sequences; 1 445 974 residues). The search parameters were: (1) 10 ppm for the precursor mass tolerance (2) 0.6 Da for fragment mass tolerance (3) variable modification: oxidation of methionine (4) fixed modification: carbamidomethylation of cysteine (5) allowing two missed cleavage sites, and (6) the minimum number of tryptic termini was 1. Scaffold (versions 1.07 and 2.01) (Proteome software) was used to validate peptide and protein identification. Peptides with a probability greater than 95% and proteins with a greater than 99% probability were accepted. Protein quantification was estimated by spectral counting as described previously (Liao et al. 2019) and proteins with a 2.0-fold change between wild type Xcc17 and clpX mutant J67 was considered as differentially expressed proteins. The cluster of orthologous group (COG) analysis of the identified proteins was conducted using the Xanthomonas Genome Browser (https://xgb.leibniz-fli.de/cgi/index.pl) against X. campestris pv. campestris strain 8004.
Statistical analysis
Statistical analysis was performed by two-tailed Student’s t test using the Microsoft Excel statistical package. For each quantification experiment, at least three times were carried out and p value less than 0.05 was considered statistically significant.
Results and discussion
The clpX gene is involved in bacterial attachment and extracellular enzyme production
Through bulk screening of the X. campestris pv. campestris mutant library generated previously (Liao et al. 2016; Liu et al. 2018) for bacterial attachment, the J67 mutant that had a reduced ability to form biofilms in the polystyrene 96-well microtiter plates was obtained. To identify the transposon–inserted gene, the transposon flanking region was rescued by the rescue cloning method as described by the manufacture (Epicentre) and sequenced by primer R6KAN-2 RP-1 (Liu et al. 2018). A blast search using the rescued DNA sequence against the Xcc17 genome (GenBank accession no. NZ_CP011946) revealed that the transposon insertion was mapped to a site within the locus_tag AAW18_RS04855 in the genome sequence of Xcc17, which predicts to encode ClpX, the ATP-binding subunit of ATP-dependent Clp protease. The X. campestris pv. campestris clpX gene encodes a protein consisting of 428 amino acid residues and has a theoretical molecular mass of 47,136 Da with a predicted pI of 5.44 (calculated using the Compute pI/MW tool from ExPASy; https://www.expasy.org/). The transposon insertion was localized at a distance 383 bp from the site of ATG translation start of clpX gene.
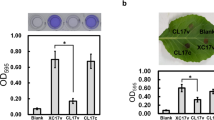
Limited information is available about the role of ClpX in bacterial attachment. In Streptococcus mutans, the clpX mutant strain revealed less biofilm mass in the presence of glucose, whereas the biofilm formation was enhanced when sucrose was provided (Kajfasz et al. 2009). In Burkholderia cenocepacia, no considerable differences in biofilm formation were observed between the clpX mutant and the parental strain (Veselova et al. 2016). In our preliminary analysis, the mutant strain J67 showed a reduced attachment on polystyrene plates compared with the parental strain Xcc17 (data not shown). To determine whether the reduced ability to form biofilms in J67 was specifically attributable to ClpX, a complemented strain named J67c was generated by introducing pRKclpX (with clpX gene cloned into pRK415) into the transposon mutant J67. The parental strain Xcc17 and the transposon mutant J67 were also transformed with empty vector pRK415 (named Xcc17v and J67v, respectively) to control for any effects of the vector itself. Biofilm formation was assayed on polystyrene microtiter plate (Fig. 1a), borosilicate glass tube (Fig. 1b), and leaf surface (Fig. 1c). It was revealed that introduction of clpX mutant J67 with pRKclpX but not pRK415 restores attachment ability to those of the parent strain. These results indicated that the reduced bacterial attachment of J67 is due to a mutation in the clpX gene.

Effects of mutation of clpX on cell attachment to polystyrene plates (a), glass tubes (b), and cabbage leaf surfaces (c) in X. campestris pv. campestris. Strains to be tested were grown overnight, cells were washed and diluted using fresh XOLN medium supplemented with glucose, and were assayed as described in the “Material and methods” section. Xcc17v: wild-type strain Xcc17 carrying empty vector pRK415; J67v: clpX mutant J67 carrying pRK415; J67c: complemented clpX mutant; Blank: XOLN medium supplemented with glucose without inoculation of bacteria. Values presented are the mean ± standard deviation (n = 3)
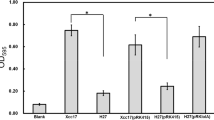
To get more understanding of ClpX in X. campestris pv. campestris, the production of a range of virulence factors including exopolysaccharide and extracellular enzymes were determined to assess whether ClpX influences the synthesis of these virulence determinants. The results revealed that the exopolysaccharide yields produced by the mutant were similar to those of the wild type (data not shown). However, the extracellular enzymes produced by the mutant were lower than the wild type. As depicted in Fig. 2, the clear zones of hydrolytic activity surrounding J67v colonies grown on plates supplemented with carboxymethyl cellulose, locust bean gum, sodium polypectate, and skim milk, were smaller than those observed for the Xcc17v and J67c. These results indicated that the clpX gene is required for the ability to synthesize extracellular enzymes (including cellulase, mannanase, pectinase, and protease).

Effects of mutation of clpX on extracellular enzyme activities in X. campestris pv. campestris. The activity of extracellular cellulase, mannanase, pectinase, and protease was evaluated using the substrate-supplemented plate assay as described in the “Material and methods”section. The tested strains included: 1 (Xcc17v, wild-type strain Xcc17 carrying empty vector pRK415); 2 (J67v, clpX mutant J67 carrying pRK415); and 3 (J67c, complemented clpX mutant). The diameter of the colony and hydrolysis zone of each strain were measured and the ratio of hydrolysis zone size-to-colony diameter is calculated. Values presented are the mean ± standard deviation (n = 3)
The observations showing reduced bacterial attachment (Fig. 1) and extracellular enzyme production (Fig. 2) in clpX mutant prompted us to test whether mutation of clpX affected the expression of genes associated with bacterial attachment and extracellular enzyme synthesis. To test the possibility, the expression of four attachment-related genes (galE, galU, ha, and yapH), along with four genes encoding extracellular enzymes (engA, manA, pelA, and prt1), was compared in strains Xcc17 and J67. Consistent with the phenotypic observations, the expressions of these tested genes were all reduced in the mutant J67 compared to the wild-type Xcc17 (Table 1), analyzed by RT-qPCR. These results indicated that a mutation in clpX affects the transcription of genes involved in bacterial attachment and extracellular enzymes. As the RT-qPCR results revealed that mutation in clpX reduced the expression of these genes, it is implying that ClpX might affect the expression of these genes at transcriptional level and that the reduced bacterial attachment and extracellular enzyme production of the clpX mutant may be attributable to the reduced expression of these genes.
ClpX has been implicated in expression of extracellular virulence factors in several organisms. In Staphylococcus aureus, the amount of both α-hemolysin and β-hemolysin as well as the activity of extracellular protease are decreased in the clpX mutant, and ClpX affects α-hemolysin production by reducing the transcription of the α-hemolysin encoding gene hla (Frees et al. 2003). In Dickeya dadantii, inactivation of the clpX led to reduce the production of pectate lyase (Li et al. 2010).
The clpX gene is essential for bacterial virulence
As ClpX was found to be involved in biofilm formation and virulence factor synthesis, experiment was conducted to evaluate whether ClpX plays a role in pathogenesis. In the pathogenicity test, the clpX mutant showed decreased virulence to the host plant cabbage. The lesion lengths caused by Xcc17 (wild type) and J67c (complementary strain) 14 days after inoculation were approximately 1.67 cm and 1.75 cm, respectively, whereas the lesion lengths caused by J67 (clpX mutant) were approximately 1.04 cm (Fig. 3). These results indicated that the mutation in the clpX gene attenuated the virulence of X. campestris pv. campestris. Inactivation of the clpX gene is shown to decrease the virulence of various bacteria, such as Bacillus anthracis (McGillivray et al. 2009), B. cenocepacia (Veselova et al. 2016), D. dadantii (Li et al. 2010), and S. aureus (Frees et al. 2003).

Effects of mutation of clpX on virulence of X. campestris pv. campestris in cabbage. a Black rot symptoms caused by X. campestris pv. campestris strains on inoculated leaves of host cabbage plant. Images were taken at 14 day post-inoculation. b Average lesion lengths caused by X. campestris pv. campestris. Values presented are the means ± standard deviations from three repeats, each with six leaves. Significance was determined using the Student t test (* indicates significance at p < 0.05)
The clpX gene is required for growth during heat stress and is important for puromycin and SDS tolerance
As the formation of a biofilm presumably functions as a protective role that allows bacteria to survive under harsh conditions (Dow et al. 2003; Buttner and Bonas 2010), the reduced biofilm formation of the clpX mutant (Fig. 1) suggested that clpX gene might implicated in stress tolerance. To examine whether ClpX contributes to stress adaptation of X. campestris pv. campestris, the sensitivity of the clpX mutant together with the wild-type and complemented strains was tested under a range of stresses, including heat (37 °C), H2O2 (3%), polymyxin B (5 mg/mL), puromycin (5 mg/ml), and SDS (5%). At physiological temperature (28 °C), the growth of all tested strains was similar (Fig. 4a, upper). When bacterial strains were grown at elevated temperature (37 °C), the growth of clpX mutant J67v was inhibited and genetic complementation restored this deficiency (Fig. 4a, down). The clpX mutant did not differ from the wild-type strain in susceptibility to H2O2 and polymyxin B (data not shown). Compared to wild-type Xcc17v, the clpX mutant J67v was more sensitive to puromycin (Fig. 4b) and SDS (Fig. 4c). Taken together, these results demonstrated that ClpX is required for tolerance to heat, puromycin and SDS stresses. Several reports have indicated that clpX mutants of bacteria are more susceptible to various stresses. In the presence of oxidative stress or at low temperature, ClpX is important for growth in S. aureus, whereas inactivation of clpX in S. aureus leads cells to growth at higher temperature or in the presence of a higher puromycin concentration than the wild-type cells (Frees et al. 2003). In B. anthracis, ClpX function protects bacteria against antimicrobial peptide killing (McGillivray et al. 2009).

Effects of mutation of clpX on stress tolerance. Heat tolerance (a) was performed with tenfold dilution of cells spotted on LB plate and incubated at 28 °C or 37 °C for 3 days. Tolerance to puromycin (b) and SDS (c) was conducted by disc diffusion assays in which exponentially growing cells were spread on LB plates and discs containing puromycin (5 mg/mL) or SDS (5%) were placed on top. After 3 days of incubation, the inhibitory-zone diameters for cultures were measured and compared. Values presented are the means ± standard deviations from three repeats. The asterisk (*) indicates p < 0.05. The cultures tested were: Xcc17v (wild-type Xcc17 with empty vector pRK415); J67v (clpX mutant J67 with pRK415); and J67c (complemented clpX mutant)
Inactivation of clpX has broad effects on the proteome of X. campestris pv. campestris
To acquire further insights into additional physiological roles of ClpX in X. campestris pv. campestris, we used quantitative proteome analysis to examine differences in protein expression profile between the wild-type Xcc17 and the clpX mutant J67. In total, 615 and 551 proteins were identified in Xcc17 and J67, respectively (Table S2). Among these proteins, the expression patterns of 211 proteins (155 proteins were downregulated while 56 proteins were upregulated in the clpX mutant) were associated with the presence of ClpX (Fig. 5a). Of these differentially expressed (fold change ≥ 2.0) proteins (1) 99 proteins were only present in the wild type (2) 35 proteins were only present in the clpX mutant, and (3) 56 and 21 proteins were highly expressed in the wild type and clpX mutant, respectively (Fig. 5a). These data suggested that approximating 35% of the identified proteins were affected by clpX in X. campestris pv. campestris. The completely list of differentially expressed proteins identified between the wild-type Xcc17 and the clpX mutant J67 can be found in Table S3. Further, the function of the 211 differentially expressed proteins was predicted and classified using COG analysis. Specifically (1) there were 17 proteins (12 downregulated and 5 upregulated) in the unclassified category; (2) the remaining 194 clpX-influenced proteins were classified into different functional categories; and (3) the majority of altered proteins including proteins related to metabolism (cluster E, G, and P), cell wall biogenesis (cluster M), chaperone (cluster O) and signal transduction (cluster T) (Fig. 5b). These differentially expressed proteins are distributed over all the functional categories of the X. campestris pv. campestris genome, indicating ClpX has a broad physiological role in X. campestris pv. campestris. Identification of such differentially expressed proteins provides new candidates for future studies that will allow assessment of their physiological roles and significance in ClpX-regulated processes.

Differentially expressed proteins identified through proteomic analysis. a Venn diagram presents the numbers of differentially expressed proteins found in Xcc17 (wild type) and J67 (clpX mutant). b COG function analysis of differentially expressed proteins. The black bar and white bar denote proteins in which the expression levels were upregulated and downregulated in Xcc17, respectively. Numbers of proteins in each category are presented on the X-axis. Detailed information concerning the differentially expressed proteins that were identified can be found in Table S2
To our knowledge, only two other reports have examined the proteome of the clpX mutant, which were performed in B. cenocepacia (Veselova et al. 2016) and S. mutans (Jana et al. 2016). Comparison of the proteomes from the wild-type strain and the clpX mutant strain revealed that at least 19 and 53 proteins had altered levels of expression in B. cenocepacia (Veselova et al. 2016) and S. mutans (Jana et al. 2016), respectively. As was the case in B. cenocepacia, the majority of altered proteins in X. campestris pv. campestris clpX mutant were involved in metabolic function and regulation. Contrary to what were seen in B. cenocepacia and S. mutans, it was found that inactivation of clpX results in a higher prevalence of downregulated proteins in X. campestris pv. campestris. While the exact reason for the presence of downregulated proteins in the clpX mutant is not known, it is speculated that some of the upregulated proteins are the substrates for ClpX. Future analysis of these candidates is necessary for identification targets recognized by ClpX.
Consistent with the observation that the clpX mutant showed reduced bacterial attachment and extracellular enzyme production, the protein level expressed by the attachment-related genes (galU, ha, and yapH) and the extracellular enzyme genes (engA and prt1) was lower in J67 by quantitative proteome analysis (Table 1). Similarly, consistent with the observation that the clpX mutant showed a reduction of virulence, the expression of several virulence-related proteins was significantly reduced in the clpX mutant, such as XC_0626, XC_1965, XC_2227, XC_2228, and XC_2699 (Table 1). It was reported that: (1) the homologues of XC_0626 and XC_1965 (XOO4035 and XOO2323) were known to have roles in the pathogenicity of X. oryzae pv. oryzae strain KACC10331 (Wang et al. 2008) (2) the homologues of XC_2227 and XC_2228 (RavS and RavR) were essential for regulation of virulence factor production and pathogenesis of X. campestris pv. campestris strain XC1 (He et al. 2009) (3) the homologue of XC_2228 of X. campestris pv. campestris strain ATCC33913 (XCC1958) and X. oryzae pv. oryzae PXO99A (PXO_01019) was important for virulence (Qian et al. 2008; Yang et al. 2012), and (4) XC_2228 and XC_2699 were required for virulence of X. campestris pv. campestris strain 8004 (Zang et al. 2007; Tao et al. 2014). The transcription of genes encoding these aforementioned proteins showed decreased expression level in the clpX mutant compared to wild type by RT-qPCR analysis (Table 1), suggesting ClpX positively regulates the expression of these virulence-related proteins transcriptionally.
In the X. campestris pv. campestris genome (da Silva et al. 2002; Qian et al. 2005; Liu et al. 2015), three Clp ATPase-encoding genes (clpA, clpX, and clpY) and two Clp protease-encoding genes (clpP and clpQ) were found, suggesting the bacteria’s genome harbors three potential ATP-dependent Clp protease systems: ClpAP, ClpXP, and ClpYQ. The proteomic analysis showed that, in the wild-type strain and the clpX mutant, the expression of ClpA was at the same level, whereas the expression of ClpY and ClpQ was increased in the clpX mutant and the expression of ClpX was significantly reduced in the clpX mutant (Table 1). The altered expression of these proteins was further validated by RT-qPCR. Further, the transcription of clpP was unaffected through RT-qPCR analysis, even the ClpP protein was not identified by proteomic analysis. These results contrast with the results obtained for B. anthracis, in which loss of clpX results in increased expression of clpP2 (Claunch et al. 2018). While the expression of ClpA and ClpP was not altered, it is worthy to notice that ClpYQ, another putative ATP-dependent Clp protease, was found to be upregulated in the clpX mutant strain, suggesting an attempt by the cells to compensate for the loss of ClpX (or ClpXP) function. At present, it is not clear why ClpX appears to influence the expression of ClpYQ, and this warrants further analysis.
Conclusion
We previously screened approximately 1000 EZ-Tn5 mutants for their ability to form biofilm and identified several biofilm-related genes that have been further demonstrated to have a role in virulence (Liao et al. 2016, 2019; Liu et al. 2018). Here, we pursued this work and identified one transposon mutant showing reduced bacterial attachment and exhibiting an insertion in clpX gene. In addition to several sequenced X. campestris pv. campestris strains (da Silva et al. 2002; Qian et al. 2005; Liu et al. 2015), the clpX gene has been annotated in other sequenced Xanthomonas species, such as X. axonopodis pv. citri (da Silva et al. 2002), X. campestris pv. vesicatoria (Thieme et al. 2005), X. gardneri (Richard et al. 2017), X. oryzae pv. oryzae (Lee et al. 2005), and X. oryzae pv. oryzicola (Bogdanove et al. 2011). Although the clpX gene exists in several Xanthomonas species, nothing is known regarding its biological functions. In this study, we provided conclusive genetic evidence demonstrating that clpX gene possesses multifarious pathogenicity-related functions (such as bacterial attachment, virulence factor synthesis, and pathogenesis) and is involved in a range of stress tolerances (heat, puromycin, and SDS) of X. campestris pv. campestris. Further, we confirmed that ClpX positively regulates the transcription of genes related to attachment and extracellular enzyme synthesis, as well as genes associated with virulence. Additionally, we employed a quantitative proteomic approach, coupling nLC to MS/MS, to compare the protein profiles between the wild type and clpX mutant strains to obtain a general picture of ClpX function. Through the preliminary comparison of the protein profiles of the two strains, it was found that clpX mutation had a considerable effect on the bacterial proteome and 211 altered proteins (a greater than twofold difference) belonging to different COG category were discerned. To the best of our knowledge, this is the first report on the functional aspects of ClpX in Xanthomonas.
References
Bogdanove AJ et al (2011) Two new complete genome sequences offer insight into host and tissue specificity of plant pathogenic Xanthomonas spp. J Bacteriol 193:5450–5464. https://doi.org/10.1128/JB.05262-11
Buttner D, Bonas U (2010) Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev 34:107–133. https://doi.org/10.1111/j.1574-6976.2009.00192.x
Chan JW, Goodwin PH (1999) The molecular genetics of virulence of Xanthomonas campestris. Biotechnol Adv 17:489–508
Chen YY, Wu CH, Lin JW, Weng SF, Tseng YH (2010) Mutation of the gene encoding a major outer-membrane protein in Xanthomonas campestris pv. campestris causes pleiotropic effects, including loss of pathogenicity. Microbiology 156:2842–2854. https://doi.org/10.1099/mic.0.039420-0
Chiang YC, Liao CT, Du SC, Hsiao YM (2017) Functional characterization and transcriptional analysis of icd2 gene encoding an isocitrate dehydrogenase of Xanthomonas campestris pv. campestris. Arch Microbiol 199:917–929. https://doi.org/10.1007/s00203-017-1370-5
Claunch KM, Bush M, Evans CR, Malmquist JA, Hale MC, McGillivray SM (2018) Transcriptional profiling of the clpX mutant in Bacillus anthracis reveals regulatory connection with the lrgAB operon. Microbiology 164:659–669. https://doi.org/10.1099/mic.0.000628
da Silva AC et al (2002) Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417:459–463
Dow JM, Crossman L, Findlay K, He YQ, Feng JX, Tang JL (2003) Biofilm dispersal in Xanthomonas campestris is controlled by cell-cell signaling and is required for full virulence to plants. Proc Natl Acad Sci USA 100:10995–11000
Erbse A et al (2006) ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439:753–756. https://doi.org/10.1038/nature04412
Frees D, Qazi SN, Hill PJ, Ingmer H (2003) Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol Microbiol 48:1565–1578
Fu JF, Tseng YH (1990) Construction of lactose-utilizing Xanthomonas campestris and production of xanthan gum from whey. Appl Environ Microbiol 56:919–923
Gottesman S (2003) Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19:565–587. https://doi.org/10.1146/annurev.cellbio.19.110701.153228
He YW, Zhang LH (2008) Quorum sensing and virulence regulation in Xanthomonas campestris. FEMS Microbiol Rev 32:842–857. https://doi.org/10.1111/j.1574-6976.2008.00120.x
He YW, Boon C, Zhou L, Zhang LH (2009) Co-regulation of Xanthomonas campestris virulence by quorum sensing and a novel two-component regulatory system RavS/RavR. Mol Microbiol 71:1464–1476. https://doi.org/10.1111/j.1365-2958.2009.06617.x
Hsiao YM, Liao HY, Lee MC, Yang TC, Tseng YH (2005) Clp upregulates transcription of engA gene encoding a virulence factor in Xanthomonas campestris by direct binding to the upstream tandem Clp sites. FEBS Lett 579:3525–3533
Hsiao YM, Liu YF, Fang MC, Song WL (2011) XCC2731, a GGDEF domain protein in Xanthomonas campestris, is involved in bacterial attachment and is positively regulated by Clp. Microbiol Res 166:548–565. https://doi.org/10.1016/j.micres.2010.11.003
Hsiao YM, Song WL, Liao CT, Lin IH, Pan MY, Lin CF (2012) Transcriptional analysis and functional characterization of XCC1294 gene encoding a GGDEF domain protein in Xanthomonas campestris pv. campestris. Arch Microbiol 194:293–304. https://doi.org/10.1007/s00203-011-0760-3
Jana B, Tao L, Biswas I (2016) Strain-dependent recognition of a unique degradation motif by ClpXP in Streptococcus mutans. mSphere. https://doi.org/10.1128/mSphere.00287-16
Kajfasz JK et al (2009) Role of Clp proteins in expression of virulence properties of Streptococcus mutans. J Bacteriol 191:2060–2068. https://doi.org/10.1128/JB.01609-08
Keen NT, Tamaki S, Kobayashi D, Trollinger D (1988) Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197
Kress W, Maglica Z, Weber-Ban E (2009) Clp chaperone-proteases: structure and function. Res Microbiol 160:618–628. https://doi.org/10.1016/j.resmic.2009.08.006
Lee BM et al (2005) The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res 33:577–586. https://doi.org/10.1093/nar/gki206
Li Y et al (2010) ClpXP protease regulates the type III secretion system of Dickeya dadantii 3937 and is essential for the bacterial virulence. Mol Plant Microbe Interact 23:871–878. https://doi.org/10.1094/MPMI-23-7-0871
Liao CT, Du SC, Lo HH, Hsiao YM (2014) The galU gene of Xanthomonas campestris pv. campestris is involved in bacterial attachment, cell motility, polysaccharide synthesis, virulence, and tolerance to various stresses. Arch Microbiol 196:729–738. https://doi.org/10.1007/s00203-014-1012-0
Liao CT et al (2016) Functional characterization and transcriptome analysis reveal multiple roles for prc in the pathogenicity of the black rot pathogen Xanthomonas campestris pv. campestris. Res Microbiol 167:299–312. https://doi.org/10.1016/j.resmic.2016.01.002
Liao CT, Chiang YC, Hsiao YM (2019) Functional characterization and proteomic analysis of lolA in Xanthomonas campestris pv. campestris. BMC Microbiol 19:20. https://doi.org/10.1186/s12866-019-1387-9
Liu YC et al (2015) Complete genome sequence of Xanthomonas campestris pv. campestris strain 17 from Taiwan. Genome Announc. https://doi.org/10.1128/genomeA.01466-15
Liu YF, Liao CT, Chiang YC, Li CE, Hsiao YM (2018) WxcX is involved in bacterial attachment and virulence in Xanthomonas campestris pv. campestris. J Basic Microbiol 58:403–413. https://doi.org/10.1002/jobm.201700591
Malik IT, Brotz-Oesterhelt H (2017) Conformational control of the bacterial Clp protease by natural product antibiotics. Nat Prod Rep 34:815–831. https://doi.org/10.1039/c6np00125d
McGillivray SM et al (2009) ClpX contributes to innate defense peptide resistance and virulence phenotypes of Bacillus anthracis. J Innate Immun 1:494–506. https://doi.org/10.1159/000225955
Porankiewicz J, Wang J, Clarke AK (1999) New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol Microbiol 32:449–458
Qian W et al (2005) Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15:757–767
Qian W, Han ZJ, Tao J, He C (2008) Genome-scale mutagenesis and phenotypic characterization of two-component signal transduction systems in Xanthomonas campestris pv. campestris ATCC 33913. Mol Plant Microbe Interact 21:1128–1138. https://doi.org/10.1094/MPMI-21-8-1128
Richard D et al (2017) Complete genome sequences of six copper-resistant Xanthomonas strains causing bacterial spot of solaneous plants, belonging to X. gardneri, X. euvesicatoria, and X. vesicatoria, using long-read technology. Genome Announc. https://doi.org/10.1128/genomeA.01693-16
Ryan RP et al (2007) Cyclic di-GMP signalling in the virulence and environmental adaptation of Xanthomonas campestris. Mol Microbiol 63:429–442. https://doi.org/10.1111/j.1365-2958.2006.05531.x
Ryan RP et al (2011) Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nat Rev Microbiol 9:344–355. https://doi.org/10.1038/nrmicro2558
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor
Schmidt R, Bukau B, Mogk A (2009) Principles of general and regulatory proteolysis by AAA+ proteases in Escherichia coli. Res Microbiol 160:629–636. https://doi.org/10.1016/j.resmic.2009.08.018
Tao J, Li C, Luo C, He C (2014) RavA/RavR two-component system regulates Xanthomonas campestris pathogenesis and c-di-GMP turnover. FEMS Microbiol Lett 358:81–90. https://doi.org/10.1111/1574-6968.12529
Thieme F et al (2005) Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J Bacteriol 187:7254–7266
Veselova MA et al (2016) The effect of mutation in the clpX gene on the synthesis of N-acyl-homoserine lactones and other properties of Burkholderia cenocepacia 370. Microbiol Res 186–187:90–98. https://doi.org/10.1016/j.micres.2016.03.009
Vicente JG, Holub EB (2013) Xanthomonas campestris pv. campestris (cause of black rot of crucifers) in the genomic era is still a worldwide threat to brassica crops. Mol Plant Pathol 14:2–18. https://doi.org/10.1111/j.1364-3703.2012.00833.x
Wang JC, So BH, Kim JH, Park YJ, Lee BM, Kang HW (2008) Genome-wide identification of pathogenicity genes in Xanthomonas oryzae pv. oryzae by transposon mutagenesis. Plant Pathol 57:1136–1145. https://doi.org/10.1111/j.1365-3059.2008.01884.x
Yang BY, Tseng YH (1988) Production of exopolysaccharide and levels of protease and pectinase activity in pathogenic and non-pathogenic strains of Xanthomonas campestris pv. campestris. Bot Bull Acad Sin 29:93–99
Yang F et al (2012) A novel two-component system PdeK/PdeR regulates c-di-GMP turnover and virulence of Xanthomonas oryzae pv. oryzae. Mol Plant Microbe Interact 25:1361–1369. https://doi.org/10.1094/MPMI-01-12-0014-R
Zang N et al (2007) Requirement of a mip-like gene for virulence in the phytopathogenic bacterium Xanthomonas campestris pv. campestris. Mol Plant Microbe Interact 20:21–30. https://doi.org/10.1094/MPMI-20-0021
Acknowledgements
This work was supported by Ministry of Science and Technology of Taiwan (Grants Nos. MOST104-2313-B-166-001-MY3 and MOST107-2313-B-166-001-MY3) to YMH, and Central Taiwan University of Science and Technology (grant No. CTU105-P-15) to HHL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lo, HH., Liao, CT., Li, CE. et al. The clpX gene plays an important role in bacterial attachment, stress tolerance, and virulence in Xanthomonas campestris pv. campestris. Arch Microbiol 202, 597–607 (2020). https://doi.org/10.1007/s00203-019-01772-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01772-3