
Abstract
ATP-dependent proteases (FtsH, Lon, and Clp family proteins) are ubiquitous in bacteria and play essential roles in numerous regulatory cell processes. Xanthomonas campestris pv. campestris is a Gram-negative pathogen that can cause black rot diseases in crucifers. The genome of X. campestris pv. campestris has several clp genes, namely, clpS, clpA, clpX, clpP, clpQ, and clpY. Among these genes, only clpX and clpP is known to be required for pathogenicity. Here, we focused on two uncharacterized clp genes (clpS and clpA) that encode the adaptor (ClpS) and ATPase subunit (ClpA) of the ClpAP protease complex. Transcriptional analysis revealed that the expression of clpS and clpA was growth phase-dependent and affected by the growth temperature. The inactivation of clpA, but not of clpS, resulted in susceptibility to high temperature and attenuated virulence in the host plant. The altered phenotypes of the clpA mutant could be complemented in trans. Site-directed mutagenesis revealed that K223 and K504 were the amino acid residues critical for ClpA function in heat tolerance. The protein expression profile shown by the clpA mutant in response to heat stress was different from that exhibited by the wild type. In summary, we characterized two clp genes (clpS and clpA) by examining their expression profiles and functions in different processes, including stress tolerance and pathogenicity. We demonstrated that clpS and clpA were expressed in a temperature-dependent manner and that clpA was required for the survival at high temperature and full virulence of X. campestris pv. campestris. This work represents the first time that clpS and clpA were characterized in Xanthomonas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regulated proteolysis is an essential process that affects several biological pathways (Mahmoud and Chien 2018). In bacteria, this process is performed by energy-dependent AAA+ (ATPases associated with cellular activities) proteases that use the power of ATP hydrolysis to unfold, and translocate substrates (Mahmoud and Chien 2018). AAA+ proteases are not only required for regulated proteolysis, they are also essential for the quality control of aberrant or aggregated proteins under certain conditions (Bittner et al. 2016).
Several AAA+ proteases, such as FtsH, Lon, and Clp family proteases (ClpAP, ClpXP, and ClpYQ [HslUV]), exist in bacteria (Bittner et al. 2016; Mahmoud and Chien 2018; Sauer and Baker 2011). All these AAA+ proteases are compartmentalized proteases that consist of two distinct functional units with separate activities, namely, ATPase and protease (Baker and Sauer 2006; Bittner et al. 2016; Mahmoud and Chien 2018). In FtsH and Lon, the ATPase and protease domains are located in a single polypeptide (Bittner et al. 2016; Mahmoud and Chien 2018; Sauer and Baker 2011). In the Clp family, the AAA+ family ATPase subunit (ClpA, ClpX, and ClpY [HslU]) and the proteolytic subunit (ClpP and ClpQ [HslV]) are encoded in distinct polypeptide chains (Bittner et al. 2016; Mahmoud and Chien 2018; Sauer and Baker 2011). ClpA and ClpX function as chaperones and bind to ClpP (serine protease) to form the ATP-dependent protease ClpAP or ClpXP, whereas ClpY (HslU) interacts with ClpQ (HslV) to form the ClpYQ (HslUV) protease (Chandu and Nandi 2004).
In the ClpAP protease, one or two AAA+ ClpA hexamers associate with the tetradodecameric ClpP to perform ATP-dependent protein degradation (Olivares et al. 2018). An adaptor protein, such as ClpS, that interacts with the N-terminal domain of ClpA and influences the ClpAP complex, may also be associated (Erbse et al. 2006). ClpAP exists in most Gram-negative proteobacteria and has been characterized extensively in the model organism E. coli (Kress et al. 2009a). ClpS is a highly conserved protein, and ClpS homologs are found not only in bacteria but also in plants (Dougan et al. 2002). ClpA, as a part of the ClpAP complex, has several functions: ATP hydrolysis, substrate recognition, protein unfolding, and translocation to its proteolytic partner ClpP (Xia et al. 2004). In the absence of the proteolytic component ClpP, ClpA functions as a molecular chaperone and catalyzes protein unfolding and limited protein remodeling (Wickner et al. 1994; Xia et al. 2004). ClpS can regulate ClpA substrate selection and is an essential component of the N-end rule pathway (Erbse et al. 2006). In E. coli, ClpS directly interacts with destabilizing N-terminal residues and delivers them to the ClpAP complex for degradation (Schmidt et al. 2009). In addition to act as an enhancer of N-end rule substrate degradation, ClpS affects catalytic steps of the ClpAP degradation cycle and modulates the ClpAP complex by controlling its ATPase activity (Torres-Delgado et al. 2020).
Xanthomonas campestris pv. campestris is an important plant pathogen that causes black rot, a systemic vascular disease of crucifers (An et al. 2020). Six predicted Clp family protein-encoding genes have been annotated in the genome of X. campestris pv. campestris (da Silva et al. 2002; Liu et al. 2015; Qian et al. 2005; Vorholter et al. 2008). They are the adaptor protein encoding gene (clpS), genes coding for AAA+ ATPase (clpA, clpX, and clpY), and genes coding for Clp protease (clpP and clpQ). Among these genes, only two (clpX and clpP) have been studied. The X. campestris pv. campestris clpX has been found to play an important role in bacterial attachment, stress tolerance, and virulence (Lo et al. 2020). The X. campestris pv. campestris clpP has been shown to be required for growth under heat stress and in the presence of puromycin and is essential for full virulence; moreover, its expression is induced by heat shock (Li et al. 2020). In this study, we aimed to characterize clpS and clpA in X. campestris pv. campestris. We performed a transcriptional fusion assay to evaluate the expression of clpS and clpA. In addition, we utilized the deletion mutants of clpS and clpA, along with their genetic complements, to investigate the functions of the corresponding proteins in the stress tolerance and pathogenicity of X. campestris pv. campestris.
Materials and methods
Strains, media, and culture conditions
The E. coli ECOS™ 101 Competent Cells [DH5α] that were used as the host for DNA cloning were purchased from Yeastern Biotech. The wild-type X. campestris pv. campestris strain 17 (Xcc17) was isolated in Taiwan (Yang and Tseng 1988). The Xcc17-derived mutant strains constructed in this study included HCS (clpS mutant) and HCA (clpA mutant) and were obtained through marker exchange. All bacterial cultures were routinely cultured in Luria–Bertani (LB) medium (Miller 1972) unless otherwise noted. E. coli and X. campestris pv. campestris strains were incubated at 37 °C and 28 °C, respectively. XOLN supplemented with glycerol (2%) was used as the basal salt medium (Fu and Tseng 1990). Ampicillin (50 μg/mL), kanamycin (50 μg/mL), gentamycin (15 μg/mL), and tetracycline (15 μg/mL) were added to the medium as required. For liquid cultures, bacterial cells were grown with shaking at 180 revolutions per minute (rpm). The agar concentration used for solid media was 1.5%.
Molecular techniques
The restriction enzymes, T4 DNA ligase, and Taq DNA polymerase used in this study were purchased from Promega, Roche, and Yeastern. DNA manipulation and E. coli DNA transformation were performed in accordance with standard methods as previously described (Sambrook et al. 1989). Genomic and plasmid DNA samples were prepared by using the Wizard® Genomic DNA Purification Kit (Promega) and the Gene-Spin™ Miniprep Purification Kit (Protech), respectively. Polymerase chain reaction (PCR) was performed as previously described (Hsiao et al. 2005) with the primers listed in Table 1. DNA sequences were determined by Mission Biotech Co., Ltd. (Taipei, Taiwan). X. campestris pv. campestris was transformed through electroporation (Wang and Tseng 1992).
Upstream region construction and promoter activity assay
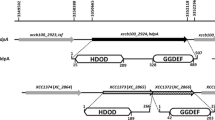
DNA fragments containing the upstream regions of clpS and clpA were PCR-amplified by using the primer pair 35PstI/476XbaI and 621PstI/946XbaI and ligated into the cloning vector yT&A (Yeastern) to generate pTclpS and pTclpA. After sequence confirmation, the insert in pTclpS and pTclpA was excised and cloned ahead of a promoterless lacZ gene (reporter) in pFY13–9 (Lee et al. 2001) to yield pFYclpS and pFYclpA. The resulting plasmids carried nt –448/–7 (442 bp) and –326/–1 (326 bp) region relative to the clpS and clpA putative translational start point, respectively (Fig. 1).

Comparative analysis of the organization of the clpS–clpA regions in the genomes of X. campestris pv. campestris and E. coli. The regions in Xcc17 and the compatible regions in Eco K-12 are shown. The arrows specify the locations and orientations of the genes, and lines show the intergenic regions. The percentage values refer to the amino acid identity shared by two homologous proteins. The inverted triangles indicate the position of pOKclpS and the GmR cartridge in the clpS and clpA mutants. The lower elements indicate the lengths and locations of the PCR-amplified fragments used for expression, mutation, and complementation analyses. The construction of the reporter constructs (pFYclpS and pFYclpA) and the complementation plasmids (pRKclpS, pRKclpA, pRKclpAK223Q, and pRKclpAK504Q) is described in the “Materials and Methods” section. The primers used to construct these plasmids are presented below the solid lines
For the promoter activity assay, the X. campestris pv. campestris wild-type Xcc17 carrying the above constructs were cultured overnight and inoculated into fresh media to an initial optical density at 550 nm (OD550) of 0.35. Then, the cultures were incubated either at 28 °C or at 37 °C. Aliquots were withdrawn at 6, 24, 30, and 48 h postincubation, and β-galactosidase activity was determined as described previously (Miller 1972). Further, the promoter activity was also measured by growing cells at 28 °C to mid-exponential phase (OD550 = 0.6) in LB medium. Then, the cultures were divided into two parts, one was further grown at 28 °C and the other was shifted to 37 °C to initiate heat shock. At various time points, samples were taken for β-galactosidase assays. The experiment was performed in duplicate and repeated at least three times. Data were represented as the average of three replicates.
Sequence analysis and homology modeling
Nucleotide and predicted amino acid sequence analyses and sequence comparison were performed online (http://www.ncbi.nlm.nih.gov). The three-dimensional structural models of Xcc17 ClpS and ClpA were based on the E. coli strain K-12 (Eco K-12) ClpS (PDB ID 2W9R) and ClpA (PDB ID 1KSF) from the RCSB PDB database. They shared 56.4% and 63.0% sequence identity with Xcc17 ClpS and ClpA, respectively. Project mode with the SWISS-MODEL workspace (https://swissmodel.expasy.org) was used for the homology modeling structure of Xcc17 ClpS and ClpA (Arnold et al. 2006). The web-based application of NGL viewer (http://proteinformatics.charite.de/ngl) was utilized for the molecular visualization of protein structure.
Mutant construction and complementation
The 135 bp PstI-EcoRI fragment in pTclpA which including internal region of clpS (nucleotide 139 ~ 273) was excised and cloned into pOK12 to construct the clpS mutant (Vieira and Messing 1991). The obtained plasmid pOKclpS that contained the internal region of the Xcc17 clpS gene was then transferred into Xcc17 via electroporation following a single crossover event. For the construction of the clpA mutant, the 2334-bp HindIII-XbaI fragment encompassing the upstream 48 bp fragment plus the entire coding region of the Xcc17 clpA was synthesized and cloned into pUC57-Mini (Protech) to yield pUCclpA. Then, the 693-bp EcoRV-PstI fragment of pUCclpA was excised and cloned into pOK12 to obtain pOKclpA. A GmR cartridge from pUCGM (Schweizer 1993) was inserted into the HincII site within the pOKclpA insert. The resultant plasmid, pOKclpAG, was further transferred into Xcc17 through electroporation allowing for double crossover. The insertion of pOKclpS or the GmR cartridge into the target gene (clpS or clpA) was verified through PCR. The confirmed clpS and clpA mutant strains were designated as HCS and HCA, respectively.
For the construction of the HCS complementation plasmid, the 546 bp DNA fragment encompassing the upstream 82 bp fragment plus the entire coding region of the Xcc17 clpS was amplified via PCR by using the primer pair 401HindIII/946XbaI and cloned into the broad-host-range vector pRK415 (Keen et al. 1988), thus providing pRKclpS (Fig. 1). For the construction of the HCA complementation plasmid, the 2334-bp HindIII-XbaI fragment of pUCclpA was excised and cloned into pRK415, thus yielding pRKclpA (Fig. 1). For the complementation of the clpS and clpA mutants, plasmids pRKclpS and pRKclpA were electroporated into HCS and HCA, respectively. The complemented strains were designated as HCS(pRKclpS) and HCA(pRKclpA). In parallel, the empty vector pRK415 was introduced into Xcc17, HCS, and HCA to generate Xcc17(pRK415), HCS(pRK415), and HCA(pRK415) for comparison.
Site-directed mutagenesis
The site-directed mutagenesis of ClpA was carried out by using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) in accordance with the manufacturer’s instructions. The mutation was constructed at the most commonly mutated residue in the Walker A motif of the AAA+ protein (Wendler et al. 2012). The invariant lysine at positions 223 and 504 was substituted with glutamine by using pUCclpA as a template for PCR and the primers listed in Table 1. After DNA sequence verification, the mutated clpA was cloned into pRK415 to obtain pRKclpAK223Q and pRKclpAK504Q (Fig. 1). The separate introduction of the resulting constructs into HCA yielded HCA(pRKclpAK223Q) and HCA(pRKclpAK504Q).
Stress tolerance assay
The temperature tolerance assay was performed in accordance with a previously described method (Lo et al. 2020). Briefly, overnight cultures of the tested strains were diluted with fresh LB broth to an initial density of OD550 = 0.5 (5 × 108 cells/mL) and then subjected to tenfold serial dilutions (5 × 107 to 5 × 105 cells/mL). Then, cell suspensions (5 µL) from each dilution were spotted on LB plates and incubated either at 28 °C or at 37 °C for 4 days. The assay was repeated independently at least three times with two replicates each time.
For survival testing, overnight cultures of the tested strains were diluted with fresh LB medium to an OD550 of 0.1, then the inoculants were incubated at 28 °C with shaking at 180 rpm. Samples were taken at 24, 48 and 72 h post-inoculation and viable-cells were counted by plating cells on LB plates after incubation of 3 days.
The tolerance of bacteria to H2O2, sodium dodecyl sulfate (SDS), and puromycin was evaluated by disk diffusion assays according to previously methods (Lo et al. 2020). Briefly, the mid-exponential-phase cultures were spread on LB plate and a disc (6 mm) containing 10 μl of H2O2 (3%), SDS (5%), or puromycin (5 mg/mL) was placed on top. The growth-inhibition zones surrounding the discs were measured after incubation at 28 °C for 3 days.
Cell extract preparation and analysis
The overnight cultures of the tested strains were inoculated into fresh XOLN–glycerol medium to an initial OD550 = 0.35 and incubated at either 28 °C or at 37 °C for 24 h. The bacterial cells were collected through centrifugation at 12 000 × g for 2 min at 4 °C. Each preparation was obtained from the same number of cells. Cell pellets were rinsed and suspended in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). Cell extracts were obtained through sonication (cycles of 4 s pulse and 5 s rest on ice for 2 min). Following sonication, cell debris and unbroken cells were removed through centrifugation at 2000 × g for 2 min at 4 °C, and the supernatant fraction was referred to as cell extracts.
Protein aggregates were isolated in accordance with a previously described procedure (Kthiri et al. 2010) with some modifications. In brief, the cell extracts were incubated with or without 0.5% Triton X-100 for 15 min at 25 °C. Subsequently, insoluble cell fractions were recovered through centrifugation at 8000 × g for 15 min at 25 °C. The insoluble cell fractions treated with Triton X-100 were referred to protein aggregates, and those without Triton X-100 treatment were obtained for comparison. The insoluble cell fractions were analyzed by using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and protein bands were stained with Coomassie brilliant blue. The amount of loading was normalized in terms of colony forming units (CFUs) and was equivalent to 4.2 × 108 CFUs/lane. Bands containing the proteins of interest were excised from the gel and subjected to mass spectrometric analysis using Thermo Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer. The mass spectra were searched against the X. campestris database from UniProt (67,706 sequences; 23,224,367 residues) by using the Mascot searching engine (Proteome Discoverer V2.2.0.388). The following parameters were set: Static modifications, Carbomidomethylation; Dynamic modifications, Acetylation, Deamidation, Oxidation; Precursor mass tolerance, 10 ppm; Fragment mass tolerance, 0.02 Da and allowance of two missed cleavages.
Virulence assay, bacterial attachment analysis and extracellular enzyme determination
The virulence of X. campestris pv. campestris strains on cabbage was determined by using the leaf clip inoculation method (Hsiao et al. 2011). Leaves were cut with scissors that had been dipped into the bacterial suspension with the adjusted cell concentration of OD550 = 1. Lesion lengths were measured, and images were taken 14 days postinoculation. Three independent experiments with six replicates were performed. The values were presented as mean lesion length ± standard deviations.
Bacterial attachment was tested by evaluating the ability of cells to adhere to the 96-well polystyrene microtiter plates (Nunc) as the previously described method (Lo et al. 2020). The production of extracellular enzymes was determined by substrate-supplemented plate assay method as described previously (Hsiao et al. 2011; Li et al. 2020). The substrates supplemented were carboxymethyl cellulose (0.5%, for cellulase), locust bean gum (0.2%, for mannanase), sodium polypectate (0.5%, for pectinase), and skim milk (1%, for protease). Each experiment was performed at least three times.
Results and discussion
Expression of clpS and clpA is induced by heat treatment
The genome of the sequenced Xcc17 (Liu et al. 2015) possesses clpS (locus_tag AAW18_RS09760) and clpA (locus_tag AAW18_RS09765), which are adjacent to each other on the chromosome (Fig. 1). The infA and aat genes, which code for translation initiation factor IF-1 and leucyl/phenylalanyl-tRNA-protein transferase, are located downstream of clpA. As shown in Fig. 1, Xcc17 and Eco K-12 contain compatible homologs with 49%–69% shared amino acid sequence identities (Blattner et al. 1997). The mutT gene (locus_tag AAW18_RS0975 in Xcc17), which codes for NUDIX hydrolase, is located upstream of clpS, and its homolog appear elsewhere in the chromosome in Eco K-12 (Blattner et al. 1997). The clpS and clpA genes, as well as their flanking genes, show similar organization in several X. campestris pv. campestris strains, such as ATCC33913, 8004, and B100, in addition to Xcc17 (da Silva et al. 2002; Qian et al. 2005; Vorholter et al. 2008).
Gene organization analysis demonstrated that clpS and its upstream gene mutT and clpA and its downstream gene infA are in opposite directions and that the clpS and clpA genes are linked together in the same orientation with a 143 bp intergenic space (Fig. 1). Considering their orientation and intergenic regions with flanking genes, clpS and clpA likely possess their own promoters. Sequence analysis revealed that the upstream regions of clpS and clpA resembled the sequence of the E. coli σ32-dependent heat-shock promoter (TTGAAA-N13-14-CCCCATNT) (Koo et al. 2009; Nonaka et al. 2006). A possible σ32 promoter with a −35 box (CGTAAA) and a −10 box (GGCCAAAT) were located at −61 and −39 (with a spacer of 14 nucleotides) relative to the clpS translation start site. A putative σ32-type promoter with a −35 box (TGGAAA) and a −10 box (CCGCATAT) separated by 13 nucleotides was present 29 nucleotides upstream of the clpA initiation codon. In consideration of the presence of the σ32-type promoter, we predicted that the expression of clpS and clpA may be influenced under heat-shock conditions.
For the evaluation of clpS and clpA expression, the upstream regions of clpS and clpA were cloned ahead of a promoterless lacZ gene in pFY13–9 (Lee et al. 2001) to yield pFYclpS and pFYclpA as described in the “Materials and Methods” section. Then, the generated reporter constructs were introduced into Xcc17. The resulting strains, Xcc17(pFYclpS) and Xcc17(pFYclpA), were used to evaluate the expression of clpS and clpA. The transcription levels of clpS and clpA was first monitored from an initial optical density at 550 nm of 0.35. The promoter activities of clpS and clpA were assayed by measuring β-galactosidase activities in Xcc17(pFYclpS) (Fig. 2a) and Xcc17(pFYclpA) (Fig. 2b) grown at 28 °C or 37 °C. In the Xcc17(pFYclpS) and Xcc17(pFYclpA) strains grown at a normal physiological temperature (28 °C), the activities were 69 and 121 U at 6 h, respectively; increased following cell growth; and then peaked at 48 h (343 and 769 U). When the strains were grown at 37 °C (heat stress condition), the levels of β-galactosidase activities increased gradually and reached their maxima of 413 U in Xcc17(pFYclpS) and 1220 U in Xcc17(pFYclpA) at 48 h, exhibiting approximately 1.6-fold increment in both cases compared to the culture growing at normal temperature. Among the four time points determined, substantially transcriptional changes were observed under heat treatment, although the differences were not big. To further evaluate the effect of temperature, cells of Xcc17(pFYclpS) and Xcc17(pFYclpA) were cultured at 28 °C to mid-log phase (OD550 = 0.6), then each of the cultures was divided into two parts, one was further grown at 28 °C and the other at 37 °C. The β-galactosidase activities were measured in cultures after 0, 5, 10, 15, 30, 60, 120, 180, and 300 min. As shown in Figs. 2c and 2d, when samples were taken at short time intervals (0, 5, 10, 15, 30, and 60 min), the increment of bacterial growth was not significant and the OD550 value of Xcc17(pFYclpS) and Xcc17(pFYclpA) was around 0.67 at normal physiological temperature (28 °C), which was almost identical to that under heat treatment (37 °C). At time points 120, 180, and 300 min, bacterial growth was observed and the OD550 values rose gradually and reached 1.67 and 1.25 in Xcc17(pFYclpS) and 1.58 and 1.35 in Xcc17(pFYclpA) at 28 °C and 37 °C, respectively, at 300 min (Fig. 2c and 2d). At 28 °C, the β-galactosidase activities expressed by Xcc17(pFYclpS) and Xcc17(pFYclpA) were around 88 U and 296 U when samples were taken at short time intervals, and were around 61 U and 195 U at the last three time points (120, 180, and 300 min), with 69% and 66% of those levels expressed at short time intervals (Fig. 2c, 2d). At 37 °C, the activities expressed by Xcc17(pFYclpS) and Xcc17(pFYclpA) were around 107 U and 308 U at all of the tested points (Fig. 2c, 2d). Although the levels of enzyme activity detected at 28 °C and 37 °C were about the same when samples were taken at short time intervals, the effect of temperature was evident at the last three time pints in which the OD550 value reached 1.0 (corresponding to late exponential growth phase, 120 min after inoculation). When the highest points were taken for comparison, the increase was about 1.93-fold for Xcc17(pFYclpS) at 120 min (Fig. 2c) and 2.05-fold for Xcc17(pFYclpA) at 300 min (Fig. 2d) after heat treatment. It is suggested that the expression of clpS and clpA was more sensitive to heat stress when cells entering the late exponential phase then those cultured in early growth phase. It was also noted that the β-galactosidase activities dropped by about 30% at 120 min after entering late exponential phase. It is possible that the upstream regions of clpS and clpA contain unidentified regulatory element(s) which are involved in their expression during different growth stages. The results of the reporter assays indicated that clpS and clpA expression was growth phase-dependent and affected by the growth temperature.

Expression of clpS and clpA in X. campestris pv. campestris. The wild-type strain Xcc17 harboring the reporter construct pFYclpS (a, c) or pFYclpA (b, d) was cultured in LB medium. The initial cultures were adjusted to OD550 = 0.35 (a, b) or grown to mid-log phase (OD550 = 0.6) (c, d). The open circles and filled circles represent the cell growth (OD550) of the reporter strain cultured at 28 °C and 37 °C, respectively. The time points at 6 h is corresponding to the log phase, 24 h and 30 h are corresponding to stationary phase, and 48 h is corresponding to late stationary phase. The open bars and filled bars represent the β-galactosidase activities (Miller units) of the reporter strain cultured at 28 °C and 37 °C, respectively. The results are provided as the average of triplicate measurements. Error bars indicate standard deviations. Significance was tested by using Student’s t test (* indicates significance at p < 0.05)
Available information on the expression of clpS and clpA is limited, and the effects of heat stress on expression differ among bacteria. For example, in Vibrio vulnificus, the cellular level of ClpA is induced under heat shock (Lee et al. 2018). In E. coli, the clpA gene is a monocistronic messenger, and the synthesis of ClpA does not increase upon heat shock (Gottesman et al. 1990; Katayama et al. 1988). In Brucella suis, the expression of clpA does not increase at high temperature (Ekaza et al. 2000). It was shown that PhoP represses the transcription of clpS but not clpA in E. coli and Salmonella enterica when the bacteria experience low cytoplasmic Mg2+ concentrations (Yeom et al. 2018). Recently, it was reported that clpS and clpA are induced during H2O2 stress, and OxyR is required for the H2O2-driven induction of clpSA in E. coli (Sen et al. 2020). The effects of OxyR and PhoP on the expression of clpS and clpA in X. campestris pv. campestris remains to be elucidated.
Bioinformatics analyses indicate that ClpS and ClpA retain key residues that are required for their functionality
The clpS open reading frame was 321 bp in length and located in the genomic sequence at position 2 227 483–2 227 803 on the Xcc17 chromosome (GenBank accession number NZ_CP011946). The predicted protein encoded by clpS, which consisted of 106 amino acids and was annotated as the ATP-dependent Clp protease adaptor ClpS, had a calculated molecular mass of 12 601 Da and a pI of 6.03. Domain organization analysis indicated that it had a ClpS domain that was located at residues 23–102 (bit score: 113.20; E-value: 1.1e−29) (Fig. 3a). A BLAST search against the proteins deposited in the Protein Data Bank (PDB) with the amino acid sequence of Xcc17 ClpS revealed that E. coli ClpS (PDB ID 2W9R) had the highest homology with Xcc17 ClpS (56% identities, 81% positives). The three-dimensional structure of Xcc17 ClpS predicted by using E. coli ClpS (PDB ID 2W9R) as the template is shown in Fig. 3b. The amino acid residues contributing to the interaction between ClpS and the N-terminal domain of ClpA in E. coli (Guo et al. 2002; Xia et al. 2004) were conserved in Xcc17 ClpS and were situated at P24, P25, Y28, E79, E82, and K84 (associated with contact A of ClpA) and D36 and Y37 (associated with contact C of ClpA) (Fig. 3b). These observations suggest that similar to that in E. coli, the Xcc17 ClpS may complex with the N-terminal domain of ClpA and function as a modulator of ClpA.

Domain organization prediction (a) and model of the three-dimensional structure (b) of X. campestris pv. campestris ClpS. a The predicted ClpS domain (PF02617) is shown and is drawn approximately to scale. b The N- and C-terminal ends are indicated as N’ and C’, respectively. The predicted amino acid residues responsible for ClpS binding to contacts A and C of the N-domain of ClpA are designated by yellow shading and green shading, respectively
The clpA gene was 2283 bp in length and was located at position 2 227 947–2 230 229 on the Xcc17 chromosome (GenBank accession number NZ_CP011946). It was predicted to encode an ATP-dependent Clp protease ATP-binding subunit ClpA, which consisted of 760 amino acids with a calculated molecular mass of 83 346 Da and a pI of 5.66. Domain analysis revealed that Xcc17 ClpA consisted of three functional domains: one N-domain and two AAA+ ATPase domains (designated as AAA+ modules D1 and D2) that were each divided into a large subdomain and a small subdomain (Fig. 4a). A BLAST search against the PDB with the amino acid sequence of Xcc17 ClpA revealed that ClpA from E. coli (PDB ID 1KSF) had the highest identity with Xcc17 ClpA (64% identities, 79% positives). The predicted three-dimensional Xcc17 ClpA structure built through homology modeling with E. coli ClpA as the template structure is shown in Fig. 4b. In E. coli, the N-domain of ClpA has multiple ClpS binding sites (Xia et al. 2004) and is essential for the docking of ClpS and required for the recognition and degradation of some substrates (Dougan et al. 2002; Erbse et al. 2008; Guo et al. 2002; Zeth et al. 2002). Several amino acid residues contribute to the interactions of the N-terminal domain of ClpA and ClpS in E. coli (Xia et al. 2004) are highly conserved in Xcc17 ClpA. They included residues that participate in ClpS binding via contact A (E23, F24, V27, E28, T81, R86, D116, and Y121) and residues that are involved in ClpS binding via contact C (S97, K99, N106, R130, and H139). In addition, three residues (H22, H29, and E63) in the Zn2+ binding motif and two residues (Y14 and F84) in the N-domain hydrophobic path for peptide binding were conserved in Xcc17 ClpA. These amino acid residues in the built three-dimensional structure of ClpA are shown in Fig. 4c (left part). The ATPase domain of the AAA+ protease consists of several classical key elements of the ATPase function that form the nucleotide binding pocket, including Walker A, Walker B, sensor 1, the arginine finger, and sensor 2 (Bittner et al. 2016; Miller and Enemark 2016; Wendler et al. 2012). The Walker A motif (GxxxxGKT/S, where x is any amino acid) is required for ATP binding and oligomerization, whereas the Walker B motif (hhhhDE, where h is any hydrophobic amino acid) is essential for ATP hydrolysis (Bittner et al. 2016; Miller and Enemark 2016). The sensor 1 and arginine finger motifs are required for ATP hydrolysis, and sensor 2 mediates the conformational changes that are associated with the cycle of ATP binding and hydrolysis (Miller and Enemark 2016). These sequence motifs were found to be highly conserved in AAA+ modules D1 and D2 in Xcc17 ClpA. The Walker A motif in D1 and D2 was situated in the regions of 217–224 (GEAGVGKT) and 498–505 (GPTGVGKT), respectively. The Walker B motif in D1 and D2 was situated in the regions of 284–289 (VLFIDE) and 562–567 (VLLLDE), respectively. The sequences and positions of the sensor 1, arginine finger, and sensor 2 motifs in D1 and D2 were as follows: (i) C321IGSTT326 and L604VMTTN609 (sensor 1), (ii) R342R343 and R646 (arginine finger), and (iii) D394RLLPDKAID403 and M702GARPM707 (sensor 2). Extensive functional studies on different AAA+ proteins have demonstrated that several amino acid residues are critical for AAA+ ATPase activity (Wendler et al. 2012). Fig. 4c (right part) denotes the conserved interacting nucleotide residues in the AAA+ module of ClpA in Xcc17, in which the effects of mutations on oligomerization and hydrolytic activity have been tested in other AAA+ proteins (Wendler et al. 2012). As depicted in Fig. 4c (right part), the residues in the AAA+ module D1 were (i) K223 (Walker A), (ii) E289 (Walker B), (iii) T326 (sensor 1), (iv) R342 and R343 (arginine finger), and (v) R395 (sensor 2), and those in the AAA+ module D2 were K504 (Walker A), D567 (Walker B), N609 (sensor 1), R646 (arginine finger), and R705 (sensor 2). Given the conserved sequence motifs and catalytic residues shared by the X. campestris pv. campestris ClpA with its homologs from a diverse set of bacteria, the Xcc17 ClpA is suggested to be a functional AAA+ ATPase.

Domain organization prediction (a) and model of the three-dimensional structure (b and c) of X. campestris pv. campestris ClpA. a The predicted N-domain, AAA+ module D1, and AAA+ module D2 are shown and are drawn approximately to scale. The large and small subdomains of each AAA+ module are depicted as elliptical and hexagonal shapes, respectively. Typical elements of the AAA+ module are highlighted in color: Walker A (red), Walker B (green), sensor 1 (orange), arginine finger (purple), and sensor 2 (brown). b Schematic of the structure of ClpA. The N- and C-terminal ends are indicated as N’ and C’, respectively. The N-domain and the two AAA+ ATPase domains (D1 and D2) are highlighted by squares in green and ovals in blue, respectively. c Close-up view of the site in a similar orientation as in part (b) in a stick and colored representation. Left part (N-domain): The conserved residues in contact A and contact C of the N-domain of ClpA are indicated by yellow and green shading, respectively. Amino acid residues that have potential roles in Zn2+ binding and peptide binding are shaded blue and orange, respectively. The colors of each corresponding residues are also given in a. Right part (ATPase domain): The symbols in front of each amino acid residue denote the highly conserved and postulated amino acid residues required for ClpA function. Asterisks (*) and triangles (▲) denote the residues conserved in Walkers A and B. Diamonds (◆) and squares (■) represent the residues conserved in sensors 1 and 2. Filled circles (●) indicate the conserved residues in the arginine finger. The amino acid residues characterized in the present study are underlined
ClpA is required for survival under heat-shock conditions
We proceeded to generate the mutants (HCS and HCA) and their complementary strains HCS(pRKclpS) and HCA(pRKclpA) as described in the “Materials and Methods” section to investigate the function of ClpS and ClpA in X. campestris pv. campestris. Considering that the expression of clpS and clpA was higher at 37 °C compared to normal temperature (28 °C), these proteins can be reasonably suggested to have a role in stress adaptation. We first characterized the effect of clpS and clpA deletions on sensitivity to heat stress. Cell suspensions from 10-fold serial dilutions of the X. campestris pv. campestris wild-type strain, clpS and clpA mutant strains, and their complementary strains were spotted on LB agar plates and incubated either at 28 °C or 37 °C. We found that at physiological temperature (28 °C), all strains spotted at all densities showed similar growth and indistinguishable colony morphologies (Fig. 5a, left part). Under heat-shock conditions (37 °C), Xcc17(pRK415), HCS(pRK415), HCS(pRKclpS), and HCA(pRKclpA) displayed similar growth behaviors, and the clpA mutant HCA(pRK415) demonstrated inhibited growth (Fig. 5a, right part). This suggests that in X. campestris pv. campestris, the deletion of clpS did not affect sensitivity to heat stress under the assay conditions, whereas the clpA gene was required for heat stress tolerance. Similar observations have been obtained for the clpA mutants of Brucella susi and Salmonella typhimurium (Ekaza et al. 2000; Sangpuii et al. 2018). The clpA mutant of B. susi shows reduced growth rates at elevated temperatures (Ekaza et al. 2000). The clpA mutant of S. typhimurium is hypersusceptible to exposure to the temperature of 42 °C (Sangpuii et al. 2018). These observations contradicted the results obtained for E. coli, in which ClpA did not appear to be a heat shock protein and clpA mutants grew well at temperatures between 25 and 42 °C (Katayama et al. 1988). Although the E. coli ClpA was not a heat-shock protein, the clpA mutant exhibited defective growth at 46 °C, and this protein appeared to have a role in cellular recovery from transient incubation at 50 °C (Thomas and Baneyx 1998).

Effects of clpS and clpA mutation on cell growth under heat-shock treatment (a) and stationary phase survival (b). a Overnight cultures of the indicated strains were spotted on LB agar plates with tenfold serial dilutions. The plates were incubated at 28 °C or 37 °C for 4 days prior to imaging. 100 indicates the inoculation concentration of bacterial cells of 5 × 108 cells/mL. The experiment was carried out least three times, and similar results were obtained. b Overnight cultures of the tested strains were diluted to an OD550 of 0.1, and survival was monitored by viable-cell counting 24 h (white bars), 48 h (gray bars), and 72 h (black bars) post-inoculation. Error bars indicate standard deviations; CFU means colony forming unit; asterisk (*) indicates significant difference (p < 0.05)
Because the level of clpS and clpA expression increased following cell growth in LB (Fig. 2a, b), it was interested to known whether ClpS and ClpA play a role in stationary phase survival. To evaluate the effect of clpS and clpA mutation on stationary-phase survival, we determined the number of viable cells after the cultures had entered stationary phase. Our results revealed that the viable counts of all tested strains including wild-type Xcc17(pRK415), two mutants [HCS(pRK415) and HCA(pRK415)], and complemented strains [HCS(pRKclpS) and HCA(pRKclpA)] are comparable at 24 h and 48 h post-inoculation (Fig. 5b). At 72 h post inoculation, the survival of clpS and clpA mutants was impaired, whereas the complementary strains shown similar survival to the wild type. These results indicate that both clpS and clpA are required for stationary-phase survival of X. campestris pv. campestris.
In bacteria, the Clp family of proteases has a multitude of functions, such as protein quality control, stress tolerance, and virulence factor expression (Malik and Brotz-Oesterhelt 2017). The clpA mutant strain of S. typhimurium shows susceptibility to HOCl (Sangpuii et al. 2018). Recently, it was reported that clpS and clpA are important for enabling E. coli to grow under H2O2 stress (Sen et al. 2020). In X. campestris pv. campestris, ClpX and ClpP, other members of the Clp family protein, are documented to be important for its survival of this bacteria under various stresses, including temperature and puromycin (Li et al. 2020; Lo et al. 2020). The X. campestris pv. campestris ClpX also has a role in SDS tolerance (Lo et al. 2020). The sensitivities of HCS and HCA to H2O2, SDS, and puromycin were assessed to examine whether clpS and clpA have roles in tolerance to other stresses. The HCS and HCA mutants did not differ from Xcc17 in terms of susceptibility to the investigated stresses (data not shown). This result was similar to the finding for Acinetobacter baumannii showing that the deletion of the clpS and clpA genes does not alter oxidative stress sensitivity (Belisario et al. 2021). Taken together, these results suggested that clpX is more important than clpA in the survival of X. campestris pv. campestris under H2O2, SDS, and puromycin treatment. We cannot exclude the contributions of ClpS and ClpA to tolerance to other stresses; further insight regarding this aspect is needed.
In the AAA+ proteins with two AAA+ ATPase modules, such as ClpA, both regions have different contributions to ATP biding/hydrolysis (Bittner et al. 2016). Mutational and functional analyses have demonstrated that the two domains in the ClpA of E. coli possess different functional roles: the first AAA+ domain in ClpA is crucial for oligomerization, whereas the second AAA+ domain is primarily responsible for ATP hydrolysis (Pak et al. 1999; Seol et al. 1995; Singh and Maurizi 1994), and the two domains operate independently even in the presence of ClpP or ClpS (Kress et al. 2009b). In E. coli ClpA, (i) the domain 1 mutant (ClpA-K220Q) cannot form a hexamer, whereas the comparable domain 2 mutant (ClpA-K501Q) associates into a hexamer; (ii) ClpA-K220Q is defective in ATPase activity and in the capability to activate protein and peptide degradation by ClpP; and (iii) ClpA-K501Q has very low ATPase activity and severely defective protein degradation activation but can activate ClpP to degrade a peptide (Singh and Maurizi 1994). We performed site-directed mutagenesis to construct mutated versions of ClpA to investigate whether the conserved motif observed in ClpA had any effect on heat stress tolerance in X. campestris pv. campestris. The most common mutated residues (K223 in AAA+ module 1 and K504 in module 2) were selected and replaced with glutamine. As depicted in Fig. 5a (left part), at 28 °C, the growth of HCA complemented with the mutated version of pRKclpAK223Q or pRKclpAK504Q did not significantly differ from that of other tested strains. However, in contrast to the complementation of HCA with the wild-type pRKclpA, the introduction of pRKclpAK223Q or pRKclpAK504Q into HCA could not restore growth behavior under heat-shock conditions (Fig. 5a, right part). This suggests that the putative residues K223 and K504 are associated with the full function of ClpA in the stress tolerance of X. campestris pv. campestris. The growth of the clpA mutant complemented with pRKclpAK223Q was similar to that of the mutant complemented with pRK415 (empty vector), whereas it was worth to note that growth was prominent in the areas spotted with 5 × 108 cells of HCA(pRK415), HCA(pRKclpA), and HCA(pRKclpAK223Q), but not in areas spotted with the same concentration of HCA(pRKclpAK504Q) at 37 °C (Fig. 5a, right part). Why the introduction of ClpA with the K504 mutation led to the failure of the clpA mutant to form colonies under heat stress treatment is unclear. The introduction of pRKclpAK504Q appeared to be detrimental to X. campestris pv. campestris, and both predicted AAA+ modules in ClpA appeared to possess different roles in this bacterium. The effects of pRKclpAK504Q in the growth of the clpA mutant and the function of each module of ClpA require further study.
The clpA mutant shows differential protein expression at high temperature
ClpA can act as a molecular chaperone in the prevention of aggregation and participates in the refolding and remodeling of proteins (Hoskins et al. 2001; Pak and Wickner 1997; Suzuki et al. 1997; Wawrzynow et al. 1996; Wickner et al. 1994). In E. coli, the amount of protein aggregation in the clpA mutant is not significantly greater than that in the wild-type cells (Dougan et al. 2002), whereas in S. typhimurium, the clpA mutant exhibits greater amounts of protein aggregates than the wild-type strain (Sangpuii et al. 2018). The affected growth of the clpA mutant at 37 °C suggested that ClpA participates in preventing the accumulation of heat-inactivated and aggregated proteins in X. campestris pv. campestris. We grew the mutant and parental strains at 28 °C or 37 °C to test whether clpA deletion would alter protein expression profiles and affect protein aggregation in heat-shocked cells. Then, protein samples were prepared and fractionated through SDS-PAGE as described in the “Materials and Methods” section. For the cultures were grown at 28 °C, the protein profiles obtained without Triton X-100 treatment did not significantly differ between the parental strain and the clpA mutant (Fig. 6, lanes 1 and 5). Similar patterns are observed when Triton X-100 treatment was administered (Fig. 6, lanes 2 and 6). When the bacteria were cultured at 37 °C, several protein bands differed between the parental strain and the clpA mutant despite the absence of the large-scale aggregation of proteins in the clpA mutant. In the absence of Triton X-100 treatment, band I was present in the wild-type Xcc17 and was missing in the clpA mutant HCA (Fig. 6, lanes 3 and 7). Under Triton X-100 treatment, band II was present in Xcc17 but not in HCA, and bands III and IV were found in HCA but were too faint to be visible in Xcc17 (Fig. 6, lanes 4 and 8). These protein bands were excised and identified using mass spectrometry. They were TolC protein (band I), hypothetical protein (band II), chemotaxis protein (band III), and low molecular weight heat shock protein (band IV). These identified proteins belong to different functional categories (Table 2). Identification of these proteins provides new targets for future studies that will allows assessment of their physiological roles and significance in clpA related functions and regulated processes.

Effects of clpA mutation on the protein production profile of X. campestris pv. campestris. The cells were cultured in XOLN medium supplemented with 2% glycerol for 24 h at 28 °C or 37 °C. Protein samples from the indicated strains were separated by using SDS-PAGE on 15% polyacrylamide gel and stained with Coomassie blue. Each lysate was loaded into the wells at the same protein concentration. The experiment was performed at least three times, and similar results were obtained. The filled triangles (◀) and open triangles (◁) indicate the bands of the proteins that were upregulated and downregulated after clpA mutation, respectively
TolC forms an outer membrane channel and is involved in export of small molecules and toxins across the outer membrane of Gram-negative bacteria (Zgurskaya et al. 2011). The higher amounts of TolC protein (band I) in heat-treated wild type than in the clpA mutant indicated that material transportation in the clpA mutant may not function properly under heat stress condition. In X. oryzae pv. oryzae, RaxC (TolC homolog) is required for AvrXa21 activity (da Silva et al. 2004). The rice XA21 protein, a receptor-like kinase, provides immunity against strains of X. oryzae pv. oryzae carrying AvrXa21 activity (da Silva et al. 2004). AvrXa21 activity requires the presence of raxA, raxB, and raxC genes that encode components of a type one secretion system (da Silva et al. 2004; Lee et al. 2006). Type one secretion system is responsible for the secretin of unfolded cognate substrates from the cytoplasm directly to the extracellular medium and is required for induction of XA21-mediated resistance in rice (Alvarez-Martinez et al. 2021; Buttner and Bonas 2010). Homologous raxA and raxB genes are not present in the sequenced X. campestris pv. campestris genomes (da Silva et al. 2002; Liu et al. 2015; Qian et al. 2005; Vorholter et al. 2008). Genome sequence analysis revealed that the type one secretion system is absent in X. campestris pv. campestris (Alvarez-Martinez et al. 2021). The physiological role of TolC and potential candidate(s) works with TolC remain to be investigated.
The conserved hypothetical protein (XCC0205) whose function is unknown in X. campestris pv. campestris belongs to Ax21 family protein. Ax21 is extensively conserved in plant pathogenic Xanthomonas and the associated genera Xylella and Stenotrophomonas (An and Tang 2018). Deletion of ax21 gene resulted in reduced biofilm formation, extracellular polysaccharide production and virulence in X. oryzae pv. oryzicola (Qian et al. 2013). Studies on Ax21 in X. oryzae pv. oryzae have shown that it is secreted in association with outer membrane vesicles (Bahar et al. 2014). In addition, phenotypic analysis demonstrated that Ax21 is required for motility and biofilm formation in X. oryzae pv. oryzae (Park et al. 2014). The Ax21 protein influences biofilm formation, motility, and virulence in Stenotrophomonas maltophilia (An and Tang 2018). The higher amounts of XCC0205 protein (band II) in heat-treated wild type than in the clpA mutant suggested that the expression of this protein might influenced in the clpA mutant under heat stress condition. Further analyses will be carried out to test whether XCC0205 functions as a virulence factor, and to examine that other phenotypes in X. campestris pv. campestris are affected by the protein.
The chemotaxis protein homolog in X. oryzae pv. oryzae is known to be required for virulence (Kumar Verma et al. 2018). The level of chemotaxis protein (band III) was increased in the Triton X-100-insoluble fraction in the clpA mutant than that in the wild type, implying that this protein forms aggregates in clpA mutant. Mutation of clpA may affect the aggregation of chemotaxis protein in heat-treated cells.
Small heat shock proteins (sHsps) are a ubiquitous family of chaperones present in all three domains of life (Haslbeck and Vierling 2015; Mogk et al. 2019). In bacteria, there are usually one or two sHsps (Haslbeck and Vierling 2015; Mogk et al. 2019). A number of bacteria encode only a single sHsp, such as the cyst forming bacterium Azotobacter vinelandii (Hsp20), cyanobacterium Synechocystis sp. PCC6803 (Hsp16.6), marine bacterium Vibrio harveyi (IbpA/B), and X. campestris pv. campestris (HspA) (Haslbeck and Vierling 2015; Obuchowski et al. 2021). Two sHsps, IbpA and IbpB, have been described in E. coli (Ratajczak et al. 2009). Deinococcus radiodurans also possess two sHsps, Hsp17.7 and Hsp20.2 (Bepperling et al. 2012). The A. vinelandii Hsp20 is essential for cyst desiccation resistance (Cocotl-Yanez et al. 2014), and Synechocystis Hsp16.6 is involved in the development of thermotolerance (Lee et al. 2000). The IbpA/B protein of V. harveyi binds to proteins aggregated in a cell during heat shock (Klein et al. 2001). The HspA in X. campestris pv. campestris is required for survival under heat stress conditions and possesses an intrinsic ability to reactivate inactivate proteins (Lin et al. 2010). The E. coli IbpA and IbpB were found in a fraction of aggregated proteins formed following heat stress and were reported to interact with endogenous polypeptides upon heat stress (Laskowska et al. 1996). IbpA and IbpB co-operate with ClpB and the DnaK system in reversing protein aggregation (Mogk et al. 2003a, b), and there is an interplay between IbpA and IbpB in promoting efficient protein disaggregation (Ratajczak et al. 2009). In D. radiodurans, Hsp20.2 is known to associate with aggregated protein and cooperate with ATP-dependent chaperones in their refolding, whereas Hsp17.7 appears to keep substrates in a refolding competent state by transient interactions (Bepperling et al. 2012). sHsps act as the first line of cellular defense against protein unfolding stress and can prevent the irreversible aggregation of denaturing proteins (Haslbeck and Vierling 2015; Mogk et al. 2019). It has been proposed that sHsps bind to aggregation-prone protein substrates to form assemblies that keep substrates from irreversible aggregation (Obuchowski et al. 2021). In vivo sHsps frequently localize to insoluble protein fractions of heat stressed cells (Mogk et al. 2003a). The level of HspA (band IV) is increased in the Triton X-100-insoluble fraction in the clpA mutant than that in the wild type, implying that HspA might coaggregate with midfolded proteins in clpA mutant under heat stress. The potential substrate proteins bound by HspA requires further determination.
ClpA is required for full virulence
Given that Clp family proteins have been documented to be important for the pathogenicity of several bacterial species (Brotz-Oesterhelt and Sass 2014), we aimed to test whether this situation is also the case in X. campestris pv. campestris. The virulence of X. campestris pv. campestris was tested on the host plant cabbage through leaf-clipping inoculation to determine whether clpS and clpA is involved in pathogenicity. The inoculants for the pathogenicity test included Xcc17(pRK415), HCS(pRK415), HCS(pRKclpS), HCA(pRK415), and HCA(pRKclpA). The length of the lesion caused by the wild-type strain was approximately 2 cm at 14 days postinoculation, and the virulence symptoms of the clpS mutant and its complementary strain were similar to those of the wild-type (Fig. 7). Although the clpA mutant could cause disease, its virulence was significantly attenuated compared with that of the wild type, and the disease symptoms caused by the complemented strain were restored toward those of the wild-type phenotype (Fig. 7). These results indicated that clpS was not related to virulence and that clpA was required for the full virulence of X. campestris pv. campestris. These results were similar to the observations reported for Pseudomonas aeruginosa, in which the clpA mutant but not the clpS mutant exhibits virulence-attenuated phenotypes (Feinbaum et al. 2012). The disruption of ClpA function has been suggested to be responsible for the altered pathogenesis of Ralstonia solanacearum (Lin et al. 2008). A recent study on A. baumannii found that clpS and clpA are important for virulence (Belisario et al. 2021).

Effects of clpS and clpA mutation on the virulence of X. campestris pv. campestris against cabbage. a Black rot symptoms caused by the indicated strains on the inoculated leaves of cabbage. Lesion lengths were measured, and images were taken on day 14 postinoculation. Scale bars = 1 cm. Similar results were obtained at least three times. b The values of average lesion lengths (cm) are presented as mean ± standard deviation. Asterisk (*) indicates significant difference (p < 0.05); ns means no significant difference
In Xanthomonas species, successful infection often depends on an arsenal of virulence factors, such as adhesins, degradative enzymes, and extracellular polysaccharides (Buttner and Bonas 2010; Chan and Goodwin 1999; Denance et al. 2016; Dow et al. 2003; Tang et al. 2021). We investigated the contribution of clpA to bacterial attachment and extracellular enzyme production to test whether the reduced virulence of the clpA mutant is correlated with the reduced production of these pathogenicity factors. No considerable differences in bacterial attachment and extracellular enzyme production were observed between the clpA mutant and the parental strain (data not shown). In A. baumannii, clpS and clpA are essential for biofilm formation (Belisario et al. 2021). In X. campestris pv. campestris, clpX is required for bacterial attachment (Lo et al. 2020). The annotation of the X. campestris pv. campestris genome has revealed that numerous virulence determinants are associated with bacterial pathogenesis and that clpA might affect other unidentified factor(s) that require further evaluation.
In addition to the above results for ClpS, the clpS mutant was observed to have no effect on stress tolerance and virulence. Even its expression was heat inducible. Whether this gene has other physiological role(s) in X. campestris pv. campestris remains to be investigated. In the case of ClpA, its mutation resulted in growth defects at high temperature and attenuated pathogenicity but did not significantly alter virulence-related factors and responses to a range of stresses. In contrast to the previous findings for ClpX and the other member of Clp ATPase family, the findings of this work showed that the mutation of clpX caused pleiotropic effects, including attachment, virulence, and stress tolerance (Lo et al. 2020). Therefore, ClpX could be reasonably suggested to play a highly essential role in X. campestris pv. campestris. On the basis of the observations showing that (i) clpA expression is heat inducible; (ii) clpA mutation affects growth at high temperatures; and (iii) the clpA mutant presents different protein patterns under heat treatment, the ClpAP protease could be inferred to have evolved to address the increased need for proteolysis caused by enhanced denaturation or aggregation under heat-shock conditions. Whether ClpA can associate with ClpP remains unknown, and the role of ClpS as a modulator that affects the function of ClpA in X. campestris pv. campestris remains unclear. The further clarification of the nature of the interaction among ClpS, ClpA, and ClpP can provide novel insights and a comprehensive description of these proteins.
Conclusion
We investigated the transcription and function of clpS and clpA in X. campestris pv. campestris. First, upstream region analysis together with a reporter assay indicated that (i) putative σ32-dependent sequences are present in the upstream region of clpS and clpA; (ii) clpS and clpA contain their own promoters and are transcribed independently; and (iii) the levels of clpS and clpA promoters under heat stress are induced approximately twofold relative to those under the control treatment. Second, through sequence comparison and homology modeling, the conserved sequence motifs were identified, and the predicted three-dimensional structures of ClpS and ClpA were determined. Third, by combining insertional inactivation and phenotypic evaluation, the mutation of clpA but not of clpS was found to cause defects in heat stress tolerance and pathogenicity, and the conserved residues of Walker A motif of the AAA+ ATPase module of ClpA were demonstrated to be required for the function of ClpA in heat tolerance. Finally, SDS-PAGE analysis revealed that the clpA mutant showed different protein profiles in response to heat-shock treatment.
The Clp family proteins are found in most domains of life, and their functional importance varies from organism to organism. The genome of X. campestris pv. campestris has been predicted to encode several common Clp family proteins, such as ClpS, ClpA, ClpX, ClpP, ClpQ, and ClpY (da Silva et al. 2002; Liu et al. 2015; Qian et al. 2005; Vorholter et al. 2008). Although these proteins have been well-described in other organisms, their functions in X. campestris pv. campestris are poorly understood. To date, only the functions of ClpX and ClpP in X. campestris pv. campestris have been documented in the literature. The importance of ClpX and ClpP in extracellular enzyme production, virulence, and heat and puromycin tolerance suggests that ClpXP is important for pathogenesis and environmental adaptation (Li et al. 2020; Lo et al. 2020). The clpX mutant of X. campestris pv. campestris also exhibits reduced bacterial attachment and increased sensitivity to SDS (Lo et al. 2020). The expression of X. campestris pv. campestris clpP is shown to be heat inducible (Li et al. 2020). A growing body of evidence, combined with the findings that we have presented here, shows that Clp family proteins can be regarded as key virulence factors in X. campestris pv. campestris. These proteins are plausible candidates for the development of new therapeutics for combating the phytopathogen X. campestris pv. campestris.
Availability of data and material
Not applicable.
References
Alvarez-Martinez CE et al (2021) Secrete or perish: the role of secretion systems in Xanthomonas biology. Comput Struct Biotechnol J 19:279–302. https://doi.org/10.1016/j.csbj.2020.12.020
An SQ, Tang JL (2018) The Ax21 protein influences virulence and biofilm formation in Stenotrophomonas maltophilia. Arch Microbiol 200:183–187. https://doi.org/10.1007/s00203-017-1433-7
An SQ et al (2020) Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol Rev 44:1–32. https://doi.org/10.1093/femsre/fuz024
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. https://doi.org/10.1093/bioinformatics/bti770
Bahar O et al (2014) The Xanthomonas Ax21 protein is processed by the general secretory system and is secreted in association with outer membrane vesicles. PeerJ 2:e242. https://doi.org/10.7717/peerj.242
Baker TA, Sauer RT (2006) ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci 31:647–653. https://doi.org/10.1016/j.tibs.2006.10.006
Belisario JC, Lee HH, Luknauth H, Rigel NW, Martinez LR (2021) Acinetobacter baumannii strains deficient in the Clp chaperone-protease genes have reduced virulence in a murine model of pneumonia. Pathogens. https://doi.org/10.3390/pathogens10020204
Bepperling A et al (2012) Alternative bacterial two-component small heat shock protein systems. Proc Natl Acad Sci U S A 109:20407–20412. https://doi.org/10.1073/pnas.1209565109
Bittner LM, Arends J, Narberhaus F (2016) Mini review: ATP-dependent proteases in bacteria. Biopolymers 105:505–517. https://doi.org/10.1002/bip.22831
Blattner FR et al (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. https://doi.org/10.1126/science.277.5331.1453
Brotz-Oesterhelt H, Sass P (2014) Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int J Med Microbiol 304:23–30. https://doi.org/10.1016/j.ijmm.2013.09.001
Buttner D, Bonas U (2010) Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev 34:107–133. https://doi.org/10.1111/j.1574-6976.2009.00192.x
Chan JW, Goodwin PH (1999) The molecular genetics of virulence of Xanthomonas campestris. Biotechnol Adv 17:489–508
Chandu D, Nandi D (2004) Comparative genomics and functional roles of the ATP-dependent proteases Lon and Clp during cytosolic protein degradation. Res Microbiol 155:710–719. https://doi.org/10.1016/j.resmic.2004.06.003
Cocotl-Yanez M, Moreno S, Encarnacion S, Lopez-Pliego L, Castaneda M, Espin G (2014) A small heat-shock protein (Hsp20) regulated by RpoS is essential for cyst desiccation resistance in Azotobacter vinelandii, Microbiology (Reading) 160:479–487 doi:https://doi.org/10.1099/mic.0.073353-0
da Silva AC et al (2002) Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417:459–463
Denance N, Lahaye T, Noel LD (2016) Editorial: genomics and effectomics of the crop killer Xanthomonas. Front Plant Sci 7:71. https://doi.org/10.3389/fpls.2016.00071
Dougan DA, Reid BG, Horwich AL, Bukau B (2002) ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9:673–683. https://doi.org/10.1016/s1097-2765(02)00485-9
Dow JM, Crossman L, Findlay K, He YQ, Feng JX, Tang JL (2003) Biofilm dispersal in Xanthomonas campestris is controlled by cell-cell signaling and is required for full virulence to plants. Proc Natl Acad Sci USA 100:10995–11000
Ekaza E, Guilloteau L, Teyssier J, Liautard JP, Kohler S (2000) Functional analysis of the ClpATPase ClpA of Brucella suis, and persistence of a knockout mutant in BALB/c mice Microbiology (Reading) 146 ( Pt 7):1605–1616 . https://doi.org/10.1099/00221287-146-7-1605
Erbse A et al (2006) ClpS is an essential component of the N-end rule pathway in E. coli. Nature 439:753–756. https://doi.org/10.1038/nature04412
Erbse AH et al (2008) Conserved residues in the N-domain of the AAA+ chaperone ClpA regulate substrate recognition and unfolding. FEBS J 275:1400–1410. https://doi.org/10.1111/j.1742-4658.2008.06304.x
Feinbaum RL, Urbach JM, Liberati NT, Djonovic S, Adonizio A, Carvunis AR, Ausubel FM (2012) Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLoS Pathog 8:e1002813. https://doi.org/10.1371/journal.ppat.1002813
Fu JF, Tseng YH (1990) Construction of lactose-utilizing Xanthomonas campestris and production of xanthan gum from whey. Appl Environ Microbiol 56:919–923
Gottesman S, Clark WP, Maurizi MR (1990) The ATP-dependent Clp protease of E. coli Sequence of clpA and identification of a Clp-Specific substrate. J Biol Chem 265:7886–7893
Guo F, Esser L, Singh SK, Maurizi MR, Xia D (2002) Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone. ClpA J Biol Chem 277:46753–46762. https://doi.org/10.1074/jbc.M208104200
Haslbeck M, Vierling E (2015) A first line of stress defense: small heat shock proteins and their function in protein homeostasis. J Mol Biol 427:1537–1548. https://doi.org/10.1016/j.jmb.2015.02.002
Hoskins JR, Sharma S, Sathyanarayana BK, Wickner S (2001) Clp ATPases and their role in protein unfolding and degradation. Adv Protein Chem 59:413–429. https://doi.org/10.1016/s0065-3233(01)59013-0
Hsiao YM, Liao HY, Lee MC, Yang TC, Tseng YH (2005) Clp upregulates transcription of engA gene encoding a virulence factor in Xanthomonas campestris by direct binding to the upstream tandem Clp sites. FEBS Lett 579:3525–3533
Hsiao YM, Liu YF, Fang MC, Song WL (2011) XCC2731, a GGDEF domain protein in Xanthomonas campestris, is involved in bacterial attachment and is positively regulated by Clp, Microbiol Res 166:548–565 doi:S0944–5013(10)00109–6 [pii]. https://doi.org/10.1016/j.micres.2010.11.003
Katayama Y, Gottesman S, Pumphrey J, Rudikoff S, Clark WP, Maurizi MR (1988) The two-component, ATP-dependent Clp protease of E. coli purification, cloning, and mutational analysis of the ATP-binding component. J Biol Chem 263:15226–15236
Keen NT, Tamaki S, Kobayashi D, Trollinger D (1988) Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197
Klein G, Laskowska E, Taylor A, Lipinska B (2001) IbpA/B small heat-shock protein of marine bacterium Vibrio harveyi binds to proteins aggregated in a cell during heat shock. Mar Biotechnol (NY) 3:346–354. https://doi.org/10.1007/s10126001-0009-2
Koo BM, Rhodius VA, Campbell EA, Gross CA (2009) Dissection of recognition determinants of E. coli sigma32 suggests a composite -10 region with an “extended -10” motif and a core -10 element. Mol Microbiol 72:815–829. https://doi.org/10.1111/j.1365-2958.2009.06690.x
Kress W, Maglica Z, Weber-Ban E (2009a) Clp chaperone-proteases: structure and function. Res Microbiol 160:618–628. https://doi.org/10.1016/j.resmic.2009.08.006
Kress W, Mutschler H, Weber-Ban E (2009b) Both ATPase domains of ClpA are critical for processing of stable protein structures. J Biol Chem 284:31441–31452. https://doi.org/10.1074/jbc.M109.022319
Kthiri F et al (2010) Protein aggregation in a mutant deficient in YajL, the bacterial homolog of the Parkinsonism-associated protein DJ-1. J Biol Chem 285:10328–10336. https://doi.org/10.1074/jbc.M109.077529
Kumar Verma R, Samal B, Chatterjee S (2018) Xanthomonas oryzae pv. oryzae chemotaxis components and chemoreceptor Mcp2 are involved in the sensing of constituents of xylem sap and contribute to the regulation of virulence-associated functions and entry into rice. Mol Plant Pathol 19:2397–2415. https://doi.org/10.1111/mpp.12718
Laskowska E, Wawrzynow A, Taylor A (1996) IbpA and IbpB, the new heat-shock proteins, bind to endogenous E. coli proteins aggregated intracellularly by heat shock. Biochimie 78:117–122. https://doi.org/10.1016/0300-9084(96)82643-5
Lee TC, Chen ST, Lee MC, Chang CM, Chen CH, Weng SF, Tseng YH (2001) The early stages of filamentous phage phiLf infection require the host transcription factor. Clp J Mol Microbiol Biotechnol 3:471–481
Lee S, Owen HA, Prochaska DJ, Barnum SR (2000) HSP16.6 is involved in the development of thermotolerance and thylakoid stability in the unicellular cyanobacterium, Synechocystis sp. PCC 6803. Curr Microbiol 40:283–287. https://doi.org/10.1007/s002849910056
Lee SW, Han SW, Bartley LE, Ronald PC (2006) From the academy: colloquium review. Unique characteristics of Xanthomonas oryzae pv. oryzae AvrXa21 and implications for plant innate immunity. Proc Natl Acad Sci USA 103:18395–18400. https://doi.org/10.1073/pnas.0605508103
Lee KJ, Jung YC, Park SJ, Lee KH (2018) Role of heat shock proteases in quorum-sensing-mediated regulation of biofilm formation by Vibrio Species, mBio. https://doi.org/10.1128/mBio.02086-17
Li CE, Liao CT, Lo HH, Hsiao YM (2020) Functional characterization and transcriptional analysis of clpP of Xanthomonas campestris pv. campestris. Curr Microbiol 77:2876–2885. https://doi.org/10.1007/s00284-020-02093-1
Lin YM et al (2008) Transposon mutagenesis reveals differential pathogenesis of Ralstonia solanacearum on tomato and Arabidopsis. Mol Plant Microbe Interact 21:1261–1270. https://doi.org/10.1094/MPMI-21-9-1261
Lin CH, Lee CN, Lin JW, Tsai WJ, Wang SW, Weng SF, Tseng YH (2010) Characterization of Xanthomonas campestris pv. campestris heat shock protein A (HspA), which possesses an intrinsic ability to reactivate inactivated proteins. Appl Microbiol Biotechnol 88:699–709. https://doi.org/10.1007/s00253-010-2776-z
Liu YC et al (2015) Complete genome sequence of Xanthomonas campestris pv. campestris strain 17 from Taiwan. Genome Announc. https://doi.org/10.1128/genomeA.01466-15
Lo HH, Liao CT, Li CE, Chiang YC, Hsiao YM (2020) The clpX gene plays an important role in bacterial attachment, stress tolerance, and virulence in Xanthomonas campestris pv. campestris. Arch Microbiol 202:597–607. https://doi.org/10.1007/s00203-019-01772-3
Mahmoud SA, Chien P (2018) Regulated proteolysis in bacteria. Annu Rev Biochem 87:677–696. https://doi.org/10.1146/annurev-biochem-062917-012848
Malik IT, Brotz-Oesterhelt H (2017) Conformational control of the bacterial Clp protease by natural product antibiotics. Nat Prod Rep 34:815–831. https://doi.org/10.1039/c6np00125d
Miller JM, Enemark EJ (2016) Fundamental characteristics of AAA+ protein family structure and function. Archaea 2016:9294307. https://doi.org/10.1155/2016/9294307
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y.
Mogk A, Deuerling E, Vorderwulbecke S, Vierling E, Bukau B (2003a) Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol 50:585–595. https://doi.org/10.1046/j.1365-2958.2003.03710.x
Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B (2003b) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278:31033–31042. https://doi.org/10.1074/jbc.M303587200
Mogk A, Ruger-Herreros C, Bukau B (2019) Cellular functions and mechanisms of action of small heat shock proteins. Annu Rev Microbiol 73:89–110. https://doi.org/10.1146/annurev-micro-020518-115515
Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA (2006) Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev 20:1776–1789. https://doi.org/10.1101/gad.1428206
Obuchowski I, Karas P, Liberek K (2021) The small ones matter-sHsps in the bacterial chaperone network. Front Mol Biosci 8:666893. https://doi.org/10.3389/fmolb.2021.666893
Olivares AO, Baker TA, Sauer RT (2018) Mechanical protein unfolding and degradation. Annu Rev Physiol 80:413–429. https://doi.org/10.1146/annurev-physiol-021317-121303
Pak M, Wickner S (1997) Mechanism of protein remodeling by ClpA chaperone. Proc Natl Acad Sci U S A 94:4901–4906. https://doi.org/10.1073/pnas.94.10.4901
Pak M, Hoskins JR, Singh SK, Maurizi MR, Wickner S (1999) Concurrent chaperone and protease activities of ClpAP and the requirement for the N-terminal ClpA ATP binding site for chaperone activity. J Biol Chem 274:19316–19322. https://doi.org/10.1074/jbc.274.27.19316
Park HJ, Lee SW, Han SW (2014) Proteomic and functional analyses of a novel porin-like protein in Xanthomonas oryzae pv. oryzae. J Microbiol 52:1030–1035. https://doi.org/10.1007/s12275-014-4442-0
Qian W et al (2005) Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res 15:757–767
Qian G et al (2013) Proteomic analysis reveals novel extracellular virulence-associated proteins and functions regulated by the diffusible signal factor (DSF) in Xanthomonas oryzae pv. oryzicola. J Proteome Res 12:3327–3341. https://doi.org/10.1021/pr4001543
Ratajczak E, Zietkiewicz S, Liberek K (2009) Distinct activities of E. coli small heat shock proteins IbpA and IbpB promote efficient protein disaggregation. J Mol Biol 386:178–189. https://doi.org/10.1016/j.jmb.2008.12.009
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd. Cold Spring Harbor Press, Cold Spring Harbor, N. Y.
Sangpuii L et al (2018) Comparative roles of clpA and clpB in the survival of S. Typhimurium under stress and virulence in poultry. Sci Rep 8:4481. https://doi.org/10.1038/s41598-018-22670-6
Sauer RT, Baker TA (2011) AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. https://doi.org/10.1146/annurev-biochem-060408-172623
Schmidt R, Zahn R, Bukau B, Mogk A (2009) ClpS is the recognition component for E. coli substrates of the N-end rule degradation pathway. Mol Microbiol 72:506–517. https://doi.org/10.1111/j.1365-2958.2009.06666.x
Schweizer HD (1993) Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. Biotechniques 15:831–834
Sen A, Zhou Y, Imlay JA (2020) During oxidative stress the Clp proteins of E. coli ensure that iron pools remain sufficient to reactivate oxidized metalloenzyme. J Bacteriol. https://doi.org/10.1128/JB.00235-20
Seol JH, Baek SH, Kang MS, Ha DB, Chung CH (1995) Distinctive roles of the two ATP-binding sites in ClpA, the ATPase component of protease Ti in Escherichia coli. J Biol Chem 270:8087–8092. https://doi.org/10.1074/jbc.270.14.8087
da Silva FG, Shen Y, Dardick C, Burdman S, Yadav RC, de Leon AL, Ronald PC (2004) Bacterial genes involved in type I secretion and sulfation are required to elicit the rice Xa21-mediated innate immune response. Mol Plant Microbe Interact 17:593–601. https://doi.org/10.1094/MPMI.2004.17.6.593
Singh SK, Maurizi MR (1994) Mutational analysis demonstrates different functional roles for the two ATP-binding sites in ClpAP protease from Escherichia coli. J Biol Chem 269:29537–29545
Suzuki CK, Rep M, van Dijl JM, Suda K, Grivell LA, Schatz G (1997) ATP-dependent proteases that also chaperone protein biogenesis. Trends Biochem Sci 22:118–123. https://doi.org/10.1016/s0968-0004(97)01020-7
Tang JL, Tang DJ, Dubrow ZE, Bogdanove A, An SQ (2021) Xanthomonas campestris pathovars. Trends Microbiol 29:182–183. https://doi.org/10.1016/j.tim.2020.06.003
Thomas JG, Baneyx F (1998) Roles of the Escherichia coli small heat shock proteins IbpA and IbpB in thermal stress management: comparison with ClpA. ClpB, and HtpG in Vivo J Bacteriol 180:5165–5172. https://doi.org/10.1128/JB.180.19.5165-5172.1998
Torres-Delgado A, Kotamarthi HC, Sauer RT, Baker TA (2020) The intrinsically disordered N-terminal extension of the ClpS adaptor reprograms its partner AAA+ ClpAP protease. J Mol Biol 432:4908–4921. https://doi.org/10.1016/j.jmb.2020.07.007
Vieira J, Messing J (1991) New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene 100:189–194
Vorholter FJ et al (2008) The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J Biotechnol 134:33–45
Wang TW, Tseng YH (1992) Electrotransformation of Xanthomonas campestris by RF DNA of filamentous phage ϕLf. Lett Appl Microbiol 14:65–68
Wawrzynow A, Banecki B, Zylicz M (1996) The Clp ATPases define a novel class of molecular chaperones. Mol Microbiol 21:895–899. https://doi.org/10.1046/j.1365-2958.1996.421404.x
Wendler P, Ciniawsky S, Kock M, Kube S (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta 1823:2–14. https://doi.org/10.1016/j.bbamcr.2011.06.014
Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi MR (1994) A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad Sci USA 91:12218–12222. https://doi.org/10.1073/pnas.91.25.12218
Xia D, Esser L, Singh SK, Guo F, Maurizi MR (2004) Crystallographic investigation of peptide binding sites in the N-domain of the ClpA chaperone. J Struct Biol 146:166–179. https://doi.org/10.1016/j.jsb.2003.11.025
Yang BY, Tseng YH (1988) Production of exopolysaccharide and levels of protease and pectinase activity in pathogenic and non-pathogenic strains of Xanthomonas campestris pv. Campestris. Bot Bull Acad Sin 29:93–99
Yeom J, Gao X, Groisman EA (2018) Reduction in adaptor amounts establishes degradation hierarchy among protease substrates. Proc Natl Acad Sci U S A 115:E4483–E4492. https://doi.org/10.1073/pnas.1722246115
Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA (2002) Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat Struct Biol 9:906–911. https://doi.org/10.1038/nsb869
Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S (2011) Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front Microbiol 2:189. https://doi.org/10.3389/fmicb.2011.00189
Funding
This study was supported by the Ministry of Science and Technology of Taiwan (Grants No. MOST 107-2313-B-166-001-MY3 and MOST 110-2313-B-166-001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Code availability
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lo, HH., Chang, HC., Liao, CT. et al. Expression and function of clpS and clpA in Xanthomonas campestris pv. campestris. Antonie van Leeuwenhoek 115, 589–607 (2022). https://doi.org/10.1007/s10482-022-01725-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-022-01725-9