
Abstract
A new triterpene ester (1) together with eight known compounds (2–9) were isolated from the leaves of Cadaba farinosa Forssk. Their chemical structures were established on the basis of physical, chemical, and spectroscopic methods (IR, 1D and 2D NMR, and mass spectral analyses) to be: lupeol-3-O-decanoate (1), lupeol (2), β-sitosterol (3), ursolic acid (4), 12-aminododecanoic (5), dillenetin-3-O-β-d-glucopyranoside (6), stachydrine (7), 3-hydroxy-stachydrine (8), and quercetin-3-O-β-d-glucopyranoside (9). That is the first report for the isolation of compound 5 from a plant source. Compounds 5, 6, and 9 were evaluated for their antioxidant activity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cadaba farinosa Forssk. (family Capparidaceae) is a slender shrub with strongly furrowed stem. The plant grows in African Sahel and Saudi Arabia (Rahman et al., 2004; Telrandhe et al., 2010a). It is a West African plant known by different names as dangarafa (Hausa) and legel (Fulani) in Nigeria; maure in Mauritania and Senegal; quinquemini in Guinea; balamji in Niger (Gaffa and Ayo, 2003); and Azan-al-arnab in Saudi Arabia (Rahman et al., 2004). The leaf juice is used as a remedy for dysentery, fever, cough, anthrax, and lungs problem in Africa, Saudi Arabia, and India (Gohar, 2002; Rahman et al., 2004). It possesses anti-diabetic, antioxidant, anthelmintic, purgative, anti-inflammatory, and hepatoprotective activities (Tenpe et al., 2006; Sanghi et al., 2007; Telrandhe et al., 2010b). Previous chemical investigations of C. farinosa revealed the presence of alkaloids, sesquiterpenes, phenolic acids, and fatty acids (Viqar Uddin et al., 1985; Yousif et al., 1984; Viqar Uddin et al., 1987; Viqar Uddin et al., 1990; Glew et al., 2010). No reports on the constituents of C. farinosa growing in Saudi Arabia, so a comprehensive study of the plant’s constituents was undertaken. This article reports the isolation and characterization of one new triterpene ester (1) together with eight known compounds (2–9) from the leaves of C. farinosa. Also documents the isolation of compound 5 for the first time from plant source. Compounds 5, 6, and 9 exhibited good antioxidant activity.
Materials and methods
General
Melting point was carried out in Electrothermal 9100 Digital Melting Point (England). IR was measured on Schimadzu Infrared-400 spectrophotometer (Japan). The UV spectra were carried out in MeOH using a perkin elmer lambda 25 UV-Vis spectrophotometer. Low-resolution mass spectra were recorded on a Finnigan MAT TSQ 7000 mass spectrometer. HRESIMS was determined with a micromass Qtof two mass spectrometer. HRFABMS was determined on a Finnigan MAT-312. UV spectra were recorded in MeOH on a Shimadzu 1601 UV-Vis spectrophotometer. 1H and 13C NMR spectra were measured on Bruker DRX 500 spectrometer (Bruker, Rheinstetten, Germany). A GCMS was performed on Clarus 500 GC/MS (perkin elmer, Shelton, CT). The software controller/integrator was turbo mass, version 4.5.0.007 (perkin elmer). An elite 5 MS GC capillary column (30 × 0.25 mm × 0.5 μm, perkin elmer) was used. The carrier gas was helium at a flow rate of 2 mL/min (32 p.s.i., flow initial 55.8 cm/s, split; 1:40). Temperature conditions were: inlet line temperature, 200 °C; source temperature, 150 °C; trap emission, 100 °C; and electron energy, 70 eV. The column temperature program was: 50 °C for 5 min, increased to 220 °C (rate, 20 °C/min), and held for 5 min. The injector temperature was 220 °C. MS scan was from 50 to 650 m/z. Vacuum liquid chromatography (VLC) was carried out on silica gel 60 (0.04–0.063 mm, Merck). Column chromatographic separations were performed over silica gel 60 (0.04–0.063 mm, Merck) and sephadex LH-20 (0.25–0.1 mm Merck). TLC analyses were carried out on pre-coated silica gel F254 aluminum sheets and RP-18 F254s glass plates (Merck). Compounds were detected by UV absorption at λmax 255 and 366 nm followed by spraying with anisaldehyde/H2SO4 reagent and heating at 110 °C for 1–2 min. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and propyl gallate (PG) as reference samples for antioxidant were purchased from sigma chemical co. (Germany).
Plant material
The leaves of C. farinosa Forssk. were collected in March 2010 from Al-Baha, Saudi Arabia. The plant was kindly identified by taxonomist at the department of pharmacognosy, college of pharmacy, King Saud University. A voucher specimen has been deposited at the herbarium of research center for medicinal aromatic and poisonous plants of the same college, under the registration number CF-3-2008.
Chemistry
Extraction and isolation
The air-dried powdered leaves (400 g) were extracted with MeOH (2.5 L × 4) at room temperature. The combined extract was concentrated under reduced pressure to afford a dark greenish brown residue (20.3 g). The latter was suspended in distilled water (200 mL) then partitioned between n-hexane (500 mL × 4), EtOAc (500 mL × 4), and n-butanol (500 mL × 3), successively. Each fraction was concentrated under reduced pressure to give n-hexane (4.3 g), EtOAc (4.0 g), n-butanol (4.1 g), and aqueous (5.8 g) fractions.
The n-hexane fraction (4.3 g) was subjected to VLC (100 g, silica gel) using n-hexane:EtOAc gradient, five fractions were obtained. Fraction-2 (0.72 g, n-hexane:EtOAc 75:25) was chromatographed on silica gel column (120 g × 50 × 3 cm) using n-hexane:EtOAc gradients to afford compounds 1 (13 mg, white amorphous powder) and 2 (18 mg, colorless crystals). Fraction-3 (0.73 g, n-hexane:EtOAc 50:50) was subjected to silica gel column (120 g × 50 × 3 cm) using n-hexane:EtOAc gradients to afford compound 3 (52 mg, white needles). Fraction-4 (0.65 g, n-hexane:EtOAc 25:75) was chromatographed over silica gel column (100 g × 50 × 3 cm) using n-hexane:EtOAc as an eluent to afford three main subfractions (A–C). Column chromatography (100 g × 50 × 3 cm) of subfraction B (0.23 g) using n-hexane:EtOAc gradient yielded compounds 4 (16 mg, White crystals) and 5 (9 mg, white crystals).
The EtOAc fraction was subjected to VLC (100 g, silica gel) using CHCl3:MeOH gradients, four fractions were obtained. Fraction-2 (0.83 g, CHCl3:MeOH 50:50) was subjected to silica gel column (120 g × 50 × 3 cm) using CHCl3:MeOH gradients to afford compounds 6 (16 mg, yellow amorphous powder) and 7 (28 mg, colorless oil). Fraction-3 (0.78 g, CHCl3:MeOH 25:75) was chromatographed over sephadex LH-20 column (150 g × 100 × 5 cm) using CHCl3:MeOH (1:9) as an eluent to obtain three main subfractions (D–F). Subfraction E (0.21 g) was subjected to silica gel column (80 g × 50 × 3 cm) using CHCl3–MeOH gradients to give compound 9 (12 mg, yellow amorphous powder). Silica gel column chromatography of subfraction-F (0.31 g) using CHCl3–MeOH gradient gave compound 8 (19 mg, colorless oil).
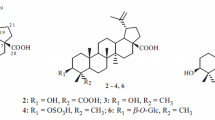
Lupeol 3-O-decanoate (1): white amorphous powder. IR (KBr): γ max 2,835, 1,695 1,664, 885, 725 cm−1. HRFABMS m/z: 581.5214 (Calcd. for C40H69O2 581.5219) [M + H]+, 410.4611 [C30H50]+, 426.6912 [C30H50O]+. 1H and 13C NMR (see Table 1).
12-Aminododecanoic acid (5): white crystals; m. p. 186–187 οC. IR (KBr): γ max 3,280, 2,950, 2,845, 1,710, 725 cm−1. HRESIMS m/z: 216.1889 [M + H]+ (calcd. for C12H26NO2 216.1885). 1H and 13C NMR (see Table 1).
Alkaline hydrolysis of compound 1
A solution of 1 (7 mg) in 3 % KOH/MeOH (4 mL) was left to stand for 15 min at room temperature then neutralized with 1 N HCl/MeOH. The solution was extracted with CHCl3. The solvent was evaporated and the residue obtained was chromatographed on a silica gel column using n-hexane:EtOAc gradient to furnish lupeol and methyl ester of decanoic acid. Lupeol was identified by EIMS analysis and co-TLC with authentic sample. The methyl ester of decanoic acid was identified by GCMS (Fig. 1).

Chemical structures of isolated compounds
Free radical scavenging activity (DPPH assay)
The method was carried out as previously described (Mohamed, 2008). The tested compounds were dissolved in HPLC MeOH to obtain a concentration of 20 μM. Then 0.5 mL of sample was mixed with 1 mL of DPPH (11.8 mg in 1 L HPLC MeOH) and allowed to stand for half an hour for any reaction to occur. The UV absorbance was recorded at 517 nm. The experiment was performed in triplicate and the average absorption was noted. The antioxidant activity was calculated using the following equation:
Results and discussion
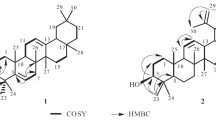
Compound 1 was obtained as white amorphous powder and its molecular formula was determined to be C40H68O2 by HRFABMS (m/z 581.5214 [M + H]+), representing seven degrees of unsaturation. The IR spectrum exhibited absorption bands at 1,695 (carbonyl) and 1,664, 885 cm−1 (exocyclic di-substituted double bond) as well as absorption band at 725 cm−1 for long aliphatic chain (Chumkaew et al., 2005; Furukawa et al., 2002). The 13C NMR spectrum indicated that 1 was a triterpenoid with a lupane skeleton (Chumkaew et al., 2005; Furukawa et al., 2002; Jain and Bari, 2010). This was established by the characteristic signals for two broad singlet signals at δ H 4.56 (H-29a) and 4.68 (H-29b) in the 1H NMR spectrum, together with the values of unsaturated carbons at δ C 109.3 (C-29) and 151.0 (C-20), as well as seven tertiary methyl signals at δ H 0.79, 0.83, 0.84, 0.93, 0.96, 1.06, and 1.68, respectively, and confirmed by the fragment ion peaks at m/z 410.4611 [C30H50]+ and 426.6912 [C30H50O]+. Moreover, a multiplet signal at δ H 4.38 was assigned to an oxymethine proton H-3. Additionally, a terminal methyl signal at δ H 0.88 (t, J = 6.7 Hz, H-10), multiplet methylene at δ H 2.38 (H-2′), and strong methylene proton signals around δ H 1.25 were indicative of the presence of a fatty acid, which was also supported by the appearance of 13C signals due to an ester carbonyl group at δ C 170.2, a long-chain of methylene groups at δ C 29.4–29.7, and a terminal methyl at δ C 14.1. The fatty acid was estimated to be composed of C10 by alkaline treatment of 1 to afford lupeol and decanoic acid methyl ester that was identified by GCMS analysis. The downfield shift of H-3 (δ H 4.38) in 1 in comparison to its corresponding signal in 2 (δ H 3.17) revealed the attachment of the fatty acid at C-3 of 1 that was confirmed by the HMBC correlation of H-3 with C-1′ (Fig. 2). On the basis of these findings, 1 was assigned as lupeol 3-O-decanoate and found to be a new natural product. This is the first report for the isolation of fatty acid ester of a triterpene from this plant.

Important COSY and HMBC correlations for compounds 1 and 5
Compound 5 was isolated as white crystals. The molecular formula for 5 was C12H25NO2 confirmed by the HRESIMS molecular ion peak at m/z 216.1889 [M + H]+ in combination with 13C NMR and HMQC data, with one degree of unsaturation due to the carbonyl carbon. It showed IR absorption bands at 3,280, 2,950, 2,845, and 725 cm−1 for the long aliphatic chain and at 1,710 cm−1 for the carbonyl moiety. Its 1H NMR displayed a downfield triplet at δ H 3.23 (J = 7.5 Hz, H-12), which was assigned to a methylene bonded to NH group and a triplet for a methylene adjacent to carbonyl (CH2–COO) at δ H 2.52 (J = 7.5 Hz, H-2), as well as strong methylene signals at δ H 1.24–1.28 for (-CH2-)8 indicated the fatty acid nature of 5. The 13C NMR and HMQC spectra of 5 revealed the presence of an acid carbonyl at δ C 176.8 (C-1), NH-bonded mehylene at δ C 50.7 (C-12), a methylene adjacent to carbonyl δ C 35.6 (C-2), three methylene carbons at δ C 26.3 (C-3), 30.3 (C-4), 30.4 (C-11), and intense signals at δ C 30.5–30.7 for the long chain of methylene groups. On the basis of these findings, 5 was assigned as 12-aminododecanoic acid and further established by the COSY and HMBC correlations (Fig. 2). Compound 5 was isolated for the first time from natural origin.
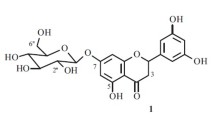
The known compounds were identified by analysis of the spectroscopic data (1D, 2D NMR, and MS) and comparison of their data with those in the literature to be: lupeol (2) (Chumkaew et al., 2005; Furukawa et al., 2002; Jain and Bari, 2010), β-sitosterol (3) (Mohamed et al., 2009), ursolic acid (4) (Seebacher et al., 2003), dillenetin-3-O-β-d-glucopyranoside (6) (Muhit et al., 2010), stachydrine (7), and 3-hydroxystachydrine (8) (Yousif et al., 1984; Blunden et al., 1983; Viqar Uddin et al., 1975; Feng et al., 2011; Afsharypuor and Jazy, 1999), and quercetin-3-O-β-d-glucopyranoside (9) (Ebada et al., 2008). This is the first report of compound 6 in family Capparidaceae. Compounds 4 and 9 were isolated for the first time from this plant. The results of antioxidant activity proved that compounds 5 and 9 exhibited strong antioxidant activity of 87.1 and 71.8 %, respectively. While, compound 6 had moderate activity of 49.1 % in comparison with propyl gallate (a known synthetic antioxidant) set as 100 % antioxidant activity. These results are in accordance with previous studies regarding the structure–activity relationship of flavonoids (Pier-Giorgio, 2000; Stanislaw and Wieslaw, 2001; Dugas et al., 2000; Cotelle et al., 1996).
References
Afsharypuor S, Jazy AA (1999) Stachydrine and volatile isothiocyanate from the unripe fruit of Capparis spinosa L. DARU 7:11–13
Blunden G, Gordon SM, Mclean WFH, Keysel GR (1983) β-Stachydrine, a novel betaine from griffithsla flosculosa. Phytochemistry 22:293
Chumkaew P, Kato S, Chantrapromma K (2005) A new triterpenoid ester from the fruits of Bruguiera parviflora. Chem Pharm Bull 53:95–96
Cotelle N, Bernier J, Catteau J, Pommery J, Wallet J, Gaydou EM (1996) Antioxidant properties of hydroxyl-flavones. Free Radic Biol Med 20:35–43
Dugas AJ Jr, Castañeda-Acosta J, Bonin GC, Price KL, Fischer NH, Winston GW (2000) Evaluation of the total peroxyl radical-scavenging capacity of flavonoids: structure activity relationships. J Nat Prod 63:327–331
Ebada SS, Ayoub NA, Singab A, Al-Azizi MM (2008) Phytophenolics from Peltophorum africanum sond. (fabaceae) with promising hepatoprotective activity. Pharmacog Mag 4:286–292
Feng X, Lu J, Xin H, Zhang L, Wang Y, Tang K (2011) Anti-arthritic active fraction of Capparis spinosa L. fruit and its chemical constituents. Yakugaku Zasshi 131:423–429
Furukawa S, Takagi N, Ikeda T, Ono M, Nafady AM, Nohara T, Sugimoto H, Doi S, Yamada H (2002) Two novel long-chain alkanoic acid esters of lupeol from Alecrim-Propolis. Chem Pharm Bull 50:439–440
Gaffa T, Ayo JA (2003) Physicochemical and sensory effects of Cadaba farinosa crude extract on cereal starches during kunun zaki production. Pak J Nutr 2:13–17
Glew RH, Kramer JK, Hernandez M, Pastuszyn A, Ernst J, Djomdi NN, Jagt DJV (2010) The amino acids, mineral and fatty acid content of three species of human plant foods in Cameroun. Food 4:1–6
Gohar AA (2002) Flavonol glycosides from Cadaba glandulosa. Z Naturforsch 57c:216–220
Jain PS, Bari SB (2010) Isolation of lupeol, stigmasterol and campesterol from petroleum ether extract of woody stem of Wrightia tinctoria. Asian J Plant Sci 9:163–167
Mohamed GA (2008) Alliuocide G, a new flavonoid with potent α-amylase inhibitory activity from Allium cepa L. ARKIVOC xi:202–209
Mohamed GA, Abdel-Lateff A, Fouad MA, Ibrahim SRM, Elkhayat ES, Okino T (2009) Chemical composition and hepatoprotective activity of Imperata cylindrica Beauv. Pharmacog Mag 4:28–36
Muhit MA, Tareq SM, Apu AS, Basak D, Islam MS (2010) Isolation and identification of compounds from the leaf extract of Dillenia indica Linn. Bangladesh Pharm J 13:49–53
Pier-Giorgio P (2000) Flavonoids as antioxidants. J Nat Prod 63:1035–1042
Rahman MA, Mossa JS, Al-Said MS, Al-Yahya MA (2004) Medicinal plant diversity in the flora of Saudi Arabia 1: a report on seven plant families. Fitoterapia 75:149–161
Sanghi DK, Jaiswal SB, Shamsudin, Joshi SB, Saini V (2007) Hepatoprotective activity of Cadaba farinosa. Int J Green Pharm 1:41–42
Seebacher W, Simic N, Weis R, Saf R, Kunert O (2003) Complete assignments of 1H and 13C NMR resonances of oleanolic acid, 18α-oleanolic acid, ursolic acid and their 11-oxo derivatives. Magn Reson Chem 41:636–638
Stanislaw B, Wieslaw O (2001) Antioxidant and antiradical activities of flavonoids. J Agric Food Chem 49:2774–2779
Telrandhe UB, Hemalatha S, Modi A (2010a) Pharmacognostic and phytochemical investigation on root of Cadaba farinosa Forsk. Int J Pharma Biosci 2:1–13
Telrandhe UB, Modi A, Uplanchiwar V, Gahane A, Hemalatha S, Goswami DV (2010b) Hepatoprotective and antioxidant activity of root of Cadaba farinosa Forsk. against carbon tetra chloride induced hepatotoxicity in rats. J Pharm Res 3:1412–1416
Tenpe CR, Upaganlawar AB, Yeole PG (2006) Antidiabetic activity of Cadaba indica in Alloxan induced diabetic rats. Ind J Nat Prod 22:14–17
Viqar Uddin A, Basha A, Atta-ur-Rahman (1975) Identification and 13C NMR spectrum of stachydrine from Cadaba fruticosa. Phytochemistry 14:292–293
Viqar Uddin A, Aziz-ur-Rahman A, Shoib AC, Marie HM, Cladry J (1985) Cadabicine, an alkaloid from Cadaba farinosa. Phytochemistry 24:2709–2711
Viqar Uddin A, Kaniz F, Azia-ur-Rahman A, Shoib A (1987) Cadabacine and cadabacine diacetate from Crataeva nurvala and Cadaba farinosa. J Nat Prod 50:1186
Viqar Uddin A, Azia-ur-Rahman A, Kaniz F, Arshad K (1990) Cadabicilone, a sesquiterpene lactone from Cadaba farinosa. Z Naturforsch 45b:1100–1102
Yousif G, Iskander GM, Eisa EB (1984) Alkaloids of Cadaba farinosa and C. Rotundifolia. Fitoterapia 55:117–118
Acknowledgments
The authors are grateful to Dr. Volker Brecht (nuclear magnetics resonance, institute fuer pharmazeutische wissenschaften, Albert-Ludwigs-Universität, Freiburg, Germany) for MS spectral measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al-Musayeib, N.M., Mohamed, G.A., Ibrahim, S.R.M. et al. Lupeol-3-O-decanoate, a new triterpene ester from Cadaba farinosa Forssk. growing in Saudi Arabia. Med Chem Res 22, 5297–5302 (2013). https://doi.org/10.1007/s00044-013-0536-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0536-1