
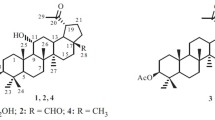
A new lupane-type triterpene, named 7α,11β-dihydroxy-2,3-seco-lup-12(13),20(29)-diene-2,3,28-trioic acid (1), along with 5 other known lupane-type triterpenoids, namely 3β-hydroxy-lup-20(29)-ene-23,28-dioic acid (2), betulinic acid (3), betulinic acid 3-O-sulfate (4), 12(13)-ene betulinic acid (5), and betulinic acid glucoside (6), was isolated from Schefflera octophylla stems and leaves. The structures of these compounds were determined by 1D and 2D NMR, MS techniques, and chemical methods. Compound 1 was the new compound, and 2, 3, 5, 6 were isolated from S. octophylla for the first time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Schefflera octophylla (Lour.) Harms (Araliaceae) is a medium-size evergreen tree up to 25 m tall and bole up to 80 cm in diameter, used as a folk remedy for the treatment of pain and inflammation. It is a principal ingredient of an herbal tea formulation widely used to treat common cold in southern China [1, 2]. In Vietnamese folk medicine, it is also used as a tonic drug, an antirheumatic agent, and for liver diseases [3]. Previous phytochemical studies on S. octophylla showed that the plant is rich in triterpenoids and triterpenoid glycosides. As part of our continuing search for bioactive constituents, a 75% EtOH extract of the stems and leaves of S. octophylla was investigated, and six lupane-type triterpenoids (1–6) were isolated. This present paper describes the structures of these compounds.

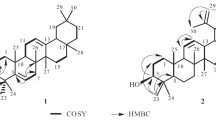
Compound 1 was isolated as colorless needles and gave a positive result in the Liebermann–Burchard test, mp 231–233°C. The IR spectrum (nujol) showed absorptions at 3450, 3075, 2975, 1698, and 1640 cm–1 assignable to hydroxyl, carboxyl, and C=CH2 functions. UV (MeOH, λmax, nm): 205. Its negative electrospray ionization mass spectrum (ESI-MS) exhibited a quasi-molecular ion peak at m/z 531.2 ([M – H]–), indicating a molecular weight of 532.2. The molecular formula was established as C30H44O8 by negative-ion mode HR-FAB-MS, showing a pseudo-molecular ion peak at m/z 532.2932 (calcd 532.2926), a compound with nine degrees of unsaturation. The 1H NMR and 13C NMR spectrum of 1 (Table 1) displayed signals for typical triterpenoid methyl groups at δH(ppm): 0.79 (3H, H3-25), 0.88 (3H, H3-26), 1.00 (3H, H3-27), 1.02 (3H, H3-23), 1.05 (3H, H3-24), and 1.65 (3H, H3-30) [δC 17.55, 14.96, 15.87, 16.56, 16.49, and 19.42, respectively, according to the HMQC experiment]. The presence of a broad vinyl methyl proton signal at δ 1.65 and two vinyl proton signals at 4.69 (d, J = 2.0 Hz) and 4.56 with 13C signals at 19.42, 150.79 and 110.10 was characteristic of an isopropenyl group of lupene triterpenes. Two proton signals, 3.33 (dd, J = 11.5, 4.5 Hz, H-7), and 3.57 (dd, J = 11.5, 4.5 Hz, H-11), were due to hydroxymethylene groups δC 72.14 and 79.64. The δH 5.17 (d, J = 3.6 Hz, H-12) proton was connected with δC 125.04 (C-12). The 13C NMR spectrum and DEPT experiments indicated the presence of 30 carbon atoms due to six methyls, seven methylenes, seven methines, and ten nonprotonated carbons. The signals included two olefinic carbons [δ 110.10 (C-29), and 150.79 (C-20)] and three carbonyl carbons [δ 177.69 (C-2), 179.03 (C-3), and 178.74 (C-28)]. The 1H detected heteronuclear multiple bond connectivity (HMBC) correlations of H2-1 to one carboxyl carbon, indicating that the carboxyl group [δ 177.69 (C-2)] was adjacent to C-1. The long-range correlation of two methyl groups (H3-23 and H3-24) to another carboxyl carbon [δ 179.03 (C-3)] suggested that the second carboxyl group was adjacent to [δ 43.50 (C-4)]. H2-16 and H2-22 have correlations to the third carboxyl carbon [δ 178.74 (C-28)], suggesting that the third carboxyl group was adjacent to C-28. Moreover, the HMBC correlations between a carbon signal at δ 72.14 (C-7) and a methyl proton signal at δ 0.88 (H3-26), as well as between a proton signal at δ 3.33 (H-7) and a carbon signal at δ 50.36 (C-5), indicated that a hydroxyl group was attached at C-7 (Fig. 1). The proton signal at δH 3.57 (H-11) has correlations with δC 144.36 (C-13), δC 41.20 (C-8) and δC 16.40 (C-10), indicating that hydroxyl group was attached at C-11 (δ 79.64). The orientation of the hydroxyl group was determined by a NOESY experiment, in which the proton signal (H-7) was found to be correlated with a methyl signal (H3-26, β-orientation) and the H-11 to have a slight correlation with a methyl signal (H3-27, α-orientation) (Fig. 1). The structure of compound 1 was elucidated as 7α,11β-dihydroxy-2,3-seco-lup-12(13),20(29)-diene-2,3,28-trioic acid.

Selected HMBC correlations and NOESY for 1.
Experimental
General Experimental Procedures. Melting points (mp) were determined using an X-4 micromelting-point apparatus (Beijing, China) and were uncorrected. UV spectra were measured on a Shimadzu UV-2501 spectrometer (Kyoto, Japan). IR spectra were obtained on KBr pellets using a Nicolet impact 410 spectrometer (Madison, USA). The 1H and 13C NMR spectra were obtained on Bruker Avance 400 and 100 MHz spectrometers (Germany) with TMS as an internal standard. SEI-MS measurements were undertaken on an HP5989A spectrometer (Palo Alto, USA). TLC and column chromatography were performed on plates precoated with silica gel F254 and silica gel (200–300 mesh; Qingdao Marine Chemical Ltd., Qingdao, China), respectively. Solvents were distilled prior to use.
Plant Material. Schefflera octophylla fresh stems and leaves were collected from Quanzhou (118°36′E, 24°58′N), Fujian Province, China, in September 2012, and the plant was identified by Prof. X. X. Xu at the School of Biomedical Sciences, Huaqiao University. A voucher specimen (No. S.O.20120915) was deposited in Huaqiao University.
Extraction and Isolation. The air-dried and roughly powdered S. octophylla stems and leaves (5 kg) was extracted three times with 75% ethanol under reflux. After removal of the solvent by evaporation, the extracts were partitioned between H2O and peltroleum ether, CH2Cl2, EtOAc, and n-BuOH, successively. The EtOAc and n-BuOH extracts were chromatographed on silica gel (300–400 mesh; 1500 g) columns respectively, eluting with a CH2Cl2–MeOH mixture (concentration gradients 1:0, 50:1, 20:1, 10:1, 5:1, 1:1, 1:5, 1:10, 1:20, 1:50, and 0:1) repeatedly. Six compounds, 1 (61 mg), 2 (139 mg), 3 (47 mg), 4 (25 mg), 5 (71 mg), and 6 (43 mg) were obtained.
Compound 1. Colorless needles, mp 231–233°C (CH2Cl2–MeOH, 10:1). IR (KBr, cm–1): 3450, 3075, 2975, 1698, and 1640. UV (MeOH, λmax, nm): 205. [α] 26D +102.3° (c 0.15, MeOH). HR-SEI-MS ([M]+ 532.2932, calcd 532.2926). 1H and 13C NMR spectral data are given in Table 1; HMBC and NOESY experiments, in Fig. 1.
Compound 2. Colorless needles, mp 260–262°C (CH2Cl2–MeOH, 20:1), positive in the Liebermann–Burchard reaction. ESI-MS m/z 486 [M]+ (C30H46O5). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 0.80 (3H, s, H-27), 0.88 (3H, s, H3-25), 0.99 (3H, s, H3-26), 1.02 (3H, s, H3-24), 1.66 (3H, s, H3-30), 3.57 (br.s, H-3α), 4.57 and 4.70 (2 × br.s, H2-29), 11.88 (2H, COOH). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 36.81 (C-1), 30.57 (C-2), 72.14 (C-3), 55.43 (C-4), 49.02 (C-5), 20.70 (C-6), 34.13 (C-7), 41.28 (C-8), 50.34 (C-9), 36.93 (C-10), 25.50 (C-11), 29.62 (C-12), 38.04 (C-13), 50.82 (C-14), 32.12 (C-15), 25.55 (C-16), 55.43 (C-17), 43.88 (C-18), 47.10 (C-19), 150.30 (C-20), 21.13 (C-21), 32.20 (C-22), 177.20 (C-23), 14.94 (C-24), 17.53 (C-25), 16.54 (C-26), 16.47 (C-27), 177.24 (C-28), 110.08 (C-29), 19.39 (C-30). Based on the above evidence, the structure of 2 was determined as 3β-hydroxy-lup-20(29)-ene-23,28-dioic acid [4].
Compound 3. White needles, mp 286–288°C (CH2Cl2–MeOH, 50:1), positive in the Liebermann–Burchard reaction. ESI-MS m/z 456 [M]+ (C30H48O3). Based on the above evidence, the structure of 3 was determined as betulinic acid [5].
Compound 4. White powder (CH2Cl2–MeOH, 10:1), mp 253–258°C. [α] 27D +4.3° (c 0.5, EtOH). IR (KBr, cm–1): 1695, 1640, 1230, 875. ESI-MS m/z 536 [M]+ (C30H47O6S). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 11.90 (1H, br.s, 28-COOH), 4.70 (1H, br.s, H-29a), 4.56 (1H, br.s, H-29b), 4.46 (1H, m, H-3α), 0.79, 0.88, 1.00, 1.05, 1.26, 1.66 (each 3H, s, tert-CH3). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 177.78 (C-28), 151.20 (C-20), 110.21 (C-29), 79.52 (C-3), 55.35 (C-17), 50.84 (C-5), 50.37 (C-9), 49.06 (C-19), 47.11 (C-18), 43.92 (C-14), 42.65 (C-8), 38.40 (C-1), 38.41 (C-4), 38.22 (C-13), 36.94 (C-10), 36.82 (C-22), 34.15 (C-7), 34.14 (C-21), 32.10 (C-16), 30.59 (C-15), 29.63 (C-23), 25.63 (C-2), 21.14 (C-12), 20.73 (C-11), 19.28 (C-30), 17.39 (C-6), 16.57 (C-24), 16.51 (C-25, 26), 14.96 (C-27). Compound 4 was characterized as betulinic acid 3-O-sulfate [6].
Compound 5. White powder (CH2Cl2–MeOH, 50:1), mp 265–267°C. ESI-MS m/z 454 [M]+ (C30H46O3). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 11.90 (1H, br.s, 28-COOH), 5.75 (1H, s, H-12), 4.69 (1H, br.s, H-29a), 4.56 (1H, br.s, H-29b), 3.56 (1H, m, H-3), 1.65 (3H, s, H-30), 1.23 (3H, s, H-27), 1.01 (3H, s, H-26), 0.98 (3H, s, H-25), 0.88 (3H, s, H-24), 0.79 (3H, s, H-23). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 177.70 (C-28), 150.80 (C-20), 110.09 (C-29), 72.15 (C-3), 55.91 (C-17), 50.84 (C-5), 50.37 (C-9), 49.06 (C-19), 47.11 (C-18), 43.92 (C-14), 42.65 (C-8), 39.40 (C-1, 4), 38.07 (C-13), 36.94 (C-10), 36.82 (C-22), 34.15 (C-7), 34.14 (C-21), 32.20 (C-16), 30.59 (C-15), 29.63 (C-23), 25.55 (C-2), 21.18 (C-12), 20.73 (C-11), 19.43 (C-30), 17.56 (C-6), 16.57 (C-24), 16.50 (C-25, 26), 14.97 (C-27). According to be above evidences, 5 was characterized as 12(13)-ene betulinic acid [7].
Compound 6. White powder (CH2Cl2–MeOH, 1:1), mp 266–268°C, positive in the Liebermann–Burchard and Molisch reactions. Acid hydroxlysis of 6 gave an aglycone and D-glucose. IR (KBr, νmax, cm–1): 3400 (OH), 1680 (C=O), 1640, 875 (C=CH2). ESI-MS m/z 630.2514 [M]+ (C36H58O9). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 15.17 (C-27), 16.18 (C-25), 16.41 (C-26), 18.17 (C-23), 19.43 (C-30), 20.80 (C-24), 20.95 (C-21), 22.66 (C-16), 25.57 (C-22), 29.10 (C-6), 29.70 (C-2), 30.58 (C-12), 32.17 (C-15), 33.27 (C-1), 34.19 (C-7), 37.14 (C-4), 37.18 (C-10), 38.04 (C-19), 38.38 (C-13), 40.9 (C-8), 42.60 (C-18), 47.07 (C-14), 49.01 (C-11), 49.48 (C-5), 49.69 (C-17), 55.90 (C-9), 80.44 (C-3), 110.10 (C-29), 150.89 (C-20), 177.69 (C-28). 13C NMR showed a set of signals due to one molecule of β-glucopyranose [100.58 (C-1), 77.44, 77.23 (C-3, 5), 73.88 (C-2), 70.96 (C-4), 61.84 (C-6)]. Based on these data, 6 was confirmed as betulinic acid glucoside [8].
References
T. V. Sung, W. Steglich, and G. Adam, Phytochemistry, 30, 2349 (1991).
Y. L. Li, P. H. B. Paul, and E. C. O. Vincent, Antiviral. Res., 68, 1 (2005).
G. Adam, M. Lischewski, and H. V. Phiet, Phytochemistry, 21, 1385 (1982).
M. J. Just, M. C. Recio, and R. M. Giner, Nat. Prod. Lett., 9, 167 (1997).
S. Siddiqui, F. Hafeez, and S. Begum, J. Nat. Prod., 51, 229 (1988).
J. Kitajima, M. Shindo, and Y. Tanaka, Chem. Pharm. Bull., 38, 714 (1990).
J. Hussain, N. U. Rehman, and H. Hussain, Fitoterapia, 83, 593 (2012).
J. Kitajima and Y. Tanaka, Chem. Pharm. Bull., 37, 2727 (1989).
Acknowledgment
This work was supported by the key Programs of Science & Technology of Fujian Province (2012Y0049).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2016, pp. 378–380.
Rights and permissions
About this article

Cite this article
Su-qiu, P., Ai-jing, S., Guo-quan, W. et al. Lupane-Type Triterpenoids from Schefflera octophylla . Chem Nat Compd 52, 432–435 (2016). https://doi.org/10.1007/s10600-016-1666-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-016-1666-8