
Abstract
Although surgical excision, chemo-, and radio-therapy are clearly advanced, tumors may relapse due to cells of the so-called “minimal residual disease”. Indeed, small clusters of tumor cells persist in host tissues after treatment of the primary tumor elaborating strategies to survive and escape from immunological attacks before their relapse: this variable period of remission is known as “cancer dormancy”. Therefore, it is crucial to understand and consider the major concepts addressing dormancy, to identify new targets and disclose potential clinical strategies. Here, we have particularly focused the relationships between tumor microenvironment and cancer dormancy, looking at a re-appreciated aspect of this compartment that is the low extracellular pH. Accumulating evidences indicate that acidity of tumor microenvironment is associated with a poor prognosis of tumor-bearing patients, stimulates a chemo- and radio-therapy resistant phenotype, and suppresses the tumoricidal activity of cytotoxic lymphocytes and natural killer cells, and all these aspects are useful for dormancy. Therefore, this review discusses the possibility that acidity of tumor microenvironment may provide a new, not previously suggested, adequate milieu for “dormancy” of tumor cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Early detection and new therapeutic approaches drastically improve cure and progression-free survival of tumor-bearing patients. In general, a detectable primary tumor is treated by chemotherapy, surgery, and/or radiation leading to its elimination from the patient or reduction in size to a few viable cells. However, survival of a small number of malignant cells, the so-called “minimal residual disease”, might arise for diverse reasons, including an incomplete surgical excision for impossible diagnosis of too small clusters of cells. Furthermore, tumor cells might survive to chemotherapeutic and radiotherapeutic treatments, depending on the variability of cell cycle length and phase of each single cell, or depending on the expression of a drug resistant phenotype. Surviving cells may have different fates, consisting of starting again to grow or entering into a “dormant state”. Thus, it is possible for tumor cells to persist for extended periods until the host dies. Otherwise, after a prolonged period of time corresponding to clinical remission, tumor cell can give rise to a secondary lesion. Those lesions may occur locally or at distant site from the pre-existing primary tumor and are known as organ metastasis. The emergence of metastasis is, indeed, the most dangerous event during the natural history of the neoplastic disease. Although only a minority of patients exhibit clinically detectable metastases at the time of diagnosis, metastases are responsible for more than 90% of patient mortality. Metastatic lesions result from a multistep process that involves progression of tumor cells into primary host tissues, intravasation into altered tumoral blood vessels, and dissemination to distant organs, and extravasation and growth after adaptation into the foreign tissue microenvironment. The term “dormancy” indicates the length of time needed to metastatic cells for adaption to secondary organs [1]. Clinical observations report that melanoma, breast, and prostate tumors may relapse into target organs many years or decades after apparent curative treatment of primary tumor. This is possible because tumor cells spread to distant organs earlier than we may expect and survive in the hostile new microenvironments. Isolation of disseminated tumor cells in the blood by liquid biopsy may help to prevent and combat this behaviour. Other clinical reports describe extraordinarily long remissions following surgical treatment of intraocular melanoma, testicular teratoma, osteogenic sarcoma, and renal carcinoma [2]. During the dormant state, tumor cells survive in the host tissues remaining confined in a low proliferating or quiescent state, resisting to drugs and immune attacks, until being recruited into a high proliferation compartment by either genetic or epigenetic changes. Dormancy may be sustained by limited blood supply; this condition of low oxygen and reduced nutrients determines an absolute increase of tumor cells number very close to zero, since the number of dividing cells may equate the number of dying cells [3, 4]. It is important to remember that many of the mechanisms proposed to explain dormancy have been identified using experimental models, although all are very plausible, up to now are not completely confirmed in tumor-bearing patients [1]. Thus, it is important to introduce new hypotheses and offer new tools to improve knowledge regarding tumor dormancy, to translate these new advances into clinic.
This review discusses the relevance of a particular aspect of tumor microenvironment, participating in harbouring dormant tumor cells, either from primary or secondary lesions. The aspect of tumor microenvironment which we are interested to introduce in the complex scenario of dormancy is represented by the extracellular acidosis, which frequently characterizes solid tumors.
Why tumor microenvironment is often characterized by low pH
Tumor cells, even when there is enough O2 to support mitochondrial function, use glycolysis to sustain their high proliferation rate, the so-called “Warburg effect” [5, 6]. This phenomenon was first reported by O.H. Warburg in the 1920s, and although the “Warburg hypothesis” of impaired mitochondrial metabolism has proven to be incorrect, an increased conversion of glucose to lactic acid and H+ in tumors has been constantly demonstrated. “Aerobic glycolysis” leads to the conversion of one molecule of glucose into two molecules of lactic acid and 2 H+ to produce 2 ATP, compared to the 36 ATP produced by oxidative metabolism [5, 6]. Thus, ATP generation from glycolysis is far less efficient, although rapidity of this pathway might represent a compensatory aspect. Furthermore, tumor cells implement glycolysis promoting an abnormal high rate of glucose uptake [7] trough the up-regulation and the enhanced activity of glucose transporters. The increased glucose uptake is useful for diagnostic purposes, as in the case of monitoring uptake of glucose analogue tracer (18F)-fluoro-2-deoxy-d-glucose (18FdG) by positron-emission tomography (PET) [8]. PET imaging of tumor-bearing patients has shown that numerous, although not all, primary and metastatic lesions express a significant glucose uptake.
Aerobic glycolysis up-regulation in malignant phenotype, that is mostly due to stable genetic or epigenetic changes [5], is important for proliferating tumor cells since glucose can be diverted into macromolecular precursors. Indeed, glucose 6-phosphate is used for glycogen and ribose 5-phosphate biosynthesis, dihydroxyacetone phosphate for triacylglycerol and phospholipid synthesis, and pyruvate for alanine and malate synthesis. Furthermore, the isoform of pyruvate kinase M2, usually highly expressed in tumor cells under activity of oncogenic transcription factor c-MYC, may slow down aerobic glycolysis, which results into accumulation of up-stream precursors for anabolic demand of proliferating cells [9]. c-MYC is also involved in promotion of many of glycolytic enzymes [10], together with Akt pathway, which enhances glycolysis through activation of hexokinase 2, phosphofructokinase 1 and 2 and expression of glucose transporters [11–13]. The generated pyruvate, together with the entry of glutamine into tricarboxylic acid (TCA) cycle, cooperates to the promotion of anabolic reactions needed for cell division. Thus, an enhanced lactic acid production during a high rate of proliferation is difficult to imagine, and only when uptake of glucose superimposes anabolic biomass formation, lactic acid starts to accumulate and extracellular pH reduces. A high glycolytic rate has an additional advantage for proliferating tumor cells, that is the reduction of radical oxidative substances and the related cytotoxic effects [14].
When oxygen tension decreases and a hypoxic microenvironment develops, stabilization of hypoxia inducible factor 1α (HIF-1α) transcription factor may regulate an elevated number of genes driving tumor cells metabolism toward an anaerobic glycolysis pathway. Such genes include glucose transporters (GLUT1 and 3) [15], glycolytic enzymes (Hexokinase I and II, phosphofructokinase-1, aldolase-A and C, phosphoglycerate kinase 1, enolase-α), pyruvate dehydrogenase kinase 1 (PDK1, that inactivates the enzyme responsible for conversion of pyruvate to acetyl-CoA [16]), and genes for lactate production (LDHA, encoding lactate dehydrogenase A, which converts pyruvate to lactate [17]) and for lactate extrusion (MTC-4, the monocarboxylate transporter-4). Lactate production in hypoxic cancer cells is also enhanced by the increased expression of PKM2 [18] and activation of PDK1. PDK1 suppresses pyruvate dehydrogenase (PDH) activity limiting the entry of pyruvate into TCA cycle. Thus, pyruvate is mostly redirected into lactate and H+ by LDH-A.
Therefore, to maintain an intracellular pH compatible with survival and/or proliferation, additional adaptations are required. The mechanism for long-term internal pH control is represented by promotion in tumor cells of multiple and redundant families of transporters, which release protons and lactate into extracellular environment, such as: Na+/H+ exchanger 1 (NHE1), HCO3 − transporters (Na+/HCO3 − co-transporters, NBCs and anion exchange protein 1, AE1), proton-linked monocarboxylate transporters (MCT1 and 4), and vacuolar H+-ATPases (V-ATPases) [19]. In addition, carbonic anhydrases IX (CAIX), a hypoxia–inducible effector, participates to the complex machinery responsible of internal pH control favouring export of protons and import of bicarbonate ions [19].
On the whole, cancer cells that adapt their metabolism to different levels of oxygen caused by either dividing process or hypoxic condition produce a significant acidification of the extracellular microenvironment. It is known that pH value of extracellular environment in several tumor histotype ranges from 6.7 to 7.1 [20]. Interestingly, extracellular acidosis maintains a high level of CAIX expression in tumor cells, in a HIF-1α-independent way [21]. Furthermore, we cannot exclude that tumor extracellular acidity may also be supported by a poor blood perfusion and an uneven distribution of lymphatic vessels, frequently very poor in the core of the mass and more evident in the periphery. As a consequence, interstitial fluid pressure is maintained elevated. It is for these considerations that acidosis of the extracellular space of tumors has been included among the most important hallmarks of tumor microenvironment [22].
How low pH may contribute to dormancy of tumor cells
Acidity induces a reduced proliferation, high resistance to apoptosis and autophagy
Whereas a coordinate series of events account for progression through the cell cycle in normal cells, in cancer cells, the deregulation of multiple control mechanisms results in different degrees of autonomy from growth-stimulatory or -inhibitory signals, making cancer cells adapted to grow quite indefinitely. However, proliferative rate of tumor cells may vary and non/slow-proliferating cells are common, which implies a high rate of cell death. Indeed, studies of human and animal tumors have demonstrated heterogeneity in mitotic index within different regions of the same tumor mass. The heterogeneity of cell proliferation is due to different factors, such as cell differentiation, variant generation with different proliferative rates, and availability of oxygen and nutrients (i.e., glucose, glutamine, and branched-chain amino acids) in the microenvironment. Thus, slow-proliferating cells may occur in solid tumors and often are responsible for relapse, due to their high resistance to clinical treatments. In this context, acidic tumor microenvironment promotes in tumor cells a low-proliferating phenotype, which does not revert even in the presence of a high level of growth factors [23]. It has also been shown by several reports that extracellular acidosis increases the percentage of cells in G0 phase [24]. Indeed, exposure to acidosis alters growth factor signaling and reduces serum ability to stimulate Raf/ERK/mTORC1 kinase pathway [25]. Aguirre-Ghiso et al. showed that dormancy in human head and neck cancer cells depends on the block of the Raf/ERK pathway together with the activation of p38 [26]. Besides, it was demonstrated that acidity inhibits AMPK [27] driving a mitotic delay through a misoriented spindle due to improper astral microtubule-cell cortex interactions [28]; indeed, it is known that cell division is achieved through a correct assembly, positioning, and orientation of the microtubule-based spindle. Extracellular acidity modulates intracellular pH that in turn directly affects cell cycle. This is a quite common mechanism for cell proliferation control in both normal and cancer cells that was demonstrated several years ago [29]. When internal pH drops following a sustained extracellular acidosis, also G2/M entry is limited by a reduced cyclin-dependent kinase 1-cyclin B1 activity [30].
A recent study showed that transforming growth factor-β2 (TGFβ2) was effective in promoting the up-regulation of p27 and p38 by the SMAD1/2/5 pathway in tumor cells undergoing a dormant state [31]. Lactic acid was shown to induce both transcription and protein secretion of TGFβ2 in glioma cells [32] and to induce TGFβ1 production in in vivo model [33]. More recently, we found that an acidic microenvironment is able to promote TGFβ secretion and signaling in mesenchymal stem cells (MSC), driving the progression to malignancy in a melanoma cell system [34] and suggesting that acidity is a strong inducer of TGFβ family members. Esomeprazole, a proton pump inhibitor activated by acidity, inhibited TGFβ expression in MSC grown in a low pH medium [34].
In addition, we have observed that a reduced pH promotes in melanoma cells a typical epithelial-to-mesenchymal transition (EMT) program characterized by a reduced level of proliferation, induction of several mesenchymal markers, resistance to apoptosis and high metalloprotease-dependent invasiveness [35]. Acidosis-exposed tumor cells were also able to favour in vivo organ colonization of intravenously injected non-acidic tumor cells, indicative of a new cooperation between tumor cell subpopulations [35]. We may suggest that acidic-adapted melanoma cells protect non-acidic cells from blood turbulence and possible interactions with immune cells, and support survival and final lodgment in host microenvironment of secondary organ. Furthermore, acidity contributes to promote in melanoma cells an elevated resistance to pro-apoptotic agents [35] and resistance to apoptosis is well ascertained to be useful for dormant tumor cells. Additional evidences indicate that low pH activates MAPK signaling in a variety of tumor cells generating a protective effect against apoptosis induced by multiple cytotoxic stresses [36, 37]. Incubation of lymphoma cells in acidic conditions (pH 6.5) blocks apoptosis promoted by glucose or glutamine deprivation, a phenomenon mediated by the acid-sensing G protein-coupled receptor (GPR65) through an up-regulation of Bcl-2 and Bcl-xL factors [38]. Thus, it is possible that the reduction of tumor mass expansion during dormancy favoured by acidity will be a consequence of a delay of the cell cycle, rather than the induction of cell death.
Another aspect associated with EMT elicited in tumor cells by acidity is represented by their metabolic adaptation. Acidic cancer cells switch from glycolysis to oxidative phosphorylation (Oxphos) to favour a generation of more bioenergetic molecules instead of biomass for cell division [27, 39]. Metformin, a standard drug for diabetes, is a very efficient drug to kill acidic melanoma cells inhibiting their metabolic program [27]. The main energetic source of acidic pH-adapted cancer cells was identified to derive from fatty acid oxidation [40]. An acidic niche is able to preserve Oxphos metabolism in tumor cells also in high glucose, indicating the absence of Crabtree effect in acidic cells [41]. Oxphos metabolism might be convenient for dormant cells which need energy to survive and resist several stressors using alternative substrates. Furthermore, it has been observed that quiescent human leukaemia stem cells use Oxphos to produce the required energy [42].
Despite the liaison between acidic phenotype and stemness is still not completely clear, an indication of a possible connection can consist in the fact that EMT induces stem-like traits in different tumor types, such as mammary tumors [43] and head and neck squamous cell carcinoma [44]. Acidity, independently from a restricted oxygen tension, promotes stem cell markers and self-renewal in glioma cells. Low pH exerts a promotion of glioma stem cells (GSC) through elaboration of HIF-2α-dependent angiogenic factors. Induction of HIF-2α and other GSC markers by acidic stress can be reverted by pH elevation, suggesting that GSC phenotype might be targeted, raising pHe [45]. Lisanti et al. [46] found that ketones and lactate stimulate stem cell markers in breast carcinoma cells, expressing an oxidative mitochondrial metabolism. This suggests that an appropriate mitochondrial antagonist, such as metformin, could eradicate cancer stem cell subpopulation. Hirsch et al. [47] showed that metformin selectively kills cancer stem cells isolated from breast cancer, and the combination between metformin and doxorubicin kills both cancer stem cells and non-stem cancer cells and prevents relapse much more efficiently than either drug alone in a xenograft mouse model. Quite recently, Petrachi et al. demonstrated that phenphormin, but not metformin, is able to affect viability and growth of melanoma cancer stem cells [48].
An additional tumor survival mechanism, also important for dormancy, is autophagy. Autophagy is a catabolic process through which cellular metabolism is maintained recycling the self-digested cytoplasmic proteins and organelles. Low levels of autophagic activity are observed under physiological conditions, while in tumor cells, autophagy promotes cell survival during the frequent nutrient restriction and hypoxia [49]. Autophagy was found to sustain the maintenance of cancer stem cell phenotype, and cooperates to chemo- and radio-resistance [50–53]. Inhibition of autophagy, on the contrary, increases chemo- and radio-sensitivity [54]. Acute and chronic exposure to low pH promotes high level of autophagic activity, and this sustains cell survival and aggressiveness, while the pharmacological inhibition of autophagy in such acidic tumor cells reduces their ability to survive [55]. Furthermore, it was demonstrated that acidosis blocks the inhibitory activity of chloroquine on autophagy, thus chloroquine does not result effective in tumor treatment [56]. Up-regulation of autophagy-mediated degradation and recycling of cellular substrates by acidic cancer cells can support metabolic pathways considering that an acidic microenvironment is generally characterized by low nutrients and oxygen.
Finally, it seems reasonable to suggest that the phenotype acquired by acidic tumor cells results into several characteristics useful for a dormant state, such as low replication rate, high resistance to apoptosis and autophagy.
Acidity promotes invasion
Acid-activated tumoral catepsin L was reported to be able to amplify proteinase cascade through activation of urokinase-type plasminogen activator (uPA) [57], which converts plasminogen to plasmin. This latter then degrades various components of the extracellular matrix (ECM), such as fibronectin, laminin, proteoglycan, and collagen [58], and activates latent collagenase and growth factors [59]. The concomitant expression of uPA and uPA receptor (uPAR) by tumor cells results in a high invasive capability [60]. uPAR is a glycosylphosphatidyl-inositol (GPI)-anchored molecule, and therefore, it lacks transmembrane domains able to transfer extracellular signals to signaling pathways. Thus, uPAR must cooperate with cell membrane molecules able to confer signaling competence on uPAR itself [61]. Upon interaction of α chain with uPAR, integrins α5β1 and αvβ3 can interact and activate Receptor Tyrosine Kinases (RTKs). Such interaction is potentiated by catalytic and non-catalytic uPA binding to uPAR and can induce RTKs activity even in the absence of the relevant ligand (EGF, PDGF, ILGF, and HGF) [61, 62] in an FAK-dependent manner [63]. In head and neck carcinoma, uPAR drives tumor growth by interacting with α5β1 integrin, a complex that activates focal adhesion kinase (FAK) and EGF receptor (EGFR), final effector of this mitogenic signal. Blocking uPAR, β1-integrin, FAK or EGFR, singly or in combination, results in tumor growth suppression and dormancy [1]. Indeed, it was reported that dormancy associates with a quite complete inhibition of the Raf-MEK-ERK pathway as in quiescent cells [64]. Therefore, it is reasonable that, even though uPAR is up-regulated in acidity, its mitogenic activity may be abrogated by loss of integrin interaction and/or reduction in EGFR cross talk. This last condition may leave uPAR to exert pro-invasive effects without promoting cell proliferation.
Furthermore, acid-activated cathepsin B and cathepsin-activated uPA/plasminogen/plasmin axis activate secreted latent metallo-proteases (MMPs) by proteolytic cleavage. MMPs are a family of structurally related zinc-dependent endopeptidases, collectively capable of degrading all components of ECM [65]. MMPs have long been associated with invasiveness, angiogenesis and tumor cell dissemination [66–68], due to their capacity to help tumor cells to cross-structural barriers, including basement membranes, to migrate into the blood, extravasate and colonize in distant host tissues. Kato et al. have shown that, in mouse metastatic B16 melanoma cells, expression of MMP-9 and in vitro invasiveness is induced by acidic pHe (pHe 5.4–6.5) [69]. Another component of basement membrane to be degraded by tumor cells to disseminate is the heparan sulphate chains. Toyoshima and Nakajima reported that heparanase has an optimal pH of 4.2, but a significant heparanase activity persists at pH 6.0–6.5, suggesting that the acidic environment of tumors may activate the degrading properties of tumor heparanases [70].
When melanoma cells were exposed to acidic condition for long time and selected subpopulations were isolated, it was found that these tumor cells exhibit a high level of invasiveness and proliferation following re-exposure to physiological pH [71]. The high level of invasiveness and proliferation of acid-adapted tumor cells after exposure to the standard pH evokes the awaking of dormant tumor cells, invasion in local host tissues, and growth.
In contrast to the several findings on the positive relationship between acidity and invasiveness, it is still under debate whether low pH may promote motility, the so-called amoeboid style of migration. Stock et al. using a model of directional cell migration in an extracellular pH gradient showed that melanoma cells hardly migrate at pHe 6.6 when adhesion was found particularly elevated [72]. Indeed, through computational molecular dynamic simulations, Paradise et al. found that acidic extracellular pH (pH 6.0 for 8 h) promotes opening of the αvβ3 headpiece [73], a conformational change supporting a stable interaction between integrin αvβ3 and its ECM biological ligands. In accordance, we experienced that acidity-exposed melanoma cells reduce their ability to heal wounds, tested by scratch test analysis, and express a reduced capacity to cross filters of Boyden chambers. Only when we exposed melanoma cells to lactic acid, migration recovers the level of control melanoma cells grown in the standard pH medium, however, without exceeding it [27].
Acidity may participate to dormancy either conferring, during the initial period of adaptation of tumor cells into the foreign microenvironment, some invasive properties useful to lodge in new sites, or favouring growth into adjacent host tissues when some genetic or epigenetic changes induce relapse.
Acidity promotes angiogenesis
Regarding angiogenesis, it was demonstrated that a transient exposure of endometrial carcinoma cells to acidosis (8 h at pH 5.5) induced a preferential VEFG121 isoform up-regulation through p38 MAPK and SAPK/JNK pathways [74]. When human pancreatic adenocarcinoma cells were incubated for different periods at pH between 6.7 and 7.4, it was found that the expression of VEGF was particularly stimulated by a 6–8 h-pH 6.7–7.1 treatment. Acidic pH also simulates NF-kB binding activity to the NF-kB site of VEGF [75, 76]. NF-kB was also found responsible of acidic pH-induced transcriptional activation of IL-8 gene, a promoter of angiogenesis [77]. However, when breast carcinoma cells were exposed to a pH 6.7 for 18 h, there was a significant decrease in VEGF mRNA expression [78]. Acidosis may also play an inhibitory role on angiogenesis exerting a potent block on HIF-1α stabilization, a key hypoxia transcription factor involved in VEGF gene activation [79]. Thus, angiogenesis regulation by pH might be either tissue specific or time of exposure-dependent, considering that acute acidosis stimulates, whereas a chronic exposure inhibits VEGF.
By studying acidosis-induced signalings in tumor cells, we observed that extracellular acidity (pH 6.7 ± 0.1) stimulates the expression and secretion of lymphangiogenic growth factor C (VEGF-C) by melanoma cells, mediated by NF-κB transcription factor activity [80]. We also demonstrated that esomeprazole, a proton pump inhibitor (PPI) activated by a low pH, inhibits VEGF-C expression in acidic melanoma cells down-regulating NF-κB [80]. VEGF-C, in addition to the well-known role of VEGF-A in relapse, might represent a new causal link between lymphangiogenesis, lymph node colonization, and progression [81]. Development of new lymphatic vessels might be also instrumental in reducing the high interstitial pressure expressed by certain tumor regions, favouring diffusion of macromolecules from blood vessels to tumor cells, which may relapse.
During a dormant period, quiescent or low-proliferating cells may take advantage to express a pro-angiogenic and/or lymphangiogenic phenotype that an acidic microenvironment may stimulate to finely control level of nutrients.
Acidity favours escaping from immune surveillance
To explain time variability of dormant state, Aguirre-Ghiso JA has proposed three major mechanisms: cellular dormancy, angiogenic dormancy, and immunosurveillance [1]. During cellular dormancy, tumor cells are G0–G1 arrested and they need to evade immune cell cytotoxicity to survive. Genetic modifications can re-activate a proliferative activity, even at low rate, in dormant cells. Despite this, the expansion of tumor lesion may not be reached, due to the effect of an active immune response and to the balance between pro- and anti-angiogenic. We believe that tumor microenvironment acidosis may sustain cellular dormancy, maintaining tumor cells unable to resume proliferation and hiding them from immune surveillance.
There is increasing evidence that cancer cells escape immune destruction by suppressing the anti-cancer immune response through maintaining a relatively low pH in their environment [82]. Indeed, the acquired acidic phenotype enables cancer cells to suppress the anti-cancer activity of immune cells promoting the secretion of chemokines to enhance T-regulatory recruitment or producing immunosuppressive cytokines [83]. Several studies have shown that acidic pHe inhibits T-cell proliferation [84], dendritic cell maturation [85], cytotoxic T lymphocyte activity (CTLs) [86], lymphokine-activated killer cells [87], and natural killer (NK) activity and viability [88]. Furthermore, lowering pHe to 6.5 has been reported to lead to a loss of function of human and murine tumor-infiltrating T lymphocytes consisting in a significant impairment of cytolytic activity, cytokine secretion, and reduction of T-cell receptor and interleukin-2 α-chain receptor expression [89]. T-cell function was found to be completely restored buffering pH to a physiological value; however when exposure to an acidic microenvironment was sufficiently prolonged, such as a chronic exposure, T cells were deeply damaged and underwent apoptosis [89].
Extracellular lactic acid generated by glycolytic cancer metabolism and accumulated in tumor microenvironment supports the suppressive effect on T-cell function exerted by low pH [83]. Mendler et al. identified that tumor lactic acidosis suppresses CTL functions via inhibition of p38 and JNK/c-Jun activation [90] and Fisher et al. demonstrated that lactic acid inhibits T-cell proliferation in a dose-dependent manner, whereas sodium lactate, which does not lead to an acidification of the medium, had no suppressive effect [84]. Moreover, prolonged incubation with high dose of lactic acid (20 mM) was shown to induce CTL death of up to 60%, while exposure to the same low pH, derived from HCl addition to the medium, was less effective. It has also been demonstrated that lactic acid generated by cancer cells promotes tumor development by inhibiting the immune response increasing arginase-1 (ARG1) expression and inducing IL-23/IL-17 secretion by tumor-associated macrophages (TAMs) of M2 phenotype [91]. Indeed, transient exposure of cells to acidic pH suppresses the ability of macrophages to mount an adequate antibacterial response elicited by Toll-like receptor 2 and 4 activation [92].
Recent studies indicate that buffering low pH at the tumor site by in vivo administration of esomeprazole, a PPI, improves tumor-infiltrating lymphocyte effector functions in melanoma lesions and delays cancer progression in tumor-bearing mice [89, 93]. Inhibition of proton pumps on tumor cells with pantoprazole, another PPI, hampers tumor-induced suppression of macrophages both in vitro and in vivo [94, 95]. Finally, neutralization of tumor pH with bicarbonate impairs the growth of cancer in mice, and this effect was associated with an increased T-cell infiltration in tumor masses [96].
Overall, several evidences sustain that low pH might provide an immunological privileged niche functional for cellular dormancy of quiescent tumor cells.
Acidity promotes resistance to drug and radio-treatment
Acidosis is able to influence the resistance of tumor cells to anticancer therapy [97], one of the major obstacle during chemotherapeutic treatment of solid tumors [98]. Cellular drug uptake and intracellular targeting are key factors for a successful anticancer treatment and both mechanisms are influenced by pHe. Extracellular acidity reduces the intracellular concentration of chemotherapeutic agents, mainly weak base drugs, through a mechanism termed “ion trapping”, and based on ion protonation of drugs [99–102]. Thus, the reversion of this pH gradient may strongly facilitate the successful treatment. Indeed, several in vitro and in vivo studies show that low pH reduces uptake and efficacy of weak base chemotherapeutics, such as anthracyclines (e.g., doxorubicin), anthraquinones (e.g., mitoxantrone), and vinca alkaloids (e.g., vincristine and vinblastine) [101, 103]. On the contrary, alkalization of extracellular environment enhances intracellular accumulation and cytotoxicity of some of these weak base drugs [104]. Accordingly, Wojtkowiak et al. demonstrated that a combined treatment with sodium bicarbonate and doxorubicin reduces more efficiently tumor volume than doxorubicin alone [105], suggesting that alkalization enhances doxorubicin efficacy. According to these results, De Milito and Fais showed that in vivo oral pretreatment with proton pump inhibitors (PPIs) of tumor-bearing animals re-sensitizes tumors to anticancer drugs [106].
Other possible mechanisms working on drug resistance in low pH have been described. Among these, it is of a great importance the ability of acidic pHe to increase the pump activity of the p-glycoprotein (pGP), a drug efflux transporter. Thews et al. observed that acidic prostate carcinoma cells express a pGP activity more than doubled of that of cells grown in a standard pH medium [107]. These authors reported that up-regulation of pGP in acidic cells was related to an increased intracellular Ca2+ level, stimulated by a protein kinase C activity [107]. It has also been observed that acidosis-induced pGP activity may be related to ERK and p38 pathways. The inhibition of both ERK and p38 abolished acidosis-induced daunorubicin efflux and re-establish daunorubicin sensitivity in rat prostate cancer cells [108]. Moreover, acidosis of the extracellular milieu may interfere in drug efficacy inducing a cell quiescence, namely, inducing a very low proliferative rate, as previously discussed [35, 97, 109]. It is known that cells are more sensitive to drugs and radiation therapy when in S-phase than in the other cell cycle phases. Tumor pH may also have indirect effects on cell sensitivity to drugs; actually, chronic exposure to an acidic pH is reported to increase expression of heat shock protein HSP27 level in tumor cells, leading to resistance to cisplatin [110].
Studying the effects of irradiation on cell killing and induction of apoptosis on low pH (pH 6.8) adapted human maxillary carcinoma cells, Ohtsubo et al. found that these cells, chronically adapted to low pHe, express a radio-insensitive phenotype [111]. To support this finding, it was found that an acidic microenvironment induces p53 expression [112], suppresses radiation-induced apoptosis [113, 114], and prolongs radiation-induced G2/M arrest, resulting in an increased DNA damage repair during the prolonged G2 arrest [115].
On the whole, acidity of extracellular microenvironment confers to dormant tumor cells a great advantage sustaining a resistant chemo- and radio-therapy phenotype, and treatments aimed to buffer extracellular acidity may revert several aggressive properties including resistance to conventional therapy.
Concluding remarks
Acidosis of tumor microenvironment of most solid tumors is well ascertained to promote an aggressive, immune-, chemo-, and radio-resistant phenotype, and in some circumstances an EMT program and stemness (Fig. 1). Therefore, acidic-adapted tumor cells are likely be able to survive to endogenous and exogenous stressors and might represent a hidden reserve of tumor cells from which a tumor could relapse and disseminate. Thus, acidosis of solid tumors may acquire a role in generating a novel niche of dormant tumor cells. Along with this view, it is reasonable for a successful treatment of cancer to build up new combined strategies focused on the inhibition of tumor cell-microenvironment crosstalk, and in particular targeting acidosis and acidic-adapted tumor cells.

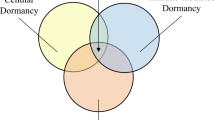
Crosstalk between the acidic microenvironment and cellular dormancy
References
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7:834–846
Wheelock EF, Weinhold KJ, Levich J (1981) The tumor dormant state. Adv Cancer Res 34:107–140
Holmgren L, O’Reilly MS, Folkman J (1995) Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1:149–153
Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N (2006) A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Ins 98:316–325
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713
Delbeke D, Coleman RE, Guiberteau MJ, Brown ML, Royal HD, Siegel BA et al (2006) Procedure guideline for tumor imaging with 18F-FDG PET/CT 1.0. J Nucl Med 47:885–895
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233
Miller DM, Thomas SD, Islam A, Muench D, Sedoris K (2012) c-Myc and cancer metabolism. Clin Cancer Res 18:5546–5553
DeBerardinis RJ (2008) Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med 10:767–777
Hirschhaeuser F, Sattler UG, Mueller-Klieser W (2011) Lactate: a metabolic key player in cancer. Cancer Res 71:6921–6925
Menon S, Manning BD (2008) Common corruption of the mTOR signaling network in human tumors. Oncogene 27(Suppl 2):S43–S51
Brand KA, Hermfisse U (1997) Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J 11:388–395
Ebert BL, Firth JD, Ratcliffe PJ (1995) Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter1 via distinct Cis-acting sequences. J Biol Chem 270:29083–29089
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switchrequired for cellular adaptation to hypoxia. Cell Metab 3:177–185
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P et al (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537
Cairns RA, Harris I, McCracken S, Mak TW (2011) Cancer cell metabolism. Cold Spring Harbor Symp Quant Biol 76:299–311
Parks SK, Chiche J, Pouysségur J (2013) Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer 13:611–623
Webb BA, Chimenti M, Jacobson MP, Barber DL (2011) Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 11:671–677
Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J, Kopacek J, Pastorekova S (2006) Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol 29:1025–1033
Calorini L, Peppicelli S, Bianchini F (2012) Extracellular acidity as favouring factor of tumor progression and metastatic dissemination. Exp Oncol 34:79–84
Pouysségur J, Franchi A, L’Allemain G, Paris S (1985) Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett 190:115–119
Avnet S, Di Pompo G, Lemma S, Salerno M, Perut F, Bonuccelli G, Granchi D, Zini N, Baldini N (2013) V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim Biophys Acta 1832:1105–1116
Balgi AD, Diering GH, Donohue E, Lam KK, Fonseca BD, Zimmerman C, Numata M, Roberge M (2011) Regulation of mTORC1 signaling by pH. PLoS One 6:e21549
Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK (2004) Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res 64:7336–7345
Peppicelli S, Toti A, Giannoni E, Bianchini F, Margheri F, Del Rosso M, Calorini L (2016) Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 15:1908–1918
Thaiparambil JT, Eggers CM, Marcus AI (2012) AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol 32:3203–3217
Gillies RJ, Deamer DW (1979) Intracellular pH changes during the cell cycle in Tetrahymena. J Cell Physiol 100:23–31
Putney LK, Barber DL (2003) Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J Biol Chem 278:44645–44649
Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, Schewe DM, Aguirre-Ghiso JA (2013) TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol 15:1351–1361
Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, Kastenberger M, Kreutz M, Nickl-Jockschat T, Bogdahn U, Bosserhoff AK, Hau P (2009) Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol 11:368–380
Ahmed S, Tsuchiya T (2004) Novel mechanism of tumorigenesis: increased transforming growth factor-beta 1 suppresses the expression of connexin 43 in BALB/cJ mice after implantation of poly-l-lactic acid. J Biomed Mater Res A 70:335–340
Peppicelli S, Bianchini F, Toti A, Laurenzana A, Fibbi G, Calorini L (2015) Extracellular acidity strengthens mesenchymal stem cells to promote melanoma progression. Cell Cycle 14:3088–3100
Peppicelli S, Bianchini F, Torre E, Calorini L (2014) Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clin Exp Metastasis 31:423–433
Xue L, Lucocq JM (1997) Low extracellular pH induces activation of ERK 2, JNK, and p38 in A431 and Swiss 3 T3 cells. Biochem Biophys Res Commun 241:236–242
Sarosi GA, Jaiswal K, Herndon E, Lopez-Guzman C, Spechler SJ, Souza RF (2005) Acid increases MAPK mediated proliferation in Barrett’s esophageal adenocarcinoma cells via intracellular acidification through a Cl-/HCO3- exchanger. Am J Physiol Gastrointest Liver Physiol 289:G991–G997
Ryder C, McColl K, Zhong F, Distelhorst CW (2012) Acidosis promotes Bcl-2 family-mediated evasion of apoptosis: involvement of acid-sensing G protein-coupled receptor Gpr65 signaling to Mek/Erk. J Biol Chem 287:27863–27875
Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG, Newgard CB, Chi JT (2013) Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 1:23
Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O (2016) Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab 24:311–323
Burd R, Wachsberger PR, Biaglow JE, Wahl ML, Lee I, Leeper DB (2001) Absence of Crabtree effect in human melanoma cells adapted to growth at low pH: reversal by respiratory inhibitors. Cancer Res 61:5630–5635
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT (2013) BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12:329–341
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ (2010) Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol 12:982–992
Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD et al (2011) Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18:829–840
Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, Wang C, Flomenberg N, Knudsen ES, Howell A, Pestell RG, Sotgia F, Lisanti MP (2011) Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 10:1271–1286
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K (2009) Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 69:7507–7511
Petrachi T, Romagnani A, Albini A, Longo C, Argenziano G, Grisendi G, Dominici M, Ciarrocchi A, Dallaglio K (2016) Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget. [Epub ahead of print] PubMed PMID:28036292.
Mizushima N, Klionsky DJ (2007) Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27:19–40
Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H et al (2013) Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 32(2261–72):1–11
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, Kallifatidis G, Kaczorowski A, Groth A, Gross W et al (2012) Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol 227:325–335
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 117:326–336
Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T, Brodie C (2009) The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 125:717–722
Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10:1533–1541
Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB et al (2012) Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res 72:3938–3947
Pellegrini P, Strambi A, Zipoli C, Hägg-Olofsson M, Buoncervello M, Linder S, De Milito A (2014) Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy 10:562–571
Goretzki L, Schmitt M, Mann K, Calvete J, Chucholowski N, Kramer M et al (1992) Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett 297:112–118
Mignatti P, Rifkin DB (1996) Plasminogen activators and matrix metalloproteinases in angiogenesis. Enzyme Protein 49:117–137
Lyons RM, Keski-Oja J, Moses HL (1988) Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 106:1659–1665
Ellis V, Pyke C, Eriksen J, Solberg H, Danø K (1992) The urokinase receptor: involvement in cell surface proteolysis and cancer invasion. Ann N Y Acad Sci 667:13–31
Smith HW, Marshall CJ (2010) Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol 11:23–36
Eden G, Archinti M, Furlan F, Murphy R, Degryse B (2011) The urokinase receptor interactome. Curr Pharm Des 17:1874–1889
Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L (2002) EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1:445–457
Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L (2003) ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res 63:1684–1695
Nagase H, Woessner JF (1999) Matrix metalloproteinases. J Biol Chem 274:21491–21494
Vihinen P, Kähäri VM (2002) Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer 99:157–166
Itoh T, Tanioka M, Matsuda H, Nishimoto H, Yoshioka T, Suzuki R et al (1999) Experimental metastasis is suppressed in MMP-9-deficient mice. Clin Exp Metastasis 17:177–181
Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S (1998) Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res 58:1048–1051
Kato Y, Nakayama Y, Umeda M, Miyazaki K (1992) Induction of 103-kDa gelatinase/type IV collagenase by acidic culture conditions in mouse metastatic melanoma cell lines. J Biol Chem 267:11424–11430
Toyoshima M, Nakajima M (1999) Human heparanase. Purification, characterization, cloning, and expression. J Biol Chem 274:24153–24160
Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ (2008) Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis 25:411–425
Stock C, Gassner B, Hauck CR, Arnold H, Mally S, Eble JA, Dieterich P, Schwab A (2005) Migration of human melanoma cells depends on extracellular pH and Na+/H + exchange. J Physiol 567:225–238
Paradise RK, Lauffenburger DA, Van Vliet KJ (2011) Acidic extracellular pH promotes activation of integrin α(v)β(3). PLoS One 6:e15746
Elias AP, Dias S (2008) Microenvironment changes (in pH) affect VEGF alternative splicing. Cancer Microenviron 1:131–139
Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y et al (2001) Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 20:3751–3756
Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK (2001) Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res 61:6020–6024
Xu L, Fidler IJ (2000) Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res 60:4610–4616
Scott PA, Gleadle JM, Bicknell R, Harris AL (1998) Role of the hypoxia sensing system, acidity and reproductive hormones in the variability of vascular endothelial growth factor induction in human breast carcinoma cell lines. Int J Cancer 75:706–712
Parks SK, Mazure NM, Counillon L, Pouysségur J (2013) Hypoxia promotes tumor cell survival in acidic conditions by preserving ATP levels. J Cell Physiol 228:1854–1862
Peppicelli S, Bianchini F, Contena C, Tombaccini D, Calorini L (2013) Acidic pH via NF-κB favours VEGF-C expression in human melanoma cells. Clin Exp Metastasis 30:957–967
Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY et al (2006) The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell 9:209–223
Lardner A (2001) The effects of extracellular pH on immune function. J Leukoc Biol 69:522–530
Choi SY, Collins CC, Gout PW, Wang Y (2013) Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol 230:350–355
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M et al (2007) Inhibitory effect of tumor cell derived lactic acid on human T cells. Blood 109:2812–2819
Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-KlieserW, Hoves S, Andreesen R et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107:2013–2021
Masson D, Peters PJ, Geuze HJ, Borst J, Tschopp J (1990) Interaction of chondroitin sulfate with perforin and granzymes of cytolytic T-cells is dependent on pH. BioChemistry 29:11229–11235
Severin T, Müller B, Giese G, Uhl B, Wolf B, Hauschildt S, Kreutz W (1994) pH-dependent LAK cell cytotoxicity. Tumour Biol 15:304–310
Liao YP, Schaue D, McBride WH (2007) Modification of the tumor microenvironment to enhance immunity. Front Biosci 12:3576–3600
Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, Santinami M, Borghi M, Fais S, Bellone M, Rivoltini L (2012) Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 72:2746–2756
Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E (2012) Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer 131:633–640
Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N (2013) Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer 133:1107–1118
Fernandez SF, Fung C, Helinski JD, Alluri R, Davidson BA, Knight PR 3rd (2013) Low pH environmental stress inhibits LPS and LTA-stimulated proinflammatory cytokine production in rat alveolar macrophages. Biomed Res Int 2013:742184
Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L (2013) The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology 2:e22058
Vishvakarma NK, Singh SM (2010) Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: Implication in antitumor activation of tumor-associated macrophages. Immunol Lett 134:83–92
Vishvakarma NK, Singh SM (2011) Augmentation of myelopoiesis in a murine host bearing a T cell lymphoma following in vivo administration of proton pump inhibitor pantoprazole. Biochimie 93:1786–1796
Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mulé JJ, Ibrahim-Hashim A, Gillies RJ (2016) Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res 76:1381–1390
Raghunand N, Gillies RJ (2000) pH and drug resistance in tumors. Drug Resist Updat 3:39–47
Liu FS (2009) Mechanisms of chemotherapeutic drug resistance in cancer therapy—a quick review. Taiwan J Obstet Gynecol 48:239–244
Mahoney BP, Raghunand N, Baggett B, Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics: I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem Pharmacol 66:1207–1218
Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C (2013) The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta 1832:606–617
Gerweck LE, Vijayappa S, Kozin S (2006) Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol Cancer Ther 5:1275–1279
Raghunand N, Mahoney BP, Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics. II. pHdependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochem Pharmacol T 66:1219–1229
Tannock IF, Rotin D (1989) Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 49:4373–4384
Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW et al (1999) Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer 80:1005–1011
Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ (2011) Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm 8:2032–2038
De Milito A, Fais S (2005) Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol 1:779–786
Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M (2006) Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia 8:143–152
Sauvant C, Nowak M, Wirth C, Schneider B, Riemann A, Gekle M, Thews O (2008) Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer 123:2532–2542
Williams AC, Collard TJ, Paraskeva C (1999) An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesis. Oncogene 18:3199–3204. doi:10.1038/sj.onc.1202660
Wachsberger PR, Landry J, Storck C et al (1997) Mammalian cells adapted to growth at pH 6.7 have elevated HSP27 levels and are resistant to cisplatin. Int J Hyperthermia 13:251–255
Ohtsubo T, Igawa H, Saito T, Matsumoto H, Park HJ, Song CW, Kano E, Saito H (2001) Acidic environment modifies heat- or radiation-induced apoptosis in human maxillary cancer cells. Int J Radiat Oncol Biol Phys 49:1391–1398
Ohtsubo T, Wang X, Takahashi A, Ohnishi K, Saito H, Song CW et al (1997) p53-dependent induction of WAF1 by a low-pH culture condition in human glioblastoma cells. Cancer Res 57:3910–3913
Lee HS, Park HJ, Lyons JC, Griffin RJ, Auger EA, Song CW (1997) Radiation-induced apoptosis in different pH environments in vitro. Int J Radiat Oncol Biol Phys 38:1079–1087
Choi EK, Roberts KP, Griffin RJ, Han T, Park HJ, Song CW et al (2004) Effect of pH on radiation-induced p53 expression. Int J Radiat Oncol Biol Phys 60:1264–1271
Park HJ, Lee SH, Chung H, Rhee YH, Lim BU, Ha SW et al (2003) Influence of environmental pH on G2-phase arrest caused by ionizing radiation. Radiat Res 159:86–93
Acknowledgements
This study was financially supported by grants from Istituto Toscano Tumori, Ente Cassa di Risparmio di Firenze.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Peppicelli, S., Andreucci, E., Ruzzolini, J. et al. The acidic microenvironment as a possible niche of dormant tumor cells. Cell. Mol. Life Sci. 74, 2761–2771 (2017). https://doi.org/10.1007/s00018-017-2496-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2496-y