
Abstract
Background
The ability of a tumor to become dormant in response to suboptimal conditions has recently been recognized as a key step in tumor progression. Tumor dormancy has been found to be implicated in several tumor types as the culprit of therapy resistance and metastasis development, the deadliest features of a cancer. Several lines of evidence indicate that the development of these traits may rely on the de-differentiation of committed tumor cells that regain stem-like properties during a dormant state. Presently, dormancy is classified into cell- and population-level, according to the preponderance of cellular mechanisms that keep tumor cells quiescent or to a balance between overall cell division and death, respectively. Cellular dormancy is characterized by autophagy, stress-tolerance signaling, microenvironmental cues and, of prime relevance, epigenetic modifications. It has been found that the epigenome alters during cellular quiescence, thus representing the driving force for short-term cancer progression. Population-level dormancy is characterized by processes that counteract proliferation, such as inappropriate blood supply and intense immune responses. The latter two mechanisms are not mutually exclusive and may affect tumor masses both simultaneously and subsequently.
Conclusions
Overall, tumor dormancy may represent an additional step in the acquisition of cancer characteristics, and its comprehension may clarify both theoretical and practical aspects of cancer development. Clinically, only a deep understanding of dormancy may explain the course of tumor development in different patients, thus representing a process that may be targeted to prevent and/or treat advanced-stage cancers. That is especially the case for breast cancer, against which the mTOR inhibitor everolimus displays potent antitumor activity in patients with metastatic disease by impeding autophagy and tumor dormancy onset. Here we will also discuss other targeted therapies directed towards tumor dormancy onset, e.g. specific inhibitors of SFK and MEK, or aimed at keeping tumor cells dormant, e.g. prosaposin derivatives, that may shortly enter clinical assessment in breast, and possibly other cancer types.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Self-sufficiency in growth signals, insensitivity to anti-proliferative signals, evading apoptosis, limitless replicative potential, sustained angiogenesis and metastatic potential are the classical “hallmarks of cancer” proposed by Hanahan and Weinberg in 2000 [1]. Two further traits were stated eleven years later: the presence of an abnormal metabolic pathway and evasion of the immune system [2]. Consistent with recent evidence, the ability to become dormant seems to allow for the acquisition of the above “hallmarks” in many tumor types. Indeed, dormancy, i.e., the state of constancy in number of tumor cells, underlies some pivotal traits of malignant tumors, which are dissemination, tissue invasion and resistance to conventional therapies, mainly due to the accumulation of mutations and the absence of proliferation. The former leads to tumor heterogeneity, the emblematic resistance-conferring feature according to the so-called clonal evolution model, whereas the latter frustrates the action of anti-proliferative agents, thus avoiding drug-induced cytotoxicity and fitting the paradigms of the cancer stem cell (CSC) model. Although these two principal theories on cancer initiation and progression, explained in further detail below, are not mutually exclusive, tumor dormancy may act as a key step in metastasis and resistance development, even when just one of them seems to be more explanatory for the case in point. Tumor dormancy represents a heterogeneous epi-phenomenon of a complex network of cues, ranging from cellular intrinsic and extrinsic processes, and from vascular to immunologic processes, driven by both genetic and epigenetic changes. In light of this, the aim of this review is to outline the state of the art on the topic, comparing and harmonizing discrepant theories to better speculate on future therapeutic approaches to tumor dormancy and effective bench-to-bedside translations to advanced-stage cancers, i.e., metastatic and/or refractory diseases.
2 Tumor dormancy as a key event in tumor progression
2.1 Tumor dormancy occurs at different levels entailing different steps in tumor progression
Tumor dormancy (TD) is a state of constancy in number of tumor cells triggered by sub-optimal conditions for tumor growth. It is also both a fundamental and heterogeneous step in cancer progression. A primary neoplastic mass can be kept constant by different cues, resulting in a quiescent or, alternatively, “dormant” state. Quiescence at the cellular level differs from that at the population level. The former results from a transient entrance in the G0 phase of the cell cycle, whereas the latter relies on a strict balance between cell division and death. In the latter case, cancer cell death can be due to a poor vascularization or to immune responses. An angiogenic dormancy can readily be distinguished from an immunological one [3]. Despite the unfavorable conditions that trigger TD, it may also be a convenient trait for a malignant neoplasm to develop. A recent gene ontology pathway analysis on tumor samples of glioblastoma patients has, for example, shown that upregulation of three antiproliferative clusters of genes may be associated with the shortest time to relapse after surgery and the worst clinical outcome during adjuvant therapy. The three identified hub genes were found to encode ferritin heavy chain 1 (FTH1), glutamate metabotropic receptor 1 (GRM1) and DNA damage inducible transcript 3 (DDIT3), all considered to be essential for glioblastoma cell dormancy [4]. These data underline the pivotal role of TD in this tumor setting: it fosters the development of drug resistance, due to accumulation of specific mutations or epigenetic changes that modify therapeutic targets, or proliferative arrest that hampers the action of anti-mitotic agents [4]. Besides, cellular dormancy enables cells of a primary tumor to disseminate, colonize host tissues and adapt to harsh microenvironments until these become less hostile [3]. In other words, cellular dormancy may allow metastatic outgrowth. The functional underpinnings of this lag phase may include activation of autophagic pathways [5], stress-tolerance signaling [6], unfolded protein responses (UPRs) [7], epigenetic modifications [8] and/or a balance between survival and pro-apoptotic signals present within the tumor niche [9]. Further details of these factors will be discussed below.
Interestingly, the two different types of dormancy are not mutually exclusive (Fig. 1), since they can occur in distinct groups of cancer cells simultaneously, or succeed each other during tumor progression [10]. In addition, a partial overlap between typologies may exist, resulting in merged dormant phenotypes. Therefore, a wide range of dormant conditions may exists, which makes the study of TD challenging. Contrasting theories on the therapeutic role of TD have, for instance, ensued from this non-univocal manifestation. On one hand, dormancy may stop tumor growth, improving the clinical outcome of cancer patients, whereas on the other hand it may lead to early dissemination of tumor cells, which lays a foundation for delayed metastasis onset and/or relapse after the removal of the primary neoplasm. Thus, whether one should try to keep dormant tumor cells asleep or directly kill them represents one of the most arduous controversies to be solved [11]. In the last section of this review we will discuss the arguments underlying these alternatives.

The heterogeneous foundations of tumor dormancy. Tumor dormancy may be triggered by specific signaling pathways and epigenetic changes that occur at the cellular level (yellow circle), adverse immune response of the host (blue circle) or inappropriate blood supply (red circle). Although those mechanisms can act separately, they often overlap each other to establish an overall dormant condition with a higher complexity in both underpinnings and consequences
2.2 Tumor dormancy on/off “switches”
Several essential properties characterize TD: dormant tumor cells are able to survive in foreign microenvironments, to develop drug resistance and to reversibly arrest their growth [11] (Fig. 2). With respect to this last trait, tumor cells can undergo a first switch, from a proliferative to a quiescent state and, unless eradicated, a second one back to the growing state. An imbalance between intrinsic and extrinsic pro- and anti-proliferative factors determines which phase a tumor cell is passing through. When this imbalance reverses, a switch occurs.

The distinctive traits of a dormant cancer cell. Dormancy of a cancer cell represents a reversible growth arrest due to interactions of several systemic and local factors. During this lag time, the cell acquires the ability to resist unfavorable conditions, such as inappropriate microenvironmental signals and anti-cancer therapies. Together, these traits represent the key feature of tumor dormancy
Several lines of clinical evidence indicate an influence of a primary neoplasm on the fate of disseminated tumor cells (DTCs) [12, 13]. Early metastatic tumor outgrowth after primary tumor removal suggests that (i) an early dissemination of cancer cells occurs while the primary mass is still growing, but they enter a latent state that keeps them undetectable and (ii) the primary tumor contributes in keeping DTCs asleep [12, 13]. Whether the switch towards dormancy occurs in cells of the primary tumor before their detachment, or while they are circulating in the bloodstream, or when they colonize foreign microenvironments still remains to be established. This transition point may, however, vary on a case-by-case basis, depending on when the general conditions become unfavorable to cancer cells. Cases of late relapse after surgery of both prostate [14] and breast cancer [15] indicate the existence of pro-dormancy cues other than those produced by the primary tumor [14, 15]. Thus, it appears that a tangled web of factors, which includes vascularization, immune surveillance and cellular processes, regulates the switches to enter and exit TD. Crosstalk between these systemic and local factors supports the existence of an overall TD state occurring in cancer cells, only conventionally classified into cellular, angiogenic and immune-mediated [12]. For the sake of simplicity, however, this terminology will be maintained in this review.
At the cellular level, the switches from proliferation to quiescence and vice versa can be monitored through changes in patterns of gene expression and protein activation. The main indicator of cellular dormancy is a low pERK (phosphorylated Extracellular signal-Regulated Kinase)/p38 ratio, whereas this ratio has been found to be high in proliferating DTCs [16]. As stated above, TD may also be governed by epigenetic mechanisms, and it has been found that distinct changes in DNA methylation, histone acetylation and micro-RNA (miRNA) expression patterns may mark the switches of a cancer cell to and from latency [8, 17, 18]. Intriguingly, it has been found that a set of dormancy-inducing miRNAs, which act by leading to silencing of the chemokine ligand (CXCL) 12-chemokine receptor (CXCR) 4 axis, can be exchanged through gap junctions [8]. Consequently, the switch towards cellular dormancy may result in a synchronized action at the population level. As both local and systemic factors can induce a dormant state, so can they induce a reactivation process. At a local level, since the dormant state can be triggered by nutrient depletion, the reactivation often follows neo-angiogenesis through a process called “angiogenic switch”. This phenomenon has e.g. been observed in a mouse model of Lewis lung carcinoma dormant micro-metastatic cells, which produce Vascular Endothelial Growth Factor (VEGF) and recruit bone-marrow-derived endothelial cell progenitors to reactivate [19]. An involvement of systemic factors has been demonstrated in mice by Elkabets et al. [20]. They found that two distinct cancer cell lines, which showed dormant phenotypes, i.e., very slow proliferation rates, when inoculated singularly in mammary fat pads, behaved differently when grafted simultaneously but at different sites. In doing so, it was found that one tumor may act as an “instigator”, keeping itself dormant but causing reactivation of the other one, the “responder”, through endocrine signals. The former was found to produce osteopontin, thereby inducing hematopoietic progenitors to secrete granulin. Granulin, in turn, led to a desmoplastic reaction by activating stromal cells and fostering collagen deposition at distant sites [20]. The resulting dense matrix represents a reactivation-inducing cue, since it enhances integrin-mediated transduction of mechanical forces into biochemical signals, thereby promoting reactivation of the “responder” [21]. Even though the chain of causative events between markers and TD still awaits verification, their profiling may be used for dormancy detection in both primary and disseminated cancer cells [3].
2.3 Tumor dormancy as cornerstone for a unified cancer development model
Growth arrest of a primary tumor or latency of pre-metastatic DTCs, both due to TD, represent key steps in tumor progression. As such, the concept of TD can be taken on by two currently debated paradigms of tumor biology, i.e., the clonal evolution and the cancer stem cell (CSC) theories. The former (Fig. 3a) postulates that every cancer cell has tumorigenic potential, since it is capable of proliferation. As in every evolutionary process, the final tumor arises from a stochastic accumulation and selection of genetic and epigenetic alterations that lead to several distinguishable clones [22]. Furthermore, it has been found that the genetic and epigenetic alteration rates become increasingly higher with disease progression, as has been shown by deep DNA sequencing of neoplasms at different stages of development [23, 24]. The latter posits that particular tumor cells with stem-like features (i.e., CSCs) stay at the apex of a hierarchical population (Fig. 3b). These cells show a long-term self-renewal ability, multipotency and resistance to apoptosis. In addition, they can give rise, through asymmetric division, to “committed” sub-sets of cancer cells with a limited proliferative ability and a limited tumorigenic potential [25]. These cells are known as transit amplifying cells, since they undergo a specific number of cell divisions and finally differentiate irreversibly [25]. Based on their unique features, CSCs have been proposed as the sole transformed cells that underlie metastatic spread and chemotherapy resistance, putatively by entering a dormant state [26]. So, while in the clonal evolution model all cells of the tumor have the same ability to contribute to tumor initiation and growth, in the CSC model only the stem sub-set can. This is the reason why tumor relapse will inevitably occur after applying a therapy that spares the stem population. As stated above, however, the two paradigms are not mutually exclusive, since one may better explain a certain disease and vice versa, or they can be applied independently to different stages of the same neoplasm. Therefore, a unified theory has recently been proposed, i.e., the plastic CSC model (Fig. 4). It explains tumor heterogeneity by combining several key points of the two theories. The main difference between the classical and plastic CSC paradigms lies in the process of differentiation: it is presumed to be unidirectional in the former, whereas the latter assumes the possibility of reverting the hierarchy through a bidirectional process mainly regulated by the niche environment and epigenetic modification patterns, which are interrelated. In this way, a differentiated cancer cell can dedifferentiate and regain stem-like features. If this cell bears a new mutation or modification in its epigenome, it can generate a clonal sub-set that differs from the progenitor cell, thereby leading to tumor heterogeneity. Alternatively, a stemness-conferring mutation can occur in a non-CSC, thereby triggering a switch to the stem-like phenotype. The new subclones, endowed with distinctive traits, e.g. the ability to metastasize and resist treatment, undergo the same selective pressure that is posited by the classical clonal evolution theory, thereby driving disease progression towards a specific outcome. TD can be contextualized in this new scenario, representing the specific step in which differentiated cancer cells accumulate genetic and epigenetic alterations, regain stemness and give rise to various clonal populations that underlie the different malignant traits [27].

Tumor dormancy in the context of clonal evolution and cancer stem cell models. a Schematic representation of cancer initiation and progression according to the clonal evolution model. A mutation or epimutation (represented as a red flash) can occur in a normal cell (white), thereby converting it into a cancer founder cell (green). The latter can proliferate generating a clonal population. Other genetic changes may occur stochastically along the generations spawning new subclones (light red, pale blue, yellow and purple). These subclones may have diverse traits compared to the original transformed cell. The selective pressure exerted by the microenvironment governs the population balance, promoting clones with specific features rather than others that show disadvantageous traits. As the cancer population expands, genomic instability and aberrations increase, giving rise to more subclones that enhance tumor heterogeneity, which in turn underlies negative clinical outcomes. In this scenario, tumor dormancy (TD) seems to be a critical state for mutation accumulation in cancer cells before they can divide again, thus generating a daughter cell with new features. b According to the cancer stem cell (CSC) model, a tumor initiating mutation should occur in a normal stem cell (white). The resulting CSC (light red) may divide symmetrically to self-renew or asymmetrically to spawn a “committed” cell (yellow). The latter has a limited number of divisions to undergo. Thereby it generates a hierarchical population of transit amplifying cells that show a progressively more differentiated phenotype. The bottom of the hierarchy is represented by the differentiated cancer cells (pale blue) without proliferative ability. The organization of the tumor mass explains the differences in treatment efficacy: only CSC-targeting therapies can eradicate cancer, whereas other treatments may spare the stem cell pool, which may consequently enable tumor re-growth. CSCs may also elude anti-mitotic drugs by entering TD, leading to cancer persistence

Tumor dormancy and the unifying plastic cancer stem cell model. Alternative putative fates (trackable by marked outlines) of a differentiated cancer cell (blue) are represented in conformity with the paradigms of the plastic cancer stem cell theory. If the balance between tumor dormancy (TD) inducers and suppressors leans toward the first ones, then the cancer cell becomes quiescent (pale blue). If the switch towards TD does not involve detachment from the primary tumor (left half), the dormant cell can regain stem-like features (light red), thereby stoking tumor growth after the switch back to the proliferative state. Alternatively, since the cancer cell may accumulate mutations (red flash) during TD, when it awakens and restores stem features, the cell (green) may acquire the potential of generating new differentiated subclones, carrying unique traits. These will be subjected to selection and enhance tumor heterogeneity and, thus, possible resistance to therapy. The right half of the figure schematically shows the fate of a differentiated cancer cell that, at the same time, enters dormancy and detaches from the primary tumor, putatively as a consequence of epithelial-to-mesenchymal transition (EMT). This cell can disseminate by entering the circulation and infiltrating distant tissues. In the meantime, dormancy may allow dedifferentiation to occur, converting the differentiated cell into a stem-like cell and spawning metastasis development. Metastatic subclones may differ from primary subclones since the new microenvironment may alter the epigenetic state of the CSCs, thus modifying their differentiation potential even in the absence of genetic mutations. This phenomenon underlies the clinical differences that exist between primary and secondary cancers
In recent years, the complex relationship between epithelial-to-mesenchymal transition (EMT), TD and the dedifferentiation of cancer cells has largely been elucidated [28,29,30]. EMT has been found to be induced by signals from the microenvironment, e.g. transforming growth factor (TGF) β2, stochastically occurring epigenetic changes and inflammation [31]. EMT is characterized by the loss of cell adhesion molecules, cell polarity and epithelial markers and, conversely, the gain of mesenchymal-like features. In vitro induction of Twist1 and Snail1, two known EMT-triggering transcription factors [32], has been found to endow breast cancer non-stem cells not only with the above listed features, but also with tumor initiating and self-renewal abilities, which are CSC-specific. In addition, the expression of breast CSC-specific markers, i.e., CD44high/CD24low, has been found to confirm that EMT, directly or indirectly, can trigger the dedifferentiation process [29]. Similar results have been obtained in other solid and hematological tumors, such as melanoma and myeloid leukemia [28].
Besides, EMT has long been considered as a pivotal step in metastatic spread, as suggested by a lineage-tracing experiment carried out in a mouse model of pancreatic intraepithelial neoplasia [33]. The authors found that cells of the primary neoplasm that have undergone EMT can differentiate, enter into the bloodstream and colonize the liver, giving rise to metastatic subclones [33]. Recent data suggest, however, that EMT may neither be a necessary nor a sufficient step to initiate tumor dissemination [34]. Specifically, it has been found that dissemination can occur also with the retention of epithelial features in a semi-mesenchymal condition, i.e., common oncogenic mutations of the human epidermal growth factor receptor 2 (HER2) cause disruption of adhesion and polarity, and hence tumor spread, without entailing a full EMT [35]. Furthermore, this detachment eases the subsequent metastasizing process, given that epithelial features, which are partially retained in mutated-HER2 expressing cells, play a major role once circulating tumor cells have reached a secondary site [35]. The importance of epithelial-like traits to complete the seeding of foreign tissues and to enable metastatic outgrowth is demonstrated by the fact that EMT, which occurs in the primary site, needs to be followed by its reverse, i.e., mesenchymal-to-epithelial transition (MET), in the secondary site [36]. As a consequence, EMT may not be considered as the sole and direct inducer of metastasis, but rather as the inducer of the switch towards TD.
Metastatic development may involve sequential steps: EMT may induce TD, which in turn may foster the switch from non-stem to stem features and allow metastasis to occur (Fig. 5). An important caveat should, however, be addressed. Since several cues are involved in inducing dormancy, EMT may not to be strictly necessary for inducing TD and, consequently, dedifferentiation and dissemination, even if it plays a role. Both in vitro and in vivo evidence underscore this notion. The expression of one of the most debated EMT-regulatory factors, Prrx1 (Paired Related Homeobox 1) in differentiated breast carcinoma cells promotes EMT, but not the recoup of stemness [37]. Conversely, its suppression has been found to foster the colonization of host tissues without promoting EMT [36, 37]. This means that (i) TD, rather than EMT, may be the direct mechanism underlying the non-CSC to CSC switch, and (ii) EMT may induce TD since an association between these processes has been reported [35], but it is neither sufficient nor necessary. That is, EMT can be uncoupled from the re-acquisition of stem-like features [37].

Putative sequential steps of cancer dedifferentiation. A differentiated cancer cell (non-cancer stem cell, non-CSC) can revert the differentiation process by passing through a dormant state. During this process, epithelial-to-mesenchymal transition (EMT) does neither seem to be a sufficient nor a direct stimulus to allow stemness recovery. It rather represents an effective tumor dormancy (TD) inducer, whereas tumor dormancy is the actual key step required to let cancer cells differentiate. Therefore, EMT may not be involved in this process, since other TD inducers can substitute for it
Overall, the above data provide strong indications for the existence of a bidirectional transition between CSCs and non-CSCs, thereby appraising the plastic CSC theory rather than the independent one. With regard to treatment resistance, the plastic CSC theory posits that also a CSC-targeting therapy may fail in eradicating the whole tumor, since differentiated subclones may endure in a dormant state, either lodged in the primary tumor site or disseminated to secondary sites. When they revive, cell intrinsic and/or extrinsic mechanisms may lead to a regain of stemness, and hence to disease relapse and metastatic outgrowth [38]. This scenario represents an alternative framework for therapy resistant cases that were previously considered to rely on CSC intrinsic features, such as chemotherapy-resistant breast cancer cells [38]. Therefore, TD may represent an important cornerstone of a plausible explanatory model of tumor progression. Funneling an increasing attention to this key step may be far-reaching, i.e., it may reinforce the unified model of tumor progression, allow a better understanding of its underpinnings and enhance the effectiveness of stem-specific therapy by comprehending how to block the dedifferentiation process [27].
2.4 Tumor dormancy as iterative process underlying cancer progression
Several lines of evidence suggest that tumor cells can enter and exit dormancy more than once in their lifespan [38,39,40]. Prostate cancer is a striking example of this iterative progression mode [41]. Indeed, the clinical course of prostate cancer is characterized by progressively shorter cycles of treatment-dormancy-relapse. Since also other cancer types, such as breast and colorectal cancer [31, 41], may show a similar behavior, TD has been proposed as the initial “steady state” of all neoplasms, i.e., the pre-diagnostic phase of a cancer in which it coexists with its host harmlessly. After this first, and usually long-lasting, period of TD, the ability to re-enter a quiescent state is progressively lost along with cancer progression, as indicated by the gradual shortening of TD periods observed in several cancer types [38,39,40]. Of note, TD duration is estimated on the basis of progression-free survival and tumor volume constancy after any therapeutic intervention.
Specifically, the diagnosis of prostate cancer at a localized stage implies prostatectomy as elective cure. Despite some cases of early metastatic relapse, the dormancy progression-free period that follows surgical removal of the prostate usually lasts 10 to 13 years [42]. The contingent metastatic onset is then counteracted with an androgen-deprivation therapy, which can stop disease progression for on average less than two years. This shorter dormancy period may end with the development of a castration-resistant prostate cancer type, which can be counteracted with docetaxel treatment. The latter induces TD by blocking its growth for a few months, after which the disease reaches its terminal stage, commonly with a mortal outcome within weeks (Fig. 6). The main explicative model for this pattern of tumor progression is still a matter of debate. Crea et al. [8] have, for example, posited that the initial sub-clinical phase of prostate cancer, which corresponds to the longest period of dormancy, may be characterized by a hierarchical organization of the cancer population, with few CSCs at the apex that are able to enter dormancy. Since the mutation rate becomes gradually higher with cancer progression, and thus with the shortening of dormancy periods, they hypothesize that as genetic alterations accumulate CSC lose their dormancy ability. In their model they also assumed that TD was a CSC prerogative, and hence a lack of differentiated cancer cells. Therefore, the shortening of dormancy periods was thought to be due to both an increase in genetic alterations and attainment of an overall higher degree of differentiation of the tumor. However, several lines of evidence call into question the plausibility of this unique explicative model. The main counterargument lies in considering TD as a non-distinctive ability of CSCs, as such phase can be undergone also by committed and differentiated tumor cells, mainly to regain stem properties, as stated above [27]. Besides that, the TD model does not provide an explanation for the occurrence of synchronous primary and metastatic diseases, thereby keeping relevance for many but not all tumor types. Since this model leaves that characteristic clinical progression pattern without a suitable explanation, new efforts should be undertaken to better understand this pattern and to elaborate a model that entirely fits current TD evidence.

Prostate cancer as an iterative model of tumor dormancy. A prostatic intraepithelial neoplasia (PIN) may persist sub-clinically for more than 10 years before being detected, during an initial “steady state” between transformed cells and the host. The figure shows the trend of the overall tumor volume in time (blue curve). Notably, the volume may fluctuate depending on medical interventions (prostatectomy, androgen-deprivation therapy and taxane-based chemotherapy) and disease responses (metastatic onset, evolution to castration-resistant prostate cancer -CRPC- and terminal mortal stage of the neoplasm). As shown, dormancy periods, characterized by constant tumor volumes, shorten during disease progression and exhibit increases in mutation rates. As yet, the functional underpinnings of this trend have not been elucidated (adapted from [8])
3 Tumor cell dormancy: Molecular mechanisms and biochemical pathways
3.1 Intrinsic and extrinsic mechanisms underlying tumor dormancy at the cellular level
As stated above, the dormant state is not considered to be brought about by a single mechanism, but rather by the interrelation of several ones: cellular pathways, neo-angiogenesis and immunological cues [43]. Even in the absence of well-defined borders among them, it is useful to study each isolated mechanism to understand its putative contribution to the whole process. TD at the cellular level is classically related to DTCs, which are cells in a quiescent state that are characterized by a lack of both proliferative and apoptotic markers [3]. Recently, it has been suggested that circulating tumor cells (CTCs) share the same features, indicating that a common dormancy mechanism may be at the root of both cell types [43]. To ascertain that cellular dormancy is occurring in tumor cells, they should be profiled for the proliferative marker Ki67, the activity of apoptotic pathways through the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and more innovative M30 expression assays. Only in case of a triple-negative outcome a cell is considered dormant, and to the best of our knowledge it can be either a DTC or a CTC. According to the aforementioned definition, cellular dormancy characterizes cells in a quiescent state, i.e., a stable non-proliferative state, similar to senescence but, contrary to the latter, reversible. Genes induced in normally quiescent stem cells, e.g. muscle, hair follicle or hematopoietic stem cells, belong to a set of genes that are also upregulated in dormant cancer cells, such as p38 and Notch [44]. Other factors and related pathways governing cellular dormancy are BMPs. These are produced by the tissue milieu and keep DTCs dormant even upon their engraftment [31] and can induce dormancy in e.g. prostate CSCs by increasing the expression of both the anti-metastatic factor N-Myc Downstream Regulated 1 (NDRG1) and the cell-cycle inhibitor p21 [44, 45]. Cellular dormancy can also be considered as a transient maladaptation to e.g. a BMP-rich microenvironment, which can be reverted by genetic and/or epigenetic changes that enhance fitness within the engrafted milieu. Conversely, BMP pathway upregulation has also been shown to block self-renewal activity and to promote differentiation of both normal stem cells and CSCs into mature cells [43].
It is important to note that, although dormancy at a single-cell level can occur in isolated cancer cells, i.e., CTCs or DTCs, so it can in constituent cells of tumor bulky masses. Since those two processes are governed by the same mechanisms, namely autophagy, stress-tolerance signaling, signals from the microenvironment and epigenetics, some authors have extended the concept of cellular dormancy to cells other than CTCs and DTCs. As a consequence, cellular processes may also affect population-level dormancy, underscoring the existence of cross-talk at the root of overall TD.
3.2 Autophagy maintains metabolic homeostasis in dormant tumor cells
When harbored in a harsh milieu, tumor cells secrete autocrine and paracrine factors that inhibit the phosphoinositide 3-kinase (PI3K) cascade, leading to quiescence and activation of the autophagic program [3]. The activation level of AKT, one of the direct downstream effectors of PI3K, has been found to inversely correlate with the probability of entering dormancy [46]. Counterintuitively, mammalian target of rapamycin (mTOR), which is a known target of AKT, has been found to be upregulated in quiescent cells of head and neck squamous cell carcinoma [5, 46]. Its activation is actually maintained independently of AKT through upregulation of RHEB, a small GTPase that is under the control of the stress-regulated transcription factor ATF6α and able to avoid apoptosis. ATF6α is, in turn, activated by a unfolder protein response (UPR), which is triggered by high p38 kinase activity. However, the majority of dormant tumors expresses high levels of ARHI, an inhibitor of the PI3K-AKT-mTOR cascade [47, 48]. Thus, the constitutive inhibition that mTOR exerts on autophagy-related (ATG) proteins vanishes during TD. Therefore, mTOR is considered a meeting point of different pathways involved in TD, thereby representing a crucial factor in the regulation of the whole process (Fig. 7). On one hand, inhibition of the PI3K pathway exerted by ARHI fosters dormancy and autophagy, whereas on the other hand, stress signals such as osmotic shock, inflammatory cytokines and irradiation activate the p38 cascade, and thus mTOR, allowing cell growth, proliferation, motility and survival.

Molecular pathways converging on mTOR to regulate tumor dormancy. In a dormant tumor cell, a high expression and activity of ARHI leads to disinhibition of autophagy via the PI3K-AKT-mTOR cascade, thus allowing tumor survival within the dormant state. This cascade may be activated by several extra-cellular factors (blue box), which interact with different types of receptors: integrins (blue) for ECM components, tyrosine kinase receptors (TKR - violet) for survival and growth factors and G protein-coupled receptors (GPCR - orange) for chemokines and hormones. On the other hand, several cancer cells may enter into a dormant state to resist stress conditions (red box), thereby activating stress-tolerance pathways. Here, the p38 pathway is represented as a trigger of the unfolded protein response (UPR), which in turn maintains mTOR activation via ATF6α and RHEB. Since mTOR is involved is several biological processes, such as cellular growth, proliferation, motility and survival, it can drive the cell fate towards awakening from tumor dormancy (TD) and regrowth. This effect can be mediated by the blockade of several ATG members, which are known autophagy and dormancy inducers
In vivo studies have been carried out in mice to shed light on those molecular pathways. By doing so, it has been found that ARHI downregulation through RNA interference may awake dormant cells of several tumor types, allowing them to re-enter the proliferative state [17, 47, 48]. Conversely, it has been found that direct inhibition of autophagy with hydroxychloroquine, hence without modifying ARHI expression, does not lead to a resume in proliferation in vitro and a reduction in tumor burden in vivo [47, 48]. ATG4, ATG7 and ATG8 are known to be pivotal to maintaining metabolic fitness in dormant cells by activating the autophagy program. In particular, it has been found that ATG7 knockout impairs breast DTC dormancy and late metastasis formation [49]. The most accredited mechanism underlying this effect includes decreased mitophagy alongside autophagy blockade, leading to an accumulation of damaged mitochondria and reactive oxygen species (ROS) in DTCs. Intriguingly, when autophagy is inhibited after proliferation recovery, it seems to be ineffective in blocking tumor progression [49].
3.3 Tumor cells enter dormancy to tolerate intra- and extra-cellular stress signals
In addition to p38 and Notch (see above), master regulators of cellular TD include the dual specificity tyrosine-phosphorylation-regulated kinase 1B (DYRK1B), which is a stress-activated protein. It can actively induce dormancy by blocking proteins of the cell-cycle checkpoint transition machinery, i.e., cyclin D1, cyclin-dependent kinase 4 (CDK4) and p27. A fundamental discovery has been made on DYRK1B in a pancreatic cancer model, i.e., when inhibited, it was found that only dormant cancer cells underwent apoptosis, whereas quiescent normal cells kept themselves alive and dormant [6, 49, 50]. This observation may pave the way to new therapies aimed at specifically targeting dormancy to prevent pancreatic tumor cell awakening, including its fatal consequences. Since also other stress signaling pathways, such as the JUN N-terminal kinase (JNK) pathway, can induce dormancy, it is likely that arresting proliferation to repair damage represents a conserved stress response during evolution. Although activation of p38 and UPR are considered stress-tolerance mechanisms that seem to be involved in triggering dormancy in tumor cells [3, 50], further investigation is required to establish whether dormancy in tumor cells acts as a stress-tolerance mechanism or not, and whether it holds other functions, such as the ability to resist therapy-induced or microenvironment-induced stress signals.
3.4 Microenvironmental regulation of tumor cell dormancy
3.4.1 Dormancy-restrictive and -permissive niches
Microenvironmental signals may determine whether a DTC is allowed to grow or is maintained in a dormant state after colonization of the target tissue. In light of these alternatives, microenvironments have been classified in dormancy-restrictive and dormancy-permissive or “dormant niches”, respectively [3]. Interestingly, a specific microenvironment is not a priori permissive or restrictive, but the kind of DTC involved in the process may also be crucial for its own development. Bone marrow has, for example, been found to be able to induce and/or preserve dormancy of DTCs derived from different types of primary tumors. Leukemia cells in the bone marrow may, for instance, be arrested in the G0 phase of the cell cycle under the influence of osteoblast-derived growth arrest-specific protein 6 (GAS6) [51]. By binding to the tyrosine-kinase receptor MER, GAS6 avoids therapy-induced apoptosis of leukemia cells by blocking their proliferation [52]. Alternatively, GAS6 can bind and activate the AXL (or UFO) receptor, which abounds on DTCs from primary prostate carcinoma cells, thereby maintaining dormancy in these cells [52]. In addition, bone marrow stromal cells are known to secrete BMP4 and BMP7, two agonists of the BMP receptor 2 (BMPR2). The latter is variably expressed on prostate DTCs that have engrafted the bone marrow following a CXCL12 gradient [53]. Its transduction pathway allows the maintenance of cells in a dormant state, which may also explain why the BMPR2-antagonist COCO is often detected in dormancy-restrictive microenvironments [3]. In particular, COCO has been found to accumulate on the surface of DTCs that colonize specific organs, thereby avoiding the dormancy-inducing action of BMPs and allowing their proliferation. An organ-specific distribution of COCO is underlined by several clinical studies showing that the COCO signature may be predictive for lung relapses, but not for brain and bone relapses, of breast cancer [31, 54].
Various studies have shown that non-stem DTCs can infiltrate foreign organs and keep themselves dormant until signals from the niche foster their conversion to CSCs and reactivation [31]. Intriguingly, a mouse model of lung adenocarcinoma has shown that metastasis onset may be correlated to stem-like feature recovery by DTCs, such as loss of Nkx2–1 [55]. Similarly, it has been found that breast cancer DTCs may survive in the bone marrow milieu when their phenotype reverts to CSCs expressing Sox family proteins [56]. However, a previous recirculation of the pre-metastatic founder cells between different microenvironments seems to be necessary for a rapid and proper final reactivation. This is because some sites play key roles as so-called “sanctuaries”, i.e., temporary stations where DTCs encounter conditions appropriate for limited expansion and the acquisition of new traits that would be required later to resume growth in the definitive metastatic site. Therefore, microenvironments may provide signals that dictate DTC fate and, as a direct consequence, the clinical outcome of the disease, i.e., early relapse, late relapse and/or relapse-free survival may be related to the dormancy-permissive or dormancy-restrictive characteristics of the colonized site. Similarly, breast DTCs with high Src activity and CXCR4 expression have been found to be able to survive in a bone microenvironment in response to local CXCL12 [15, 55]. These cells occupy a hematopoietic stem cell (HSC)-like niche, in which the CXCR4 pathway guarantees homing and quiescence, whereas the Src pathway confers resistance to pro-apoptotic signals, such as Trail [57]. Another striking example of a dormancy-inducing microenvironment is a TGF β2-rich bone marrow for head and neck squamous cell carcinoma DTCs [58].
On the other hand, bone marrow may act as a dormancy-restrictive microenvironment for metastatic breast cancer cells that highly express vascular cell adhesion molecule 1 (VCAM1) [59]. VCAM1 allows breast DTCs to recruit osteoclast progenitors by binding to their α4β1 integrins. After differentiation, osteoclasts break down the bone matrix, hence leading to DTC reactivation and metastasis. Given that reabsorption of the calcified matrix that lodges DTCs causes their awakening, also physical stimuli may govern the switches from and towards dormancy, putatively via cytoskeletal modifications. In support of this notion, breast cancer DTCs have been found to restore proliferation in response to IL11 and Jagged only within bone, due to osteolysis induction, whereas they stay dormant in colonized brain and lung tissues, where physical changes do not occur in response to the above factors [57,58,59]. Similarly, metastatic outgrowth following skeletal trauma points at a key role played by bone remodeling in reactivating DTCs via TNFα, IL1β, IL6 and prostaglandin E2 [60].
Urokinase plasminogen activator (uPA) is another promoter of tumor regrowth. After binding to its receptor uPAR, the complex recruits α5β1 integrin to activate ERK signaling. A subsequent increase in the pERK/p38 ratio leads to an overt proliferation of the tumor cells. The uPAR expression level on the tumor cell membrane, and thereby the ability to exit dormancy, is regulated by extracellular matrix-integrin interactions. Its increase in ovarian cancer cells has e.g. been found to be due to αvβ3 integrin-fibronectin binding, whereas interactions between αvβ3 integrin and vitronectin have been found to result in a decrease in uPAR expression on the tumor cells, thus favoring their quiescence [43, 61].
3.4.2 Tissue stiffness regulates entrance and escape from the dormant state
Extracellular matrix stiffness has been found to play a crucial role in dictating DTC fate in both in vitro and in vivo studies [21]. For instance, hepatocellular carcinoma cells that colonize rigid microenvironments may activate TGF β1 signaling, thereby restoring growth and proliferation, whereas less stiff microenvironments appear to support dormancy [21]. Similarly, an integrin β1-mediated proliferative switch of dormant DTCs has been found to occur in response to fibrosis in a mouse model of breast cancer [21]. Several 3D-models and in vivo studies support a pivotal role of type I collagen (Col-I) in dictating the proliferative fate of DTCs [21, 60,61,62]. The downstream effector pathways involve integrin β1, collagen receptor DDR1, SFK (Src Family Kinase), MEK, ERK and MLCK activation, and shRNA-based or pharmaceutical inhibition of these cascades has been found to prevent metastatic outgrowth [21, 62, 63].
Matrix periostin is an additional cue that, other than VCAM1 expression, TGF β1 or integrin β1 signaling activation, drives tumor cell escape from dormancy. Periostin fosters Wnt signaling, a known pathway required for restoring the self-renewal ability of DTCs and, consequently, metastatic outgrowth of e.g. breast carcinoma [64]. Specifically, periostin is produced by stromal fibroblasts in response to TGF β1 and by endothelial tips of de novo formed blood vessels [55, 65], and acts as a scaffold to ease the presentation of Wnt ligands to DTCs [31].
3.4.3 Role of basement membranes and blood vessels in tumor dormancy
The basement membrane acts as a pivotal effector of signaling that promotes survival, functional differentiation, growth arrest and resistance against cytotoxic agents of both normal and transformed epithelial cells, thereby representing a central component of the dormant niche. When cells of the primary tumor detach from the mass to enter the circulation, they also lose contact with their “native” basement membrane. Following hematogenous dissemination, DTCs may seed to target organs, but before they can do so they encounter the vascular basement membrane of the tissue-borne blood vessels. Accordingly, live imaging studies and metastasis assays have shown that DTCs often reside on the vascular basement membrane of the vessels of the target organs [11, 65]. Therefore, the fundamental concept of perivascular niches, i.e., niches surrounding the microvasculature, also extends to tumors. In particular, survival, therapy resistance and dormancy regulation are the main processes that are conveyed by the vascular basement membrane. Of note, an important caveat should be issued, i.e., while contact between mature blood vessels and DTCs drives the latter towards a quiescent state, endothelial tips of neo-formed vascular sprouts secrete periostin and TGF β1, but not thrombospondin 1, to create a microenvironment that facilitates reactivation of dormant DTCs and even accelerates their proliferation [66]. In addition, the pro-tumorigenic function of the nascent endothelium is thought to rely on its ability to trigger a T helper 2-mediated inflammatory response, which has been found to accelerate metastatic outgrowth in tumor models [67]. This effect may be due mainly to macrophage polarization to the pro-tumor M2 phenotype, which plays a major role in micro-metastatic niche formation [66, 68, 69]. Conversely, besides the lack of periostin and TGF β1 expression, the endothelium of mature vessels peculiarly secretes thrombospondin 1 in its basement membrane. This angiocrine factor is indeed known to act as a dormancy inducer and putatively as a tumor suppressor, thereby providing a molecular explanation for the differences observed between mature and newly formed blood vessels in DTC fate determination [55, 65]. It is also worth noting that the perivascular niche not only provides a treatment safe haven to DTCs, but also increases their proliferative potential, i.e., several anticancer drugs (such as taxol, cyclophosphamide and doxorubicin) can modify the signaling pathways of endothelial cells, making them paradoxically suitable to sustain the survival and proliferation of DTCs. The release of low doses of TNF (tumor necrosis factor) by lung endothelial cells is a clear example of this phenomenon. TNF has been found to promote the growth of some cancer types via VEGF induction [70]).
Since the vascular endothelium is a well-known component of the stem cell niche and a putative key constituent of the dormant niche, this raises the possibility that these two are actually one and the same [11]. In agreement with this hypothesis, evidence other than the common involvement of the vascular influence suggests the existence of only one type of niche [67, 68]. Firstly, it has been found that the primary tumor size correlates with the number of CTCs, but not DTCs detected in a specific target organ, which is approximatively constant [69, 70]. Thus, it seems that CTCs can seed to only a limited number of niches in a target organ, as has e.g. been found in late metastasis in the brain where they are exclusively perivascular as determined by real-time imaging [71]. Those niches appear to be defined in both numbers and dimensions. Second, as the stem cell niche acts as a fate determinant for daughter cells of stem cells, a differentiated DTC that seeds a target niche may be epigenetically reprogrammed to recover stemness [8]. According to the above data, a strong overlap between stem cells and dormant niches is credible. Third, evidence suggests that stem cells and DTCs are tethered to their niches by the same molecular factors. In particular, inhibition of the CXCL12-CXCR4 signaling axis has been found to result in mobilization of both hematopoietic stem cells and prostate DTCs from the bone marrow into the circulation [72]. In light of these observations, the above hypothesis seems plausible, even though further studies are required to confirm, or even refute, it [73].
The above data suggest that perivascular niches interact with their harboring cells through both common and tissue-specific mechanisms. In addition to differences in the molecular constitution of the niches that define the vascular beds of each tissue, also intra-tissue variability may exists. In view of this, it may be more apt to refer to sub-niches as diverse microenvironments within the same tissue that differ from one another by the molecular mechanisms through which they govern the fate of the hosted cells. As an example, a very recent study has pinpointed three distinct perivascular milieus of the bone marrow that exert different effects on hematopoietic stem cells: the proper perivascular, the periarteriolar and the megakaryocytic sub-niches, hence questioning the existence of a unique niche within the bone marrow [74]. Eventually, it should be kept in mind that both cells of the primary tumor and DTCs are involved in a bidirectional exchange of signals with their lodging milieus. In particular, tumor cells homing within niches and sub-niches have obviated the influence of their key constituents, such as basement membranes and microvasculature, whereas cell-derived factors and extracellular matrix compositions may drive the fate of tumor cells towards proliferation or dormancy.
3.5 Influence of inflammatory states on dormancy and awakening of tumor cells
It has amply been shown that inflammation may increase the risk of tumor recurrence after remission, as in case of late metastasis in smoking breast cancer patients [75]. Similarly, surgery-related inflammatory states have been found to trigger early outgrowth, ranging from 6 to 12 months, of previously occult metastases, especially in breast cancer patients [13]. During recent years, several efforts have been made to understand the molecular mechanisms underlying this awakening process, which is apparently driven by neutrophils [76]. In particular, neutrophils seem to release EMT-inducing cytokines via Zinc finger E-box-binding homeobox 1 (Zeb1) activation in mammary carcinoma cells intravenously injected in recipient mice [76]. More recently, it has been shown that neutrophil extracellular traps (NETs) may play a major role in the awakening of dormant cancer cells. Indeed, in mouse models of metastatic breast and prostate carcinoma, NET formation appears to be pivotal during acute inflammation to awake DTCs [77]. In particular, it has been found that the DNA scaffold of NET allows the accumulation of neutrophil proteases, leading to proteolytic remodeling of the extracellular matrix protein laminin. The generation of these new laminin epitopes activates integrin signaling and the pro-proliferative FAK-ERK-MLCK cascade, thereby awakening DTCs [77]. Myeloid cells are important players in resuming DTC proliferation. It has e.g. been found that mice orthotopically transplanted with mammary cancer that underwent surgery may experience local and systemic inflammatory responses with myeloid cell mobilization in conjunction with both local and systemic metastatic outgrowth [13]. In line with these preclinical data, clinical data support the preventive role of anti-inflammatory drugs on the occurrence of metastasis in patients undergoing tumor resection [78].
3.6 Epigenetic origin of tumor cell dormancy
The concept that both tumorigenesis and cancer heterogeneity do not require immediate genetic changes to take place is becoming broadly accepted now, as reviewed elsewhere [18]. Similarly, tumor entrance into a dormant state may rely on non-genetic modifications, i.e., microenvironment-driven variations in the epigenetic program. The latter is fundamental to the fate not only of normal differentiating cells, but also of cancerous cells, as these latter cells have been found to “keep memory” of their original epigenetic pattern in several well-documented cases [27, 75]. Therefore, the state of differentiation of the tumor founder cell does matter, since the malignant transformation of normal stem cells is generally associated with a more aggressive phenotype than that of differentiated tumor cells derived from lineage-committed cells [79, 80]. A sizable number of epigenetic changes has been found to occur during tumor progression, often underlying a dedifferentiation program that worsens the clinical outcome of previously differentiated neoplasms [27]. During both tumor onset and progression, disruption of DNA methylation, histone modification, chromatin remodeling, and alterations in the expression of non-coding RNAs (ncRNAs) occur stochastically. Along the same line, epigenetic alterations that affect dormancy-associated master regulators may represent the main driving force to dictate whether a cell has to undergo a lag phase or, conversely, to enter a proliferative phase. Epigenetic variations may indeed be the most suitable molecular substrates of TD: they are stochastic, therefore supporting evidence of sudden and unpredictable TD entrance or escape and, possibly more important, reversible. These epigenetic variations usually precede the establishment of genetic mutations, which generally lead to an irreversible condition that may not be compatible with a reversible mode of cancer progression involving TD [8, 18]. Moreover, genetic mutations may not explain adaptation and reactivation of engrafted DTCs in the pre-metastatic site, since they are less likely to be established in a phase of replicative rest. Instead, the pre-metastatic microenvironment may send signals to the DTCs, directly affecting metastasis-associated signaling networks as well as the epigenetic state of these cells [8, 31]. To recapitulate, epigenetic changes may be the main driving force of short-term cancer progression.
Independent evidence supports the above theory on epigenetic preponderance in governing TD. Gawrzak et al. [81], for instance, have highlighted a pivotal role of mitogen- and stress-activated kinase 1 (MSK1) in maintaining dormant DTCs of breast cancer through a genome-wide short hairpin RNA (shRNA)-based screening in a xenografted mouse model. MSK1 is a downstream effector of p38 MAPK, which can regulate the chromatin state through histone 3 phosphorylation (on Serine 10 and 28), thereby easing its acetylation on Lysine 9 and 27. Promoters of key differentiation genes, such as GATA3 [82] and FOXA1 [83], are activated by these modifications. In DTCs, MSK1 is essential for dormancy, since its depletion has been found to lead to increased bone homing and early metastatic outgrowth of breast cancer cells. Conversely, when MSK1 is re-expressed in highly proliferating MSK1-knockout cells, TD gets restored again [81].
Other epigenetic regulators include BMI1 (B lymphoma Mo-MLV insertion region 1 homolog), which is known to orchestrate drug resistance in several tumor types by regulating cell-cycle inhibitors like p16Ink4a and p19Arf [84]. In addition, reversible changes in DNA methylation and histone acetylation patterns have been found to govern iterative cycles of dormancy and recurrence in ovarian carcinoma cells [17]. When the epigenetic state allows the expression of TIMP3 (tissue inhibitor of metalloproteinases 3) and E-cadherin, two known anti-angiogenic factors, the tumor cell is “switched off” to a dormant state, whereas their repression leads to its reactivation [85]. Therefore, the silencer enzymes DNA methyl transferase 1 (DNMT1) and histone deacetylases can be therapeutically inhibited to allow TIMP3 and E-cadherin expression, and to avoid reactivation of ovarian cancer cells [86]. For instance, 5-azacytidine is a DNMT1 inhibitor that decreases the expression level of G0-to-G1 exit genes and increases the expression level of TD inducers, including TIMP3 and E-cadherin [3, 83].
Other epigenetic mechanisms include altered ncRNA expression patterns. Upregulation of stem-cell specific miRNAs has e.g. been found to underlie chemoresistance development and stemness recovery, two distinctive processes that usually occur within a dormant state, in prostate and breast cancer cells [8, 82]. Other tumor types, such as glioblastoma and osteosarcoma, can similarly become quiescent in response to the over-expression of a particular set of miRNAs, called dormancy-associated miRNAs: miR-190, -580 and -588 [87]. Besides these miRNAs, metastatic breast cancer cells can be driven into quiescence by miR-127, -197, -222, and -223, which are produced by tumor-associated stroma cells and transmitted to cancer cells via gap-junctions and exosomes [87, 88]. All of them are known to target CXCL12 messenger RNA (mRNA). The net effect of this post-transcriptional regulation is a decrease in chemokine concentration, thereby leading to TD or to mobilization of DTCs, as can be deduced from previously reported data on CXCL12 signaling [8, 11]. The above examples underscore the crucial role of epigenetic modifications in governing TD.
4 Population-level tumor dormancy
4.1 Population-level dormancy entails a precise balance between cell proliferation and death
Besides the cellular mechanisms of TD, incipient primary tumors and micro-metastases can be kept clinically undetectable because of an en-masse latency. This “macroscopic” TD relies on an offsetting ratio between cell division and death that impedes a net increase in the number of tumor cells [3, 89]. The main causes of tumor cell death result from blood supply shortage due to so-called angiogenic dormancy, and immune responses of the host causing immune-mediated dormancy. These dormancy mechanisms will be further outlined below, but it is worth noting here that also a lack of fibroblast recruitment, and of the consequent fibrosis, may curb tumor growth [21, 31].
The tumor-host complex may be compared with a typical ecological system, in which the Verhulst logistic equation describes the tumor population growth over time (Fig. 8) [9]. According to this logistic, the population size increases exponentially in the first period when resources exceed demands, but then growth decelerates to asymptotically approach the carrying capacity value, K. This value represents the maximum size that the population can reach within a specific environment. However, while in ecological models K is generally fixed, in tumor biology it can vary according to tumor features, mainly angiogenic potential and immunogenic properties. Actually, a high angiogenic potential and a low ability to trigger immune responses are associated with a variable K, which increases progressively with tumor size. Opposite tumor features may keep K constant, thus fixing a threshold size that cannot be overcome by the tumor mass, since cell division and death become counterbalanced [9].

Graphical representation of the Verhulst logistic equation applied to tumor growth. Comparing the tumor mass with a typical ecological system, the growth of tumor burden leads to the exhaustion of resources, so that there should be a fixed maximum capacity value K. This would only be true in an “ideal” situation with poor angiogenic potential and high immunogenicity. Actually, in most cancer cases, increased neo-angiogenesis and immunosuppressive properties are encountered. These are the main factors that allow a progressive increase in K value with tumor size expansion
4.2 Angiogenic and post-vascular dormancy
As stated above, if a tumor population, both primary and disseminated, lacks angiogenic potential, it cannot grow beyond a certain threshold size (generally 1 mm) due to nutrient and oxygen deprivation and, thus, remains clinically undetected [3]. Oxygen, for instance, diffuses at maximum 50 μm, and beyond that the intracellular oxygen pressure decreases under 1 mmHg, thus causing metabolic and, subsequently, structural damage that drives the cell towards death [42, 85]. Therefore, an angiogenic switch should occur to allow tumor expansion, i.e., tumor cells, especially DTCs, have to breach the barrier of angiogenic competency, meaning expression of pro-angiogenic cues to recruit and maintain blood supply to sustain ongoing proliferation [41]. Counterintuitively, vessels may also play a major role in the pre-angiogenic phase, since angiocrine factors can modulate TD at the cellular level, as outlined above. Therefore, a nascent vessel not only supplies the tumor mass with nutrients, but its endothelium also releases TGFβ1, periostin and epoxyeicosatrienoic acids (EETs) to foster tumor proliferation and, in the case of micro-metastasis, their outgrowth [3, 65, 86]. Nevertheless, cellular mechanisms are likely to precede population-level mechanisms, but they necessarily crosstalk [41]. It has e.g. been reported that down-regulation of Myc within tumor cells may block angiogenesis by allowing the expression and secretion of thrombospondin 1, a known anti-angiogenic factor [43].
Interestingly, neo-angiogenesis may be regulated by both local and systemic signals. In the case of micro-metastatic lesions within a hypoxic milieu, DTCs that are endowed with angiogenic potential have been found to express HIF1α (hypoxia-inducible factor 1α) and specific miRNAs to recruit endothelial cells through VEGF secretion [31]. In addition, those DTCs have been found to release metalloproteinases that degrade the extracellular matrix to allow a better diffusion of pro-angiogenic molecules [43] Conversely, systemic cues are usually anti-angiogenic, such as angiostatin and endostatin, as well as those produced by the primary tumor, such as prosaposin. In fact, prosaposin induces fibroblasts to produce thrombospondin 1, which exerts anti-angiogenic effects [90]. In case of a decrease in prosaposin concentration, caused e.g. by therapies targeting the primary tumor, micro-metastatic lesions may outgrow, thus supporting evidence of early metastatic onset after treatment of a primary neoplasm [41].
To attain angiogenic competency, changes in gene expression patterns should take place in tumor cells. In the case of DTCs, modifications of the epigenetic program after engraftment into a new milieu may underlie these changes in gene expression patterns [41]. Nevertheless, even once gained angiogenic potential, these changes can be exhibited by the tumor or not depending on interrelations with other angiogenesis regulators from either local or systemic sources. If pro-angiogenic factors secreted by competent DTCs are not surmounted by anti-angiogenic cues of different origins, then new vessels will sprout to support micro-metastatic outgrowth. On the contrary, if pro-angiogenic factors do not prevail, a new latent state, called post-vascular dormancy, may be established [9]. That state is supposed to be macroscopically identical to the classical angiogenic one, thus preventing tumor masses to become clinically relevant even if endowed with angiogenic competency. The difference between angiogenic and post-vascular dormancy does not seem to rely on clinical manifestation, but rather on the underlying mechanisms that require different approaches to be studied and, eventually, for the design of new targeted therapies.
4.3 Immuno-mediated tumor dormancy
The role of immune cells in inducing dormancy was first unintentionally noted during transplantation of organs from disease-free donors who had previously suffered from melanoma. The recipients developed melanoma metastasis in the transplanted organ after systemic immunosuppression [91]. Subsequently, four cases of breast cancer transmission to transplant recipients from a single donor with occult diagnosis have been reported. The latency time to the outgrowth varied from 16 months to 6 years, and the only survivor was a recipient with discontinued immunosuppression [92]. Recently, we also reported the onset of an aggressive non-Hodgkin lymphoma of donor origin in a kidney-transplant recipient [93]. These findings suggest that donor tissues may harbor DTCs, which persist in a latent state because of the activity of the donor’s competent immune system, but may reactivate later-on taking advantage of the immunocompromised condition of the recipient.
To shed light on the regulatory mechanisms exerted by immune cells in TD, an immune-mediated maintenance of dormancy model, also called “equilibrium”, has been proposed in which tumor cells and tumor-reactive immune cells interact with each other without being eliminated or dominated [94]. During this dormant state, the main actors are represented by CD8+ and CD4+ T cells. In mouse models it has been found that depletion of one of these two lymphocytic subtypes allows the escape of tumor cells from quiescence [95]. In fact, CD4+ T cells may lead to TD via angiogenesis inhibition, thus establishing crosstalk with angiogenic dormancy. In a pancreatic cancer xenograft model in immunocompromised mice, it was noticed that injection of CD4+ T cells arrested tumor progression independent of CD8+ T cells [96]. Instead, CD4+ T cells were found to inhibit TNF receptor 1 (TNFR1) signaling and angiogenesis through the release of CXCL9 and CXCL10 [96]. Moreover, CD4+ T helper 1 (TH1) cells were found to produce interferon-γ (IFN-γ) that through its pleiotropic signaling pathways, was found to lead to different effects. Firstly, it caused upregulation of tumor MHC (major histocompatibility complex) class I expression, so that the cancer cells could become more accessible targets for CD8+ T cells [96]. Another IFN-γ effect is its direct stimulation of CD8+ T cells, thereby increasing their cytotoxic action. Since it is known to induce the irreversible state of senescence instead of quiescence, IFN-γ may be considered as not only a dormancy inducer, but also as a DTC killer with therapeutic implications [3, 92]. The expression of this intercellular mediator relies, however, on a multitude of other factors, one of which is the level of adenosine. Within solid tumors, the adenosine level is usually high and, as such, promotes the growth of the tumor mass [97]. Experimental blockade of the adenosine receptor A2B (using the receptor antagonist aminophylline) in bladder and breast tumors has led to an increased production of IFN-γ and, consequently, to a boost of its related effects [93, 94].
On the other hand, CD8+ T cells have been shown to carry out a preponderant function in maintaining TD in an animal model of methylcholanthrene-induced sarcoma [9598]. This study revealed that the main strategy to keep malignant cells in permanent dormancy is CD8+ T cell-mediated cytotoxicity and cytostasis, due to degranulation and excretion of apoptosis-inducing enzymes, namely perforin and granzymes. The latent tumors indeed manifested a high apoptotic fraction, establishing the above-mentioned “equilibrium” with proliferation. In addition, a decrease in CD8+ T cells has been shown to shorten the latent period that usually precedes metastatic outgrowth in spontaneous mouse uveal melanoma models, demonstrating once again the crucial role of those cells in maintaining immunological TD [99]. Although CD8+ T cells represent the major antitumor component of the immune system, some tumor cells avoid T cell-induced apoptosis through immuno-editing, necessitating the participation of natural killer (NK) cells, which may act by eliminating tumor cells too [94]. In fact, it has been found that also immune-escape from NK activity through repression of NK cell-activating ligands may lead to DTC survival, putatively in a dormant state, whereas complete depletion of NKs was found to lead to a resumed proliferation of latency-competent DTCs [54, 55].
Despite the antitumor functions of T and NK cells, some immune components may promote DTC reactivation, although their exact interactions with other microenvironmental cues are still unclear. Some evidence suggests that certain immune cell types may infiltrate into the tumor microenvironment and indirectly facilitate the escape from dormancy [43]. Those immune components include myeloid-derived suppressor cells, regulatory T (Treg) cells and a subset of tumor-associated macrophages (TAMs), whose contribution is based on secretion of mitogens, proangiogenic compounds, metalloproteinases and cytokines, which in turn favor cellular proliferation, angiogenesis and immunosuppression [100]. TAMs may have a pro-tumor M2 phenotype and produce pro-inflammatory and angiogenic molecules, such as VEGF, metalloproteinases and prostaglandin E2, which are known to foster tumor growth. TAMs have, for example, been found to play a major role in metastatic lung colonization of bladder cancer cells [101]. In fact, bladder DTCs secrete endothelin-1 to chemoattract macrophages. Interestingly, TAMs release trophic and immunosuppressive factors in the microenvironment, thus fostering metastatic outgrowth [101]. Treg cells, instead, play a preponderant role in suppressing the immune response, so that they can dampen cytotoxic CD8+ T cell function against cancer cells and inhibit immunotherapeutic strategies that stimulate T cell responses [42, 96]. It has indeed been found that Treg cells can indirectly deplete tryptophan and release TGF-β, which compromises proper proliferation and activation of T cells and promotes tumor reactivation [100].
5 Therapeutic tumor dormancy approaches against metastasis development
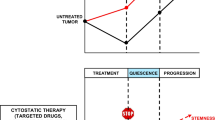
5.1 Tumor dormancy: A desirable condition to reach or a key step in cancer progression to prevent?
Clinical and experimental evidence suggests that targeting dormant DTCs may prevent metastasis, but whether trying to keep them asleep or awake them before time is due is still controversial [11, 97]. At a cellular level, direct inhibition of SFK with AZD0530 has been found to prevent the proliferative switch of murine mammary DTCs if continuously administered, while the application of a selective MEK1/2 inhibitor, i.e., AZD6244, has been found to result in their definitive apoptosis [63]. Such a combination therapy may be effective for both late stage cancers and recurrence prevention. Interventions at population-level TD, instead, may comprise angiogenesis blockade with classical anti-VEGF targeted therapies or inhibition of metalloproteinases, so that they cannot degrade the DTC surrounding matrix [11, 102]. Alternatively, dormant components, such as thrombospondin 1 and other glycoproteins, may be specifically delivered to maintain DTCs in a latent state for a longer time [90]. However, delivering those molecules may be troublesome, given their high molecular weight. Thus, increasing their endogenous production by tumor-associated cells may represent a more effective way to enhance their concentration within the niche. To date, instead, experimental approaches are focusing on a small prosaposin-derived peptide that has been synthetized and injected in immunocompromised mice bearing human breast DTCs. In doing so, it has been found that it can systemically increase thrombospondin 1 production and that, compared to a scramble peptide, this prosaposin derivative can cause a significant reduction in lung metastasis onset in these mice [42, 99, 130].
Besides, immunotherapy represents another implementation of this strategy, focusing on the immunological rather than cellular or angiogenic TD. It may be aimed at increasing the T cell-mediated apoptosis rate of tumor cells to counterbalance their proliferation. For this purpose two methods based on in vivo animal work have been developed. The first one entails radiotherapy-induced overexpression of MHC class I on tumor cells to increase their immunogenicity [104]. The second one involves direct stimulation of CD8+ T cells via upregulation of different pathways, such as the IFN-γ pathway [105]. Recent observations suggest, however, that therapeutic escape and relapse occur after tumor antigen and/or MHC class I loss due to immuno-editing. This is an adaptive process based on a Darwinian-like selection, i.e., DTCs with an immunogenic phenotype are promptly killed by T cells, so that the only cells to survive are those endowed with a non-immunogenic phenotype. As a result, these “adapted” cells should be targeted through another strategy, putatively including NK cells instead of only CD8+ T cells [94]. Nonetheless, therapeutic dormancy maintenance has several technical and practical drawbacks. First, altered expression of relevant factors may be associated with toxicity. Sustained systemic overexpression of thrombospondin 1 may e.g. also inhibit physiological angiogenesis. Second, “therapeutic” TD will not be irreversible. Intense pro-proliferative conditions, such as wounding and inflammation, may awaken DTCs, with nocuous consequences. This is because TD is governed by a dynamic balance of pro- and anti-dormancy factors. Third, such a chronic therapy may have a low patient compliance, even for those who apparently have a good prognosis [11]. On the other hand, killing DTCs may be preferable since less practical troubles may arise than chronic maintenance of TD, and relapse may be avoided. In clinical practice, drugs aimed at eradicating DTCs should be administered alongside the common adjuvant therapies in the period around surgical removal of the primary neoplasm, when indicated. Therefore, patient compliance could be higher, or their enrolment in clinical trials easier. Moreover, elimination of DTCs from biopsies of previously colonized organs may represent a short-term reliable indicator of therapeutic effectiveness [11].
With regard to technical details of a therapy aimed at killing dormant DTCs, a putative attempt could entail sensitization of dormant DTCs to conventional treatments. A Smoothened (SMO) antagonist, PF-04449913, has e.g. been shown to abrogate myeloid leukemia stem cell dormancy, thus sensitizing them to tyrosine kinase inhibitors [106] or conventional chemotherapeutics through GLI2 pathway blockade [107]. This approach may also be suitable to treat early-stage metastatic diseases, i.e., just after the metastasis onset, thereby making enrollment in clinical trials even less stringent. Taking into account that in a large number of chemo-resistant dormant cancers the ability to resist therapy-induced death is mainly conferred by the dormant niche rather than cell intrinsic mechanisms, an effective combinatorial approach may be preferable, also to target different tumor sub-populations. As suggested by several clinical trials (NCT00990054, NCT01120457, NCT00943943, NCT00906945, NCT01027923, NCT01160354) in patients with acute myeloid leukemia (AML), molecular tethers can be targeted with a first drug to detach DTCs from their milieu and deprive them from their microenvironmental protection. Then the second drug, e.g. a conventional chemotherapeutic, may be more effective in eradicating the tumor cells. In case of AML, osteopontin and CXCL12 signaling, which are known to tether both HSCs and DTCs to the bone marrow perivascular niche, can be inhibited by blocking their receptors, α4β1 integrin and CXCR4, respectively, with specific antagonists as reviewed elsewhere [108]. Mobilization of leukemic DTCs sensitizes them to chemotherapy but, in the meantime, there is the risk of exacerbating HSC mobilization and/or depletion, thereby amplifying chemotherapy side effects [109]. Alternatively, a difficult but potentially game-changing attempt may involve the identification of target genes that specifically induce dormant DTC death, as in case of DYRK1B inhibition that is known to kill quiescent pancreatic cancer cells, but not the normal ones [6].
5.2 Clinical translation of experimental data on tumor dormancy
Consistent with both preclinical and clinical evidence of the suppressive control exerted by primary tumors over metastatic deposits, clinical efforts to prevent early metastatic outgrowth immediately after the removal of the primary mass have been undertaken, especially in conjunction with adjuvant regimens [43]. Clinical breakthroughs are, however, still needed to obviate the inability of classical adjuvant therapies in eradicating tumor cells that disseminated prior to surgery [31]. A complete bench-to-bedside translation has so far been reached for only a few therapies. The first antitumor drug based on TD precepts to be approved for clinical use was sunitinib, a VEGFR inhibitor, directed against advanced-stage renal-cell carcinoma [110]. By impeding endothelial cell recruitment, sunitinib “switches off” the carcinoma into angiogenic dormancy. As such, this agent may be tested in patients at high risk for relapse, but without current evidence of disease, to prevent metastasis onset from DTCs. Alternatively, therapeutic angiogenic TD may be pursued using a metronomic strategy, that is frequent administration of low-dose chemotherapy. This approach may drive the growing tumor mass toward a quiescent state via thrombospondin 1 induction and may show fewer side effects than classical chemotherapy, as suggested by the metronomic use of cyclophosphamide and dexamethasone against advanced castration-resistant prostate cancer [42, 106].
Another strategy aimed at targeting tumor dormancy may rely on mTOR inhibitors. Everolimus has, for example, been shown to ameliorate survival and reduce bone metastatic outgrowth in patients with luminal-type breast cancer when combined with aromatase inhibitors [111]. Indeed, everolimus has been found to decrease osteoclast activation by DTCs and their own maintenance through the autophagy pathway, thus decreasing chances of DTC survival, even if in a quiescent state [112]. There is also a glimmer for a further preventive use of these adjuvant therapies in other types of breast and non-breast cancers, counteracting TD and subsequent metastatic outgrowth.
6 Conclusions and perspectives
TD may be considered as a platform allowing for a full development of the “hallmarks” of cancer. In fact, tumors can be endowed with only some of these features, and progress to develop the missing ones. The multistep process of tumorigenesis is widely considered to be a stochastic process by which several changes in gene expression consecutively affect a founder cell and its progeny. The initiation process, which is broadly accepted to rely on mutational hits affecting the expression of proto-oncogenes and/or tumor suppressor genes, may vary on an individual basis. During the progression process, tumor cells may undergo additional changes in gene expression that, together, may result in a drift towards the overall “hallmarks” of cancer. This scenario is supported by numerous, but sometimes ambiguous, lines of evidence. The stochastic multistep tumorigenesis model may, thus, suffer from some caveats. In fact, epigenetic changes, even if reversible, are de facto master regulators of short-term tumor progression, since they may affect gene expression in response to inherent or environmental factors, are inherited by the progeny, and occur at a high rate even in the absence of proliferation, as has extensively been reported [18, 110, 113]. While genetic mutations are more likely to occur in proliferating cells rather than resting ones, epi-mutations also affect, possibly even mainly, dormant cells. Thus, the longer the lag time, the higher the probability for cells to finally re-enter into the cell cycle with a distorted epigenetic program [8, 17, 18,110, 113]. This suggests that TD may represent a transitory phase during which a developing tumor refines its features to acquire the overall “hallmarks” of cancer.
When changes in microenvironment and/or immune response occur spontaneously or as a consequence of therapy, the subsequent acquisition of cancer “hallmarks” may vary accordingly, and a new latency period may be required to adapt to the new situation via additional epigenetic modifications. In addition, genetic mutations may succeed epigenetic modifications in reactivated proliferating cells, or even in cells in a population-level dormant state. As such, genetic mutations may confer further distinctive long-term features to the tumor cells. These considerations are consistent with the existence of an initial steady state in the vast majority of tumors, and with that of iterative cycles, exemplified by the above described treatment-dormancy-relapse cycles of prostate cancer. In light of this perspective, TD may play a key role in both primary and secondary tumor development. It may be assumed that a primary tumor is fully adapted to its milieu, which is rich in growth factors by chance. Therefore, the tumor cells may proliferate, even if they are not able to grow in the absence of these cues (contrary to self-sufficiency in growth signals, which is a distinctive trait of cancer). When cells of such a tumor detach and disseminate, they may engraft in a growth factor-deficient microenvironment. As a consequence, these incompetent DTCs may enter into a quiescent state until a specific epimutation endows them with the ability of being self-sufficient in growth signals, thereby restoring their proliferative capacity and allowing the accumulation of genetic mutations and the acquisition of a new phenotype, closer to the final stage of cancer development.
In brief, since it is unlikely for a novel tumor to manifest all the distinctive traits of a developed tumor mass, it has to undergo iterative dormant phases to adapt to changing contexts through epigenetic modifications that confer new characteristics to it. As such, variability in nascent tumors may also underlie differences in TD duration and outcome. One current caveat is the need of in vitro and in vivo TD models to fill the gap to clinical practice. To this end, CTCs and DTCs from patients should be functionally characterized in terms of tumorigenic, stemness, dormancy and metastatic capacities. This information may yield new short-term indicators of therapy efficacy to be used in the clinic. Identification of new biomarkers, gene signatures and functional mediators of TD and tumor reactivation, in addition to improvements in single cell genomic and epigenomic analyses, will allow the execution of more detailed tumor lineage tracing studies. All this is of prime relevance since TD may often, if not always (i.e., in case of synchronous metastases), represent a pivotal step in cancer progression and metastatic dissemination and, finally, acquisition of the overall cancer “hallmarks”. Only detailed knowledge on these issues may allow the development of effective strategies to block the disease and to improve the prognosis of the patients.
Abbreviations
- ATG:
-
Autophagy-Related
- BMI1:
-
B lymphoma Mo-MLV insertion region 1 homolog
- BMP:
-
Bone Morphogenetic Protein
- BMPR2:
-
BMP receptor 2
- BRCA1:
-
Breast Cancer type I susceptibility protein
- CDK4:
-
Cyclin-Dependent Kinase 4
- CSC:
-
Cancer Stem Cell
- CSF2:
-
Colony Stimulating Factor 2
- CTC:
-
Circulating Tumor Cell
- CXCL:
-
Chemokine Ligand
- CXCR:
-
Chemokine Receptor
- DNMT1:
-
DNA Methyl Transferase 1
- DTCs:
-
Disseminated Tumor Cells
- DYRK1B:
-
Dual specificity tyrosine-phosphorylation-regulated kinase 1B
- EETs:
-
Epoxyeicosatrienoic acids
- EMT:
-
Epithelial-to-Mesenchymal Transition
- GAS6:
-
Growth Arrest-Specific protein 6
- HER2:
-
Human epidermal growth factor receptor 2
- HIF 1α:
-
Hypoxia-Inducible Factor 1α
- IFN-γ:
-
Interferon-γ
- JNK:
-
JUN N-terminal Kinase
- K:
-
Carrying capacity
- MET:
-
Mesenchymal-to-Epithelial Transition
- MHC:
-
Major Histocompatibility Complex
- miRNA:
-
Micro RNA
- mRNA:
-
Messenger RNA
- mTOR:
-
Mammalian Target Of Rapamycin
- ncRNA:
-
Non-Coding RNA
- NK:
-
Natural Killer
- NDRG1:
-
N-Myc Downstream Regulated 1 protein
- (p)ERK:
-
(phosphorylated) Extracellular signal-Regulated Kinase
- PI3K:
-
Phosphoinositide 3-Kinase
- pRB:
-
Protein Retinoblastoma
- Prrx1:
-
Paired Related Homeobox 1
- TAMs:
-
Tumor-Associated Macrophages
- TD:
-
Tumor Dormancy
- TGF:
-
Transforming Growth Factor
- TH1 :
-
T Helper 1 cell
- TIMP3:
-
Tissue Inhibitor of Metalloproteinases 3
- TNF:
-
Tumor Necrosis Factor
- TNFR1:
-
TNF Receptor 1
- Treg :
-
Regulatory T cell
- uPA:
-
Urokinase Plasminogen Activator
- UPR:
-
Unfolded Protein Response
- VCAM1:
-
Vascular Cell Adhesion Molecule 1
- VEGF:
-
Vascular Endothelial Growth Factor
References
D. Hanahan, R.A. Weinberg, The hallmarks of cancer. Cell 100, 57–70 (2000)
D. Hanahan, R.A. Weinberg, Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011)
M.S. Sosa, P. Bragado, J.A. Aguirre-Ghiso, Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 14, 611–622 (2014)
L. Tong, L. Yi, P. Liu, I.R. Abeysekera, L. Hai, T. Li, Z. Tao, H. Ma, Y. Xie, Y. Huang, S. Yu, J. Li, F. Yuan, X. Yang, Tumour cell dormancy as a contributor to the reduced survival of GBM patients who received standard therapy. Oncol. Rep. 40, 463–471 (2018)
D.M. Schewe, J.A. Aguirre-Ghiso, ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. U. S. A. 105, 10519–10524 (2008)
X. Deng, D.Z. Ewton, E. Friedman, Mirk dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 69, 3317–3324 (2009)
P. Walter, D. Ron, The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011)
F. Crea, N.R. Nur Saidy, C.C. Collins, Y. Wang, The epigenetic/noncoding origin of tumor dormancy. Trends Mol. Med. 21, 206–211 (2015)
P. Hahnfeldt, The host support niche as a control point for tumor dormancy: Implications for tumor development and beyond. Adv. Exp. Med. Biol. 734, 19–35 (2013)
J.W. Uhr, K. Pantel, Controversies in clinical cancer dormancy. Proc. Natl. Acad. Sci. U. S. A. 108, 12396–12400 (2011)
C.M. Ghajar, Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 15, 238–247 (2015)
D. Chen, Y. Jiao, S. Torquato, A cellular automaton model for tumor dormancy: Emergence of a proliferative switch. PLoS One 9, e109934 (2014)
J.A. Krall, F. Reinhardt, O.A. Mercury, D.R. Pattabiraman, M.W. Brooks, M. Dougan, A.W. Lambert, B. Bierie, H.L. Ploegh, S.K. Dougan, R.A. Weinberg, The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 10, eean3464 (2018)
J. Pfitzenmaier, W.J. Ellis, E.W. Arfman, S. Hawley, P.O. Mclaughlin, P.H. Lange, R.L. Vessella, Telomerase activity in disseminated prostate cancer cells. BJU Int. 97, 1309–1313 (2006)
X.H.-F. Zhang, Q. Wang, W. Gerald, C.A. Hudis, L. Norton, M. Smid, J.A. Foekens, J. Massagué, Latent bone metastasis in breast Cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 (2009)
J.A. Aguirre-Ghiso, P. Bragado, M.S. Sosa, Metastasis awakening: Targeting dormant cancer. Nat. Med. 19, 276–277 (2013)
T. Lyu, N. Jia, J. Wang, X. Yan, Y. Yu, Z. Lu, R.C. Bast, K. Hua, W. Feng, Expression and epigenetic regulation of angiogenesis-related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics 8, 1330–1346 (2013)
H. Easwaran, H.-C. Tsai, S.B. Baylin, Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727 (2014)
D. Gao, D.J. Nolan, A.S. Mellick, K. Bambino, K. McDonnell, V. Mittal, Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319, 195–198 (2008)
M. Elkabets, A.M. Gifford, C. Scheel, B. Nilsson, F. Reinhardt, M.A. Bray, A.E. Carpenter, K. Jirström, K. Magnusson, B.L. Ebert, F. Pontén, R.A. Weinberg, S.S. McAllister, Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J. Clin. Invest. 121, 784–799 (2011)
D. Barkan, L.H. El Touny, A.M. Michalowski, J.A. Smith, I. Chu, A.S. Davis, J.D. Webster, S. Hoover, R.M. Simpson, J. Gauldie, J.E. Green, Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 70, 5706–5716 (2010)
M. Greaves, C.C. Maley, Clonal evolution in cancer. Nature 481, 306–313 (2012)
L.A. Loeb, Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 11, 450–457 (2011)
D.J. Weisenberger, K.D. Siegmund, M. Campan, J. Young, T.I. Long, M.A. Faasse, G.H. Kang, M. Widschwendter, D. Weener, D. Buchanan, H. Koh, L. Simms, M. Barker, B. Leggett, J. Levine, M. Kim, A.J. French, S.N. Thibodeau, J. Jass, R. Haile, P.W. Laird, CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 38, 787–793 (2006)
A.Z. Ayob, T.S. Ramasamy, Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 25, 20 (2018)
B.T. Tan, C.Y. Park, L.E. Ailles, I.L. Weissman, The cancer stem cell hypothesis: A work in progress. Lab. Investig. 86, 1203–1207 (2006)
N.D. Marjanovic, R.A. Weinberg, C.L. Chaffer, Cell plasticity and heterogeneity in cancer. Clin. Chem. 59, 168–179 (2013)
S.A. Mani, W. Guo, M.J. Liao, E.N. Eaton, A. Ayyanan, A.Y. Zhou, M. Brooks, F. Reinhard, C.C. Zhang, M. Shipitsin, L.L. Campbell, K. Polyak, C. Brisken, J. Yang, R.A. Weinberg, The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008)
A.-P. Morel, M. Lièvre, C. Thomas, G. Hinkal, S. Ansieau, A. Puisieux, Generation of breast Cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888 (2008)
K. Weidenfeld, D. Barkan, EMT and Stemness in tumor dormancy and outgrowth: Are they intertwined processes? Front. Oncol. 8, 381 (2018)
F.G. Giancotti, Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764 (2013)
D.D. Tran, C.A.S. Corsa, H. Biswas, R.L. Aft, G.D. Longmore, Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast Cancer recurrence. Mol. Cancer Res. 9, 1644–1657 (2011)
A.D. Rhim, E.T. Mirek, N.M. Aiello, A. Maitra, J.M. Bailey, F. McAllister, M. Reichert, G.L. Beatty, A.K. Rustgi, R.H. Vonderheide, S.D. Leach, B.Z. Stanger, EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012)
O. Karaosmanoğlu, S. Banerjee, H. Sivas, Identification of biomarkers associated with partial epithelial to mesenchymal transition in the secretome of slug over-expressing hepatocellular carcinoma cells. Cell. Oncol. 41, 439–453 (2018)
Y. Kang, K. Pantel, Tumor cell dissemination: Emerging biological insights from animal models and Cancer patients. Cancer Cell 23, 573–581 (2013)
S.K. Muthuswamy, B. Xue, Cell polarity as a regulator of Cancer cell behavior plasticity. Annu. Rev. Cell Dev. Biol. 28, 599–625 (2012)
O.H. Ocaña, R. Córcoles, Á. Fabra, G. Moreno-Bueno, H. Acloque, S. Vega, A. Barrallo-Gimeno, A. Cano, M.A. Nieto, Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (2012)
X. Li, M.T. Lewis, J. Huang, C. Gutierrez, C.K. Osborne, M.F. Wu, S.G. Hilsenbeck, A. Pavlick, X. Zhang, G.C. Chamness, H. Wong, J. Rosen, J.C. Chang, Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 100, 672–679 (2008)
R.J. Bowater, P.E. Lilford, R.J. Lilford, Estimating changes in overall survival using progression-free survival in metastatic breast and colorectal cancer. Int. J. Technol. Assess. Health Care 27, 207–214 (2011)
F. Sclafani, D. Gonzalez, D. Cunningham, S. Hulkki Wilson, C. Peckitt, J. Tabernero, B. Glimelius, A. Cervantes, A. Dewdney, A. Wotherspoon, G. Brown, D. Tait, J. Oates, I. Chau, TP53 Mutational Status and Cetuximab Benefit in Rectal Cancer: 5-Year Results of the EXPERT-C. Trial. J. Natl. Cancer Inst. 106, dju121 (2014)
N.S. Ruppender, C. Morrissey, P.H. Lange, R.L. Vessella, Dormancy in solid tumors: Implications for prostate cancer. Cancer Metastasis Rev. 32, 501–509 (2013)
P. Ost, D. Reynders, K. Decaestecker, V. Fonteyne, N. Lumen, A. DeBruycker, B. Lambert, L. Delrue, R. Bultijnck, T. Claeys, E. Goetghebeur, G. Villeirs, K. De Man, F. Ameye, I. Billiet, S. Joniau, F. Vanhaverbeke, G. De Meerleer, Surveillance or metastasis-directed therapy for oligometastatic prostate cancer recurrence: A prospective, randomized, multicenter phase II trial. J. Clin. Oncol. 36, 446–453 (2018)
J.A. Hensel, T.W. Flaig, D. Theodorescu, Clinical opportunities and challenges in targeting tumour dormancy. Nat. Rev. Clin. Oncol. 10, 41–51 (2013)
T.H. Cheung, T.A. Rando, Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14, 329–340 (2013)
A. Kobayashi, H. Okuda, F. Xing, P.R. Pandey, M. Watabe, S. Hirota, S.K. Pai, W. Liu, K. Fukuda, C. Chambers, A. Wilber, K. Watabe, Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 208, 2641–2655 (2011)
H. Jo, Y. Jia, K.K. Subramanian, H. Hattori, H.R. Luo, Cancer cell-derived Clusterin modulates the phosphatidylinositol 3’-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol. Cell. Biol. 28, 4285–4299 (2008)
A.P. Adam, A. George, D. Schewe, P. Bragado, B.V. Iglesias, A.C. Ranganathan, A. Kourtidis, D.S. Conklin, J.A. Aguirre-Ghiso, Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 69, 5664–5672 (2009)
Z. Lu, R.Z. Luo, Y. Lu, X. Zhang, Q. Yu, S. Khare, S. Kondo, Y. Kondo, Y. Yu, G.B. Mills, W.S.L. Liao, R.C. Bast, The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest. 118, 3917–3929 (2008)
L. Vera-Ramirez, S.K. Vodnala, R. Nini, K.W. Hunter, J.E. Green, Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 9, 1944 (2018)
E. Friedman, The kinase mirk/dyrk1B: A possible therapeutic target in pancreatic cancer. Cancers 2, 1492–1512 (2010)
G. Wu, Z. Ma, W. Hu, D. Wang, B. Gong, C. Fan, S. Jiang, T. Li, J. Gao, Y. Yang, Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 8, e2700 (2017)
Y. Shiozawa, A.M. Havens, K.J. Pienta, R.S. Taichman, The bone marrow niche: Habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia 22, 941–950 (2008)
A. Del Gobbo, N. Fusco, M. Barella, G. Ercoli, A. Sciarra, A. Palleschi, F. Pagni, C. Marchiò, M. Papotti, S. Ferrero, CXCL12 expression is a bona fide predictor of recurrence in lung neuroendocrine tumours; a multicentric study with emphasis on atypical carcinoids - a short report. Cell. Oncol. 41, 687–691 (2018)
H. Gao, G. Chakraborty, A.P. Lee-Lim, Q. Mo, M. Decker, A. Vonica, R. Shen, E. Brogi, A.H. Brivanlou, F.G. Giancotti, The BMP inhibitor Coco reactivates breast Cancer cells at lung metastatic sites. Cell 150, 764–779 (2012)
C.M.C. Li, V. Gocheva, M.J. Oudin, A. Bhutkar, S.Y. Wang, S.R. Date, S.R. Ng, C.A. Whittaker, R.T. Bronson, E.L. Snyder, F.B. Gertler, T. Jacks, Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Genes Dev. 29, 1850–1862 (2015)
S. Malladi, D.G. MacAlinao, X. Jin, L. He, H. Basnet, Y. Zou, E. De Stanchina, J. Massagué, Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016)
C. Tulotta, P. Ottewell, The role of IL-1B in breast cancer bone metastasis. Endocr. Relat. Cancer 25, R421–R434 (2018)
P. Bragado, Y. Estrada, F. Parikh, S. Krause, C. Capobianco, H.G. Farina, D.M. Schewe, J.A. Aguirre-Ghiso, TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 15, 1351–1361 (2013)
R. Sharma, R. Sharma, T.P. Khaket, C. Dutta, B. Chakraborty, T.K. Mukherjee, Breast cancer metastasis: Putative therapeutic role of vascular cell adhesion molecule-1. Cell. Oncol. 40, 199–208 (2017)
D.M. Sosnoski, R.J. Norgard, C.D. Grove, S.J. Foster, A.M. Mastro, Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 32, 335–344 (2015)
S. Hapke, H. Kessler, N. Arroyo de Prada, A. Benge, M. Schmitt, E. Lengyel, U. Reuning, Integrin alpha(v)beta(3)/vitronectin interaction affects expression of the urokinase system in human ovarian cancer cells. J. Biol. Chem. 276, 26340–26348 (2001)
S. Valastyan, R.A. Weinberg, Tumor metastasis: Molecular insights and evolving paradigms. Cell 147, 275–292 (2011)
L.H. El Touny, M.J. Hoenerhoff, J.E. Green, L.H. El Touny, A. Vieira, A. Mendoza, C. Khanna, M.J. Hoenerhoff, J.E. Green, Combined SFK / MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Invest. 124, 156–168 (2014)
R. Sriram, V. Lo, B. Pryce, L. Antonova, A.J. Mears, M. Daneshmand, B. McKay, S.J. Conway, W.J. Muller, L.A. Sabourin, Loss of periostin/OSF-2 in ErbB2/Neu-driven tumors results in androgen receptor-positive molecular apocrine-like tumors with reduced Notch1 activity. Breast Cancer Res. 17, 7 (2015)
S.E. Wheeler, A.M. Clark, D.P. Taylor, C.L. Young, V.C. Pillai, D.B. Stolz, R. Venkataramanan, D. Lauffenburger, L. Griffith, A. Wells, Spontaneous dormancy of metastatic breast cancer cells in an all human liver microphysiologic system. Br. J. Cancer 111, 2342–2350 (2014)
C.M. Ghajar, H. Peinado, H. Mori, I.R. Matei, K.J. Evason, H. Brazier, D. Almeida, A. Koller, K.A. Hajjar, D.Y.R. Stainier, E.I. Chen, D. Lyden, M.J. Bissell, The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–817 (2013)
D.G. DeNardo, J.B. Barreto, P. Andreu, L. Vasquez, D. Tawfik, N. Kolhatkar, L.M. Coussens, CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009)
B.-Z. Qian, J.W. Pollard, Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010)
L.M. Nusblat, M.J. Carroll, C.M. Roth, Crosstalk between M2 macrophages and glioma stem cells. Cell. Oncol. 40, 471–482 (2017)
Y. Jing, N. Ma, T. Fan, C. Wang, X. Bu, G. Jiang, R. Li, L. Gao, D. Li, M. Wu, L. Wei, Tumor necrosis factor-alpha promotes tumor growth by inducing vascular endothelial growth factor. Cancer Investig. 29, 485–493 (2011)
Y. Kienast, L. Von Baumgarten, M. Fuhrmann, W.E.F. Klinkert, R. Goldbrunner, J. Herms, F. Winkler, Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 16, 116–122 (2010)
S. Singh, U.P. Singh, W.E. Grizzle, J.W. Lillard, CXCL12–CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Invest. 84, 1666–1676 (2004)
Y. Shiozawa, E.a. Pedersen, A.M. Havens, Y. Jung, A. Mishra, J. Joseph, J.K. Kim, L.R. Patel, C. Ying, A.M. Ziegler, Others, Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Invest. 121, 1298–1312 (2011)
I. Bruns, D. Lucas, S. Pinho, J. Ahmed, M.P. Lambert, Y. Kunisaki, C. Scheiermann, L. Schiff, M. Poncz, A. Bergman, P.S. Frenette, Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 20, 1315–1320 (2014)
S. Bekaert, M. Fillet, B. Detry, M. Pichavant, R. Marée, A. Noel, N. Rocks, D. Cataldo, Inflammation-generated extracellular matrix fragments drive lung metastasis. Cancer Growth Metastasis 10, 1–11 (2017)
J.M. De Cock, T. Shibue, A. Dongre, Z. Keckesova, F. Reinhardt, R.A. Weinberg, Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 76, 6778–6784 (2016)
J. Albrengues, M.A. Shields, D. Ng, C.G. Park, A. Ambrico, M.E. Poindexter, P. Upadhyay, D.L. Uyeminami, A. Pommier, V. Küttner, E. Bružas, L. Maiorino, C. Bautista, E.M. Carmona, P.A. Gimotty, D.T. Fearon, K. Chang, S.K. Lyons, K.E. Pinkerton, L.C. Trotman, M.S. Goldberg, J.T.-H. Yeh, M. Egeblad, Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, 4227 (2018)
M. Retsky, R. Demicheli, W.J.M. Hrushesky, P. Forget, M. De Kock, I. Gukas, R.A. Rogers, M. Baum, V. Sukhatme, J.S. Vaidya, Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: New findings and a review. Curr. Med. Chem. 20, 4163–4176 (2013)
I. Ben-Porath, M.W. Thomson, V.J. Carey, R. Ge, G.W. Bell, A. Regev, R.A. Weinberg, An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 (2008)
K. Eppert, K. Takenaka, E.R. Lechman, L. Waldron, B. Nilsson, P. van Galen, K.H. Metzeler, A. Poeppl, V. Ling, J. Beyene, A.J. Canty, J.S. Danska, S.K. Bohlander, C. Buske, M.D. Minden, T.R. Golub, I. Jurisica, B.L. Ebert, J.E. Dick, Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 17, 1086–1093 (2011)
S. Gawrzak, L. Rinaldi, S. Gregorio, E.J. Arenas, F. Salvador, J. Urosevic, C. Figueras-Puig, F. Rojo, I.D.B. Barrantes, J.M. Cejalvo, M. Palafox, M. Guiu, A. Berenguer-Llergo, A. Symeonidi, A. Bellmunt, D. Kalafatovic, A. Arnal-Estapé, E. Fernández, B. Müllauer, R. Groeneveld, K. Slobodnyuk, C.S.-O. Attolini, C. Saura, J. Arribas, J. Cortes, A. Rovira, M. Muñoz, A. Lluch, V. Serra, J. Albanell, A. Prat, A.R. Nebreda, S.A. Benitah, R.R. Gomis, MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+breast cancer. Nat. Cell Biol. 20, 211–221 (2018)
M.L. Asselin-Labat, K.D. Sutherland, H. Barker, R. Thomas, M. Shackleton, N.C. Forrest, L. Hartley, L. Robb, F.G. Grosveld, J. van der Wees, G.J. Lindeman, J.E. Visvader, Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 9, 201–207 (2007)
M.A. Augello, T.E. Hickey, K.E. Knudsen, FOXA1: Master of steroid receptor function in cancer. EMBO J. 30, 3885–3894 (2011)
R. Bhattacharya, S. Banerjee Mustafi, M. Street, A. Dey, S.K.D. Dwivedi, Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis 2, 225–239 (2015)
B.H. Lee, S. Yegnasubramanian, X. Lin, W.G. Nelson, Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 280, 40749–40756 (2005)
L. Wang, J. Wang, MicroRNA-mediated breast cancer metastasis: From primary site to distant organs. Oncogene 31, 2499–2511 (2012)
P.K. Lim, S.A. Bliss, S.A. Patel, M. Taborga, M.A. Dave, L.A. Gregory, S.J. Greco, M. Bryan, P.S. Patel, P. Rameshwar, Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 71, 1550–1560 (2011)
V. Sundararajan, F.H. Sarkar, T.S. Ramasamy, The versatile role of exosomes in cancer progression: Diagnostic and therapeutic implications. Cell. Oncol. 41, 223–252 (2018)
M. Makita, T. Sakai, A. Ogiya, D. Kitagawa, H. Morizono, Y. Miyagi, K. Iijima, T. Iwase, Optimal surveillance for postoperative metastasis in breast cancer patients. Breast Cancer 23, 286–294 (2016)
J. Lawler, Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 6, 1–12 (2002)
R.M. MacKie, R. Reid, B. Junor, Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N. Engl. J. Med. 348, 567–568 (2003)
Y.A.H. Matser, M.L. Terpstra, S. Nadalin, G.D. Nossent, J. de Boer, B.C. van Bemmel, S. van Eeden, K. Budde, S. Brakemeier, F.J. Bemelman, Transmission of breast cancer by a single multiorgan donor to 4 transplant recipients. Am. J. Transplant. 18, 1810–1814 (2018)
L. Biavati, F. Rossari, E. Orciuolo, M.I. Ferreri, C. Zucchinetti, U. Boggi, E.M. Ciancia, F. Caracciolo, E. Benedetti, G. Buda, E. Ciabatti, F. Guerrini, S. Grassi, S. Galimberti, M. Petrini, Role of circulating DNA in the precocious diagnosis of lymphoma: An example in a transplant-related aggressive lymphoma. Clin. Oncol. Res. 2, 1–5 (2019)
M.H. Manjili, The inherent premise of immunotherapy for cancer dormancy. Cancer Res. 74, 6745–6749 (2014)
C.M. Koebel, W. Vermi, J.B. Swann, N. Zerafa, S.J. Rodig, L.J. Old, M.J. Smyth, R.D. Schreiber, Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007)
N. Müller-Hermelink, H. Braumüller, B. Pichler, T. Wieder, R. Mailhammer, K. Schaak, K. Ghoreschi, A. Yazdi, R. Haubner, C.A. Sander, R. Mocikat, M. Schwaiger, I. Förster, R. Huss, W.A. Weber, M. Kneilling, M. Röcken, TNFR1 signaling and IFN-γ signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 13, 507–518 (2008)
A. Ohta, A metabolic immune checkpoint: Adenosine in tumor microenvironment. Front. Immunol. 7, 109 (2016)
D. Panigrahy, M.L. Edin, C.R. Lee, S. Huang, D.R. Bielenberg, C.E. Butterfield, C.M. Barnés, A. Mammoto, T. Mammoto, A. Luria, O. Benny, D.M. Chaponis, A.C. Dudley, E.R. Greene, J.-A. Vergilio, G. Pietramaggiori, S.S. Scherer-Pietramaggiori, S.M. Short, M. Seth, F.B. Lih, K.B. Tomer, J. Yang, R.A. Schwendener, B.D. Hammock, J.R. Falck, V.L. Manthati, D.E. Ingber, A. Kaipainen, P.A. D’Amore, M.W. Kieran, D.C. Zeldin, Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest. 122, 178–191 (2012)
J. Eyles, A.-L. Puaux, X. Wang, B. Toh, C. Prakash, M. Hong, T.G. Tan, L. Zheng, L.C. Ong, Y. Jin, M. Kato, A. Prévost-Blondel, P. Chow, H. Yang, J.-P. Abastado, Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest. 120, 2030–2039 (2010)
W. Zou, Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 6, 295–307 (2006)
N. Said, S. Smith, M. Sanchez-Carbayo, D. Theodorescu, Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. J. Clin. Invest. 121, 132–147 (2011)
A. Borodovsky, V. Salmasi, S. Turcan, A.W.M. Fabius, G.S. Baia, C.G. Eberhart, J.D. Weingart, G.L. Gallia, S.B. Baylin, T.A. Chan, G.J. Riggins, 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 4, 1737–1747 (2013)
S.-Y. Kang, O.J. Halvorsen, K. Gravdal, N. Bhattacharya, J.M. Lee, N.W. Liu, B.T. Johnston, A.B. Johnston, S.A. Haukaas, K. Aamodt, S. Yoo, L.A. Akslen, R.S. Watnick, Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and Tsp-1. Proc. Natl. Acad. Sci. U. S. A. 106, 12115–12120 (2009)
S. Demaria, N. Kawashima, A.M. Yang, M.L. Devitt, J.S. Babb, J.P. Allison, S.C. Formenti, Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 11, 728–734 (2005)
M. Kmieciak, K.K. Payne, X.-Y. Wang, M.H. Manjili, IFN-γ Rα is a key determinant of CD8+ T cell-mediated tumor elimination or tumor escape and relapse in FVB mouse. PLoS One 8, e82544 (2013)
F. Rossari, F. Minutolo, E. Orciuolo, Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 11, 1–14 (2018)
A. Sadarangani, G. Pineda, K.M. Lennon, H.J. Chun, A. Shih, A.E. Schairer, A.C. Court, D.J. Goff, S.L. Prashad, I. Geron, R. Wall, J.D. McPherson, R.A. Moore, M. Pu, L. Bao, A. Jackson-Fisher, M. Munchhof, T. VanArsdale, T. Reya, S.R. Morris, M.D. Minden, K. Messer, H.K.A. Mikkola, M.A. Marra, T.J. Hudson, C.H.M. Jamieson, GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 13, 98 (2015)
P.S. Becker, Dependence of acute myeloid leukemia on adhesion within the bone marrow microenvironment. Sci. World J. 2012, 856467 (2012)
B. Boyerinas, M. Zafrir, A.E. Yesilkanal, T.T. Price, E.M. Hyjek, D.A. Sipkins, Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 121, 4821–4831 (2013)
C. Kollmannsberger, D. Soulieres, R. Wong, A. Scalera, R. Gaspo, G. Bjarnason, Sunitinib therapy for metastatic renal cell carcinoma: Recommendations for management of side effects. Can. Urol. Assoc. J. 1, S41–S54 (2007)
J. Baselga, M. Campone, M. Piccart, H.A. Burris, H.S. Rugo, T. Sahmoud, S. Noguchi, M. Gnant, K.I. Pritchard, F. Lebrun, J.T. Beck, Y. Ito, D. Yardley, I. Deleu, A. Perez, T. Bachelot, L. Vittori, Z. Xu, P. Mukhopadhyay, D. Lebwohl, G.N. Hortobagyi, Everolimus in postmenopausal hormone-receptor–positive advanced breast Cancer. N. Engl. J. Med. 366, 520–529 (2012)
P. Hadji, R. Coleman, M. Gnant, Bone effects of mammalian target of rapamycin (mTOR) inhibition with everolimus. Crit. Rev. Oncol. Hematol. 87, 101–111 (2013)
L.M. Glode, A. Barqawi, F. Crighton, E.D. Crawford, R. Kerbel, Metronomic therapy with cyclophosphamide and dexamethasone for prostate carcinoma. Cancer 98, 1643–1648 (2003)
Y. Takahashi, G. Sawada, J. Kurashige, R. Uchi, T. Matsumura, H. Ueo, Y. Takano, S. Akiyoshi, H. Eguchi, T. Sudo, K. Sugimachi, Y. Doki, M. Mori, K. Mimori, Paired related homoeobox 1, a new EMT inducer, is involved in metastasis and poor prognosis in colorectal cancer. Br. J. Cancer 109, 307–311 (2013)
S.V. Shmelkov, J.M. Butler, A.T. Hooper, A. Hormigo, J. Kushner, T. Milde, R.S. Clair, M. Baljevic, I. White, D.K. Jin, A. Chadburn, A.J. Murphy, D.M. Valenzuela, N.W. Gale, G. Thurston, G.D. Yancopoulos, M. D’Angelica, N. Kemeny, D. Lyden, S. Rafii, CD133 expression is not restricted to stem cells, and both CD133 + and CD133 - metastatic colon cancer cells initiate tumors. J. Clin. Invest. 118, 2111–2120 (2008)
T. Lotan, J. Hickson, J. Souris, D. Huo, J. Taylor, T. Li, K. Otto, S.D. Yamada, K. Macleod, C.W. Rinker-Schaeffer, c-Jun NH2-Terminal Kinase Activating Kinase 1/Mitogen-Activated Protein Kinase Kinase 4-Mediated Inhibition of SKOV3ip.1 Ovarian Cancer Metastasis Involves Growth Arrest and p21 Up-regulation. Cancer Res. 68, 2166–2175 (2008)
A.C. Ranganathan, A.P. Adam, L. Zhang, J.A. Aguirre-Ghiso, Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biol. Ther. 5, 729–735 (2006)
A.W. Lambert, D.R. Pattabiraman, R.A. Weinberg, Emerging Biological Principles of Metastasis Cell 168, 670–691 (2017)
Y. Kang, P.M. Siegel, W. Shu, M. Drobnjak, S.M. Kakonen, C. Cordón-Cardo, T.A. Guise, J. Massagué, A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003)
P.D. Bos, X.H.F. Zhang, C. Nadal, W. Shu, R.R. Gomis, D.X. Nguyen, A.J. Minn, M.J. Van De Vijver, W.L. Gerald, J.A. Foekens, J. Massagué, Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009)
A.M. Clark, S.E. Wheeler, C.L. Young, L. Stockdale, J. Shepard Neiman, W. Zhao, D.B. Stolz, R. Venkataramanan, D. Lauffenburger, L. Griffith, A. Wells, A liver microphysiological system of tumor cell dormancy and inflammatory responsiveness is affected by scaffold properties. Lab Chip 17, 156–168 (2017)
H. Gao, G. Chakraborty, Z. Zhang, I. Akalay, M. Gadiya, Y. Gao, S. Sinha, J. Hu, C. Jiang, M. Akram, E. Brogi, B. Leitinger, F.G. Giancotti, Multi-organ site metastatic reactivation mediated by non-canonical Discoidin domain receptor 1 signaling. Cell 166, 47–62 (2016)
D.T. Scadden, Nice neighborhood: Emerging concepts of the stem cell niche. Cell 157, 41–50 (2014)
J.M. Butler, H. Kobayashi, S. Rafii, Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 10, 138–146 (2010)
N.N. Rahbari, M. Aigner, K. Thorlund, N. Mollberg, E. Motschall, K. Jensen, M.K. Diener, M.W. Büchler, M. Koch, J. Weitz, Meta-analysis shows that detection of circulating tumor cells indicates poor prognosis in patients with colorectal cancer. Gastroenterol. 138, 1714–1726 (2010)
X.-L. Ma, Z.-L. Xiao, L. Liu, X.-X. Liu, W. Nie, P. Li, N.-Y. Chen, Y.-Q. Wei, Meta-analysis of circulating tumor cells as a prognostic marker in lung cancer. Asian Pac. J. Cancer Prev. 13, 1137–1144 (2012)
M. Berdasco, M. Esteller, Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 19, 698–711 (2010)
I.P. Torres Filho, M. Leunig, F. Yuan, M. Intaglietta, R.K. Jain, Noninvasive measurement of microvascular and interstitial oxygen profiles in a human tumor in SCID mice. Proc. Natl. Acad. Sci. U. S. A. 91, 2081–2095 (1994)
P. Bhat, G. Leggatt, N. Waterhouse, I.H. Frazer, Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 8, e2836 (2017)
C. Cekic, D. Sag, Y. Li, D. Theodorescu, R.M. Strieter, J. Linden, Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J. Immunol. 188, 198–205 (2012)
S. Sharma, T.K. Kelly, P.A. Jones, Epigenetics in cancer. Carcinogenesis 31, 27–36 (2010)
A. Chatterjee, E.J. Rodger, M.R. Eccles, Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 51, 149–159 (2018)
Acknowledgements
We thank Dr. Andrea Tuccoli and Prof. Flavio Coceani for helpful discussions and suggestions.
Author information
Authors and Affiliations
Contributions
F.R. and C.Z. reviewed the state of the art on the topic and wrote the article, focusing on biological and molecular data. G.B. and E.O curated the clinical aspects of the review. E.O. coordinated the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rossari, F., Zucchinetti, C., Buda, G. et al. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis - clinical implications. Cell Oncol. 43, 155–176 (2020). https://doi.org/10.1007/s13402-019-00467-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-019-00467-7