
Abstract
Leukemia stem cells (LSCs) are a subpopulation cells at the apex of hierarchies in leukemia cells and responsible for disease continuous propagation. In this article, we discuss some cellular and molecular components, which are critical for LSC survival. These components include intrinsic signaling pathways and extrinsic microenvironments. The intrinsic signaling pathways to be discussed include Wnt/β-catenin signaling, Hox genes, Hh pathway, Alox5, and some miRNAs, which have been shown to play important roles in regulating LSC survival and proliferation. The extrinsic components to be discussed include selectins, CXCL12/CXCR4, and CD44, which involve in LSC homing, survival, and proliferation by affecting bone marrow microenvironment. Potential strategies for eradicating LSCs will also discuss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has become apparent that the initiation and propagation of some types of leukemia are driven by leukemia stem cells (LSCs), and LSCs share some biological features with normal hematopoietic stem cells (HSCs) [1–3]. LSCs self-renew and give rise to leukemia through certain degree of cellular differentiation. They are also believed to cause disease relapse and drug resistance [1–3]. Therefore, the development of effective therapeutic strategies for eradicating LSCs is essential for improving patient survival or even curing the diseases.
In general, leukemia includes a broader group of neoplasms, which usually initiates in bone marrow (BM) and results in high numbers of abnormal white blood cells in peripheral blood [4]. These abnormal cells also infiltrate various organs and tissues, and often have some defects in differentiation and compromised immune function. There are four major types of leukemia [4]: chronic myeloid leukemia (CML), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemia (ALL); there are a number of less common leukemias as well. CML is a myeloproliferative disorder with a high number of well differentiated neutrophils in peripheral blood and myeloid cells in BM. AML is a genetically heterogeneous clonal disorder characterized by rapid growth of abnormal myeloid linage cells accumulated in BM and blood. CLL comes from a group of white blood cells called lymphocytes, typically progresses more slowly than other types of leukemia, and most commonly affects older adults. Acute lymphoblastic leukemia (ALL), also called acute lymphocytic leukemia or acute lymphoid leukemia, is the most common malignancy in children. ALL includes precursor B cell acute lymphoblastic leukemia/lymphoma (B-ALL) and T cell acute lymphoblastic leukemia/lymphoma (T-ALL).
AML stem cell is the first one to prove the hypothesis of cancer stem cells (CSCs) [5] by using separated population of blasts from AML to transfer disease into immune-deficient mice [6]. Subsequently, LSCs also are testified in CML and ALL. Unlike CML and AML, the biological characteristics of ALL stem cells are not well studied to date. In this review, we will mainly focus on the biological characteristics of CML and AML stem cells, the roles of critical regulators and microenvironment for LSC self-renewal and survival. We will also describe current and prospective therapeutic strategies to target LSCs.
Biology of LSCs
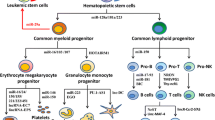
During normal hematopoietic differentiation, stem and progenitor cells may accumulate some kinds of somatically genetic abnormalities, including specific gene mutations, chromosome number alterations, and chromosomal translocations. Genetic alterations are frequently detected in leukemia. These genetic lesions alter the mechanisms of self-renewal, proliferation, and differentiation eventually resulting in the development of leukemias. For example, in patients with CML, approximately 95 % of cases associate with the chromosome translocation of t(9;22)(q34;q11), also called philadelphia chromosome (Ph) [7]. Ph chromosome is also detected in 15–30 % of adult patients with ALL, ~5 % children with ALL, and ~5 % of patients with AML [8]. This genetic abnormality results in the formation of a fusion constitutive tyrosine kinase, BCR/ABL. BCR/ABL1 alone is sufficient to cause CML in mice by expressing the fusion oncogene in a specific subpopulation of cells BM, and this cell population is required for BCR/ABL1 to induce CML-like disease in mice. By using separated population of cells from CML patients or CML-like disease mice transduced by BCR/ABL1 [9], it has been demonstrated that a subpopulation of leukemia cells called LSCs are responsible for CML continuous growth and propagation (Fig. 1). In CML patients, the Ph chromosome is believed to originate from a HSC for it can be clonally detected both in myeloid and lymphoid cells [10]. Both of LSCs and normal HSCs reside in a rare cell population among CD34+CD38− cells [11, 12]. Importantly, new biomarkers that help to distinguish LSCs from normal HSCs have begun to be discovered; for example, dipeptidylpeptidase IV (CD26) has been found to be specifically expressed on the surface of LSCs in CML [13]. In a murine model of BCR/ABL1-induced CML-like disease, Lin−Sca-1+c-Kit+ (LSK) cells have been demonstrated to function as LSCs [9]. During CML progression, LSCs do not reside in one cell population. For example, in the absence of treatment, CML has three clinical phases: chronic phase, accelerated phase, and blastic phase. In chronic phase, phenotypically LSCs exhibit some characteristics similar to those of normal HSC including morphology, biomarkers, and gene profiling [14]. In CML blastic phase (BC CML), granulocyte–macrophage progenitor (GMP) cells have been shown to acquire the properties of LSCs [15] (Fig. 1). In blastic phase, leukemia progenitor cells are thought to accumulate more genetic or epigenetic changes, which disrupt LSC differentiation and further alter critical signal pathways that regulate leukemia cell survival and growth.

Schematic illustration of the normal and leukemic hematopoietic hierarchies. Hematopoietic cells are organized in a hierarchy that is sustained by a small population of self-renewing hematopoietic stem cells (HSCs). HSCs differentiate to progenitors in a lineage-restricted manner. Progenitors retain limited self-renewal capacity which in turn produce functionally mature blood cells (middle column). If HSCs or progenitors accumulate transforming mutations resulting in acquisition of uncontrolled self-renewal capacity and giving rise to aberrant developmental hierarchy, finally leading high count abnormal white cells in blood and bone marrow, these cells are determined as LSCs. In AML, LSC can be initiated from HSC, MPP, and GMP (left column). In CML chronic phase, it is believed that LSC is generated at HSC level; in blastic phase, LSC possess some biologic characteristics of GMP (right column)
Unlike LSCs in CML, AML stem cells are diversified and heterogeneous. AML LSCs originate from different steps of hematopoietic progenitors [6] (Fig. 1), and were initially proposed to establish cancer stem cell (CSC) concept [16]. In that study, a subpopulation of CD34+CD38− human AML cells were shown to serially transplant leukemia in a mouse xenograft model. In contrast, more committed progenitors (CD34+CD38+) lack such potential [6, 17]. Recent studies showed that a subpopulation within CD34+CD38−CD90+ cells in human cord blood contains a non-HSC multipotent progenitor (MPP), which is postulated to the origination of human AML [18] (Fig. 1). This result was further confirmed in an AML1/ETO-transduced AML murine model, in which AML1/ETO was expressed in a clonal HSC population, but the MPP population was responsible for AML development [19]. The diversities of LSC in AML may be related to the somatically genetic abnormalities occurring in myeloid progenitors. Some studies performed in murine models generated by retroviral transformation provide direct evidence that some oncogenes favor transforming particular cell populations to turn them into LSCs. For example, rearrangements of the mixed-lineage leukemia (MLL) gene contribute to approximately 10 % of adult AML, which are also detected in more than 70 % of infant leukemia and many cases of secondary acute leukemias [20]. Studies from one of these rearrangements, MLL/AF9, show that MLL/AF9 can turn GMP cells to LSCs, whereas, the combination of HoxA9 and Meis1 (myeloid ecotropic viral integration site 1 homolog, a Hox cofactor) transforms LSK cells to cause AML [21].
Critical signaling mechanisms for LSC survival
Survival mechanisms of LSC survival are not well understood. Available studies indicate that LSCs share some signaling pathways with their normal stem cell counterparts for survival and proliferation. However, LSCs also require some pathways that are unique and specific to LSCs in self-renewal and proliferation [1–3]. Here we selectively describe some signaling pathways that play a critical role in survival regulation of LSCs.
Wnt signaling
The Wnt glycoproteins are highly expressed in BM and play an important role in the self-renewal and long-term maintenance of normal HSCs [22]. To date, several members of Wnt family have been identified, including Wnt1, 3a, 5a, and 1b. Wnt signaling is mediated through their intracellular components. Canonical Wnt signaling is initiated by the binding of a Wnt protein with its receptor, frizzled (Fz) family, and a co-receptor of the low-density-lipoprotein-receptor-related-protein family (LRP5 or LRP6) [23]. In signal transduction, β-catenin acts as a major molecule in this pathway [24, 25]. Upon activation, β-catenin proteins accumulate and locate to nucleus, then displace co-repressors in the groucho-related gene family (GRG) bound to T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, resulting in the induction of expression of target genes such as c-Myc, c-Jun and cyclinD1 [26] (Fig. 2). Increasing evidence supports that Wnt signaling plays an important role in the development of AML and CML. For example, Wnt/β-catenin pathway is highly up-regulated in both AML [27–29] and CML [30, 31]. In mouse models of AML induced by co-expression of Hoxa9 and Meis1a oncogenes or by fusion oncoprotein MLL/AF9, it has been showed that Wnt/β-catenin signaling pathway is required for self-renewal of LSCs that are derived from either HSC or GMP [21]. During leukemogenesis, β-catenin is essential for AML initiation from HSC or GMP [21]. Similarly in CML, it has been demonstrated that β-catenin and Wnt pathway associated genes are highly expressed in LSCs compared to normal HSCs [32]. Studies performed in a BCR/ABL1-transduced CML murine model show that donor cells with depletion of β-catenin primarily impairs the ability of BCR/ABL1 to cause CML in adoptive hosts and LSCs cannot be serially transplanted to cause CML, indicating that LSC self-renewal and long term maintenance rely on β-catenin [30, 31]. Furthermore, the up-regulation of β-catenin is not converted by BCR/ABL tyrosine kinase inhibitor (TKI), which highlights its importance as a therapeutic target in combination with TKI. All these previous studies support that β-catenin plays a critical role in LSC persistence, and Wnt/β-catenin pathway offers an attractive therapeutic strategy for targeted therapies of CML and AML [21, 30].

Wnt family members bind to their receptors, Frizzled protein alone or with the low-density lipoprotein-receptor-related proteins (LRPs). Frizzled receptors recruit intracellular signaling molecules, the Dishevelled (Dvl) family including Dvl-1, Dvl-2, and Dvl-3, for further intracellular signaling transduction. Dvls function as molecular adaptors on Frizzled receptors due to their protein-protein interaction domains and/or heterotrimeric G-proteins. Beta-catenin is associated in a cytoplasmic complex together with the adenomatous polyposis coli (APC) protein, the cytoplasmicserine/threonine kinase GSK-3β, and axin. Axin is dephosphorylated by the protein phosphatase 2A (PP2A). A complex of axin with one of the three isoenzymes of casein kinase I (CKIα, δ, or ε) phosphorylates β-catenin on serine 45. This step is independent of (GSK-3beta) and initiates the phosphorylation-degradation cascade of β-catenin. In the following step, beta-catenin is phosphorylated at serine 33/37 by GSK-3β. The phosphorylated β-catenin is, subsequently, recognized by the E3 ubiquitin ligase and thereby, targeted for ubiquitination and subsequent degradation by the proteasome. In the presence of Wnt signaling, the phosphorylating activity of GSK-3β is inhibited leading to the stabilization of β-catenin. The APC-GSK-3beta-axin activity is dissociated and unphosphorylated β-catenin accumulates in the cytoplasm, translocates into the nucleus, where it interacts with TCF and LEF transcription factors binding on the promoter of Wnt target genes and mediates their transcription
Hox family genes
Gene expression analysis and functional studies have shown that homeobox family genes (Hox) play important roles in the regulation of early stages of hematopoiesis, including self-renewal of HSCs, and also impact leukemic transformation [33–37]. Multiple Hox genes have been identified as partners of chromosomal translocations in leukemias [38]. Gene profiling studies demonstrate that Hox genes and their cofactors are globally deregulated in cells from leukemia patients [38–40], indicating that Hox genes and their cofactors play important roles in a wide spectrum of human leukemia. The clustered Hox family genes are an evolutionarily highly conserved group of genes that encode DNA-binding transcription factors [41]. Hox genes exhibit a high degree of homology to the clustered homeotic genes (HOM-C) of Drosophila melanogaster [38]. The highly conserved homeobox sequence motif is comprised with a 60 amino-acid helix–turn–helix DNA-binding domain, which is the common element defining this family genes [38]. Hox genes are particularly regulated by their upstream regulators, caudal-type homeobox genes (Cdx) genes, Cdx1, Cdx2, and Cdx4. Cdx genes are involved in Wnt and bone morphogenic protein (BMP) signaling [42]. The crosstalk between Hox genes and Wnt signaling suggest the roles of Hox genes in survival regulation of LSCs. Substantial evidence shows that aberrant expression of Hox genes contributes to the pathogenesis of myeloid malignancies, especially self-renewal of LSCs [39, 43]. For example, in CML, HoxA9 is required for LSC self-renewal and its expression correlates with a poor prognosis [40]. In most AML patients, deregulation of Hox genes were detected particularly due to the aberrant expression of Cdx2 [44]. Studies using murine models also demonstrate the roles of Hox gens in the pathogenesis of AML. For example, fusions of the HoxA9 or HoxD13 with NUP98 can sufficiently recapitulate AML in mouse [45, 46]. Overexpression of individual Hox family members, including HoxB3 [47], HoxB8 [48], or HoxA10 [49], by retroviral transduction or retroviral insertion mutagenesis also generates AML in murine models. Except for Hox family genes directly involving in leukemogenesis, they are also deregulated in other oncogene-transduced leukemias. The expression of Hox genes is significantly up-regulated in MLL-rearrangements involved leukemias [50, 51].
Without any doubt, elucidation of the roles of Hox genes in leukemogenesis helps to understand the mechanisms for LSC self-renew and differentiate, and ultimately for developing new strategies for targeting LSCs [52].
Hedgehog (Hh) pathway
The Hedgehog (Hh) pathway plays important roles in numerous developmental processes, including determination of cell fate, patterning, proliferation, survival, and differentiation [53]. Mammals have three Hedgehog homologues, Sonic Hh (SHh), Desert Hh (DHh), and Indian Hh (IHh) [53]. Recent studies point out that Hh signaling regulates adult stem cells in the maintenance and regeneration of adult tissues [54]. Hh aberrant activation is associated with the development of several types of solid tumors [55–58], and regulates the proliferation of CSCs and increases tumor invasiveness [54].
In normal hematopoiesis, Hh proteins are produced in BM niche and bind to their receptors, patched (Ptch) 1 and 2. In the absence of Hh proteins, Ptch inhibits the activity of smoothened (Smo), a membrane protein associated with G protein-coupled receptors. Current study suggests that Ptch functions as a pump to remove oxysterols from Smo, resulting in inhibiting Smo activity [59]. Upon Hh binding, the pump function of Ptch is switched off. The accumulation of oxysterols allows Smo to be activated or stay on cell membrane for a longer period of time. Smo released from Ptch inhibition results in activation of Gli family transcription factors. Activated Gli members translocate into nucleus and activate the transcription of Hh target genes (Fig. 2). It has been shown that Hh signaling preserves and increases the short-term repopulating capacity of human HSCs, and deletion of the Hh gene impairs their renewal [60].
In neoplasms, Hh has been implicated in the growth of B cell lymphoma [61] and multiple myeloma [62]. Abnormal Hh signaling has also been associated with CML and AML [63]. In a BCR/ABL1-transduced CML murine model, the Hh pathway genes (such as Gli1 and Ptch) are up-regulated in LSCs compared to normal HSCs. Similar to the findings in the murine disease model, these genes have also been shown to be up-regulated in CD34+ cells from CML patients compared to CD34+ cells from healthy donors [64]. In addition, Hh signaling plays a role in regulating LSCs of CML, because loss of Smo caused the depletion of LSCs [60]. In contrast to CML, Hh signaling is not required for the development of AML induced by MLL/AF9 in a murine model [65]. However, targeting Smo with a small molecule attenuates AML by modulating cell cycle progression and self-renewal of LSCs, and also improves AML therapy through sensitizing dormant LSCs to chemotherapy and overcoming drug resistance derived from BM microenvironment [66].
Alox5 pathway
The arachidonate 5-lipoxygenase (Alox5) pathway represents a unique signaling mechanism that is required specifically for survival regulation of LSCs of CML with a minimal role in normal HSCs. The Alox5 gene encodes an important catalytic enzyme known as 5-lipoxygenase (5-LO) that is responsible for the biosynthesis of leukotriene A4 (LTA4) from arachidonic acid [67, 68]. 5-LO is activated by 5-lipoxygenase-activating protein (FLAP), a nuclear membrane-bound protein that binds to arachidonic acid, for leukotriene synthesis. Alox5 is primarily expressed in polymorphonuclear leukocytes, peripheral blood monocytes, macrophages, and mast cells [67, 69]. Phosphorylation of Alox5 at Ser271 and Ser663 by MAPKAP2 and ERK family kinase increases Alox5 enzymatic activity in vivo [70, 71]. Alox5 plays a role in numerous physiological and pathological processes, including oxidative stress response, inflammation, and cancer [72]. Alox5 overexpression is detected in some cancer cells and is associated with poor diagnosis [67, 73]. Microarray studies of human CML cells showed that Alox5 was highly expressed in CD34+ CML cells [74], suggesting that Alox5 plays a role in regulating LSCs in CML patients. The essential role of Alox5 in CML LSCs was originally discovered from a study using a murine model of BCR/ABL1-induced CML [75]. In that study, the mRNA level of Alox5 was shown to be up-regulated in LSCs compared to normal HSCs, and the up-regulation was not restored by imatinib, a BCR/ABL kinase inhibitor. Furthermore, BCR/ABL1 failed to induce CML in the absence of Alox5, and inhibition of Alox5 function by an Alox5 inhibitor or in combination with imatinib significantly prolonged the survival of CML mice [75]. On the other hand, Alox5 has also been shown to be highly up-regulated in AML by RUNX1-ETO9a in cooperating with KLF6, although it is still unknown whether Alox5 plays a role in AML-initiating cells [76].
The role of microRNAs
Recent studies have suggested that microRNAs (miRNAs) play roles in leukemogenesis. For example, the dysregulation of miRNAs was found in human AML and CML [77–79]. Han et al. recently reported the role of miR-29a in HSC and LSC of AML [80]. They found that miR-29a was expressed at high levels in all tested AML samples. Furthermore, it has the capability to convert non-self-renewing myeloid progenitors into self-renewing populations. Overexpression of miR-29a in hematopoietic progenitors leads to biased myelopoiesis and a myeloproliferative disorder (MPD), eventually that progresses to AML [80], suggesting that miR-29a can transform AML by converting myeloid progenitors into self-renewal LSCs. The level of miRNA-150 is significantly lower in both CML and AML patient cells than in healthy BM cells [78, 81]. miRNA-150 exerts its effects partially by targeting the transcription factor MYB. MYB plays a role in maintaining self-renewal of hematopoietic progenitor cells, and its expression is decreased in fully mature myeloid cells. During the pathogenesis of AML, lower level of miRNA-150 leads to high level of MYB protein, resulting in acquisition of self-renewal capacity of LSC counterpart progenitors [78, 81]. Several studies have confirmed that a large number of miRNAs exhibit altered expression in AML [77]. To date, only a small fraction has been confirmed as functional mediators of AML or CML development or maintenance. Well understanding the mechanisms of microRNAs in hematopoiesis and leukemogenesis will help in establishing the strategies for therapies of hematopoietic system diseases.
Microenvironment and LSCs
The extrinsic components mediated by bone marrow microenvironment play critical roles in leukemogenesis, LSC survival, and drug resistance [2]. Bone marrow homing is an important biological process for HSC function, and is governed by a cascade of molecular interactions. First, migrating cells are captured from the fluid stream onto BM tissue endothelium, during which the most effective mediators are the selectins, including E-selectin (CD62E, expressed on endothelium), P-selectin (CD62P, expressed on platelets and on endothelium) and L-selectin (CD62L, expressed on leukocytes and hematopoietic stem cells). Several other molecules including CD44 and some integrins [e.g. LPAM-1 (α4β7)] are also involved by mediating the tethering and rolling adhesive interactions [82]. After intimately contacting with the endothelium, cells are exposed to chemical signals (principally chemokines, but also cytokines and other inflammatory agents) present in the local milieu, resulting in the activation-dependent up-regulation of integrin adhesiveness that leads to firm adhesion, followed by endothelial transmigration. The major mediators of firm adherence are the integrins, very-late activation protein 4 (VLA-4), and lymphocyte function-associated antigen 1 (LFA-1). Finally, HSCs home and locate to BM niches.
Besides regulating HSC functions, BM niche may similarly regulate LSCs. In fact, studies from different research groups have demonstrated that both the osteoblastic and vascular niches are critical for LSC survival, proliferation, and differentiation [2, 83–86]. By comparing the homing of the human leukemia cells with normal cord blood CD34+ cells, the transplanted leukemia and cord blood cells initially localized on the surface of osteoblasts in the epiphysial region and then expanded to the inner vascular and diaphysial regions [87]. Furthermore, after transplantation, leukemia cells initially migrated toward the CXCL12-positive vascular niches in the BM, which is similar to the homing mechanism for normal HSCs [88, 89]. CXCR4 level is significantly elevated in leukemic cells from patients with AML [90], and CXCR4 expression is associated with poor outcome [91, 92]. The high level CXCR4 enhances LSC homing to BM microenvironment by interacting with its ligand, CXCL12 (expressed on the surface of BMSCs). Blocking CXCR4 with antibody can dramatically decrease the engraftment of primary AML cells into NOD/SCID mice, but did not significantly affect engraftment of normal human progenitor cells [93]. Except for providing a leukemic niche for LSC survival and leukemic hematopoiesis, BM MSCs also mediate LSC resistance to targeted therapy. For example, it has been shown that BM MSCs render CML LSCs apoptosis triggered by TKI treatment through N-Cadherin and Wnt/β-catenin signaling [94]. In addition, inhibition of CXCR4 with its antagonist, such as Plerixafor, can enhance the efficacy of other drugs anti-leukemic effects [95, 96].
CD44 has been shown to play a critical role in conducting LSCs homing to BM niches and maintaining a primitive state [97]. As shown in a CML murine model, CD44 deficiency reduced homing of donor cells to recipient BM, resulting in decreasing LSC engraftment and impairing the induction of CML by BCR/ABL1. This defect was rescued by intra-femoral injection of or co-expression of human CD44 into the CD44 null donor cells [98]. These studies indicate that CD44 is required for LSC homing, suggesting that BM microenvironment plays a critical role in regulating self-renewal and survival of LSCs. Integrins are also required for LSC homing to the BM niches. AML cells migrate to BM microenvironment through interaction between VLA-4 on leukemic cells and fibronectin on MSCs [99], and integrin ligation triggers activation of β-integrins that promote LSC survival, which is associated with an increased phosphorylation of Akt in a PI3K-dependent manner [100].
Genetic changes in BM microenvironment play a role in leukemic transformation in some situations. For example, when phosphatase and tensin homolog (Pten) is deficient in both donor hematopoietic cells and recipient microenvironment, myeloproliferation and progression to leukemia/lymphoma occur. In contrast, when Pten-deficient hematopoietic cells are transplanted into a wild-type BM microenvironment, recipient mice remained leukemia-free [101]. In addition, dysfunction of retinoblastoma (Rb) protein or retinoic acid receptor gamma (RARγ) in the BM microenvironment contributed to development of preleukemic myeloproliferative disease [102, 103]. Mechanistically, BM microenvironment with Rb or RARγ deficiency promoted the expansion of HSCs and progenitors, which may be resulted from loss of inhibitory signals provided normally by the osteoblastic niche [102, 103]. Furthermore, osteoblast-specific deletion of Dicer1, a gene required for RNA and microRNA processing, caused cytopenia, multilineage dysplasia, and increased apoptosis of primitive hematopoietic progenitors in mice [104], phenotypically resembling myelodysplastic syndromes (MDS). Importantly, in some animals, the dyspoietic changes lead to the development of clonal myeloid sarcomas and AML, suggesting that perturbations of niche signaling selects for the acquisition of secondary genetic changes in the neighboring HSCs [104].
Together, these findings underscore the importance of interactions between LSCs and BM microenvironment in functional regulation of LSCs, and emphasize the presence of additional genetic mutations within the BM microenvironment.
Targeting LSCs
The findings outlined above suggest the following strategies for eradicating LSCs: (1) inhibition of signaling in LSC survival pathways; (2) inhibition of LSC self-renewal; (3) targeting of extrinsic components in microenvironment.
In the case of BCR/ABL1-induced CML, BCR/ABL kinase itself serves as a rational target for treating CML patients. The first BCR/ABL tyrosine kinase inhibitor (TKI) is imatinib mesylate that specifically inhibits BCR/ABL kinase activity by binding close to the ATP binding site in the BCR/ABL1 kinase domain. Later on, several other notable TKIs, such as dasatinib, nilotinib, bosutinib, and ponatinib, have been developed to treat CML with imatinib resistance. However, it has been generally recognized that a TKI alone will not completely eradiate LSCs to cure CML. We have shown that LSCs persisted in CML mice after the treatment with imatinib or dasatinib [9], suggesting that combined therapy with a TKI and another LSC-targeting agent is a legitimate strategy for treating CML. Several molecules are potential anti-LSC targets, including Alox5, Wnt/β-Catenin pathway genes, Hh, Hox family genes, etc. Specifically, selective Alox5 inhibitors have been shown to reduce proliferation and induce apoptosis of CML cells both in vitro and in vivo [75, 105]. It is to emphasize that Alox5 pathway is specifically required for the survival of CML LSC, and an Alox5 inhibitor, an anti-inflammatory drug called Zileuton (Zyflo) is currently being tested in a clinical phase I trial in combination with TKI for testing an inhibitory effect on LSCs in CML. In addition, the Wnt/β-catenin pathway is required for self-renewal of LSCs [30, 31, 58], and inhibition of Wnt signaling reduced survival of LSCs in AML and tumor cell viability in lymphoma and myeloma cell lines in vitro [106]. Furthermore, inhibition of Wnt signaling resulted in a decreased tumor growth and prolonged overall survival [106]. It should be mentioned that the inhibition of the Wnt/β-catenin signaling could be “risky” because the signaling also plays critical role in tissue homeostasis [107].
As described above, Hox family genes play critical roles in the survival regulation of LSCs, serving as potential targets for leukemia therapy. On the other hand, some studies have demonstrated that several genes (such as Notch, Wnt, and rearranged MLLs), which play important roles in leukemogenesis and LSC survival regulation, function upstream of Hox genes. Targeting these genes or pathways will decrease Hox gene expression and affect LSC survival [108]. In addition, MLL-rearrangements enhance the expression of Hox genes through DOT1, suggesting a strategy for suppressing Hox gene expression in LSCs by targeting ODT1.
Hh pathway is required for the maintenance of normal HSCs and LSCs in CML. Although, inhibition of Hh signaling can be achieved by direct binding of cyclopamine or vismodegib to Smo. Itraconazole (Sporanox) targets Smo through a mechanism distinct from cyclopamine and vismodegib [109]. Itraconazole inhibits mutant forms of Smo, which are resistant to vismodegib and other cyclopamine-competitive antagonists [110]. Inhibition of Ptch and Gli3 (5E1) with antibodies also leads to inhibition of the pathway [111, 112]. In addition, arsenic trioxide (Trisenox) inhibits Hh signaling by interfering with Gli transcription and protein function [113, 114] (Fig. 3).

In the absence of Hh, Ptch inhibits the activation of Smo by removing oxysterol from Smo, resulting in Smo inactivity and degradation. In the presence of high level of Hh ligand binding with Ptch, the pump function of Ptch is turned off allowing oxysterols to accumulate around Smo, leading to Smo accumulation at the plasma membrane. This accumulation of sterols allows SMO to become active or stay on the membrane for a longer period of time. Activated Smo stimulates Hh pathway, ultimately leading to the transcription of Gli target genes, Gli1, Ptch1, Hhip, Bcl2, CyclinD, and Snail
Although LSCs and normal HSCs share some common signal pathways, LSCs appear to also utilize some unique pathways for regulation of their survival and proliferation. For example, the NF-κB pathway is continuously activated in LSCs and plays a critical role in survival regulation of LSCs, as inactivation of NF-κB with chemical compounds eradiated LSCs [115]. In addition, constitutive activation of Akt is also commonly observed in AML, and inhibition of Akt activity by using PI3K inhibitors, such as wortmanin or LY294002, impaired LSC survival [116, 117]. Furthermore, some anti-apoptotic proteins, such as Bcl-xl and Bcl-2, are highly expressed in leukemic progenitor cells, and inhibition of the Bcl-2 family members with BH3-mimetic ABT preferentially inhibited survival of LSCs in AML mice [118].
Alternative approaches
Without any doubt, specific targeting of LSCs while sparing normal HSCs is an attractive approach to eradicating LSCs, although there is still a long way to go in this direction. Alternatively, both LSCs and normal HSCs in patients can be ablated and replaced by transplanted donor HSCs. In addition, induced pluripotent stem cells (iPSCs) provide a way of producing functional HSCs using somatic cells from a patient to overcome some shortcomings in HSC transplantation, including limited allogeneic donors, graft versus host disease (GVHD), and host versus graft disease (HVGD). iPSCs have been shown to have the capabilities of self-renewal, large-scale expansion, and differentiation into all three germ layers [119], and it is hopeful that functional HSCs can be generated from iPSCs for human use (Fig. 4). In fact, HSCs derived from iPSCs can be transplanted into patients with genetic hematologic disorders [120]. However, the challenges remain, partially because it is difficult to reproduce the microenvironment necessary for the development of various hematopoietic cell lineages during embryogenesis [120]. Also, recapitulation of an adult HSC niche in vitro has also been complicated by the fact that not all cell types in BM niches are known [120]. Nevertheless, we are closer to understanding the biology of LSCs, and have begun to develop effective strategies for targeting them.

Schematic illustration of the process to generate functional hematopoietic stem cells (HSCs). Fibroblasts are obtained from patient and induced to iPS with genetic or epigenetic modifications. iPS are guided to fully functional HSCs
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- B-ALL:
-
B cell acute lymphoblastic leukemia/lymphoma
- BM:
-
Bone marrow
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- CSC:
-
Cancer stem cell
- GMP:
-
Granulocytes-macrophage progenitors
- GVHD:
-
Graft versus host disease
- HVGD:
-
Host versus graft disease
- HSC:
-
Hematopoietic stem cell
- LSC:
-
Leukemia stem cell
- T-ALL:
-
T cell acute lymphoblastic leukemia/lymphoma
- Ph:
-
Philadelphia chromosome
- TKI:
-
Tyrosine kinase inhibitor
References
Lane SW, Gilliland DG (2010) Leukemia stem cells. Semin Cancer Biol 20(2):71–76
Konopleva MY, Jordan CT (2011) Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol 29(5):591–599
Chan WI, Huntly BJ (2008) Leukemia stem cells in acute myeloid leukemia. Semin Oncol 35(4):326–335
Alison MR, Islam S, Wright NA (2010) Stem cells in cancer: instigators and propagators? J Cell Sci 123(Pt 14):2357–2368
Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3(7):730–737
Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M (2003) Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med 138(10):819–830
Faderl S, Garcia-Manero G, Thomas DA, Kantarjian HM (2002) Philadelphia chromosome-positive acute lymphoblastic leukemia—current concepts and future perspectives. Rev Clin Exp Hematol 6(2):142–160 (discussion 200–202)
Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S (2006) Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci USA 103(45):16870–16875
Fialkow PJ, Denman AM, Jacobson RJ, Lowenthal MN (1978) Chronic myelocytic leukemia. Origin of some lymphocytes from leukemic stem cells. J Clin Invest 62(4):815–823
Eisterer W, Jiang X, Christ O, Glimm H, Lee KH, Pang E, Lambie K, Shaw G, Holyoake TL, Petzer AL et al (2005) Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia 19(3):435–441
Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE (1997) Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci USA 94(10):5320–5325
Herrmann H, Sadovnik I, Cerny-Reiterer S, Rulicke T, Stefanzl G, Willmann M, Hoermann G, Bilban M, Blatt K, Herndlhofer S et al (2014) Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 123(25):3951–3962
Ravandi F, Estrov Z (2006) Eradication of leukemia stem cells as a new goal of therapy in leukemia. Clin Cancer Res 12(2):340–344
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A et al (2004) Granulocyte–macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351(7):657–667
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367(6464):645–648
Wang JC, Dick JE (2005) Cancer stem cells: lessons from leukemia. Trends Cell Biol 15(9):494–501
Majeti R, Park CY, Weissman IL (2007) Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 1(6):635–645
Miyamoto T, Weissman IL, Akashi K (2000) AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA 97(13):7521–7526
Bernt KM, Armstrong SA (2011) Targeting epigenetic programs in MLL-rearranged leukemias. Hematology 2011:354–360
Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA (2010) The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327(5973):1650–1653
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423(6938):409–414
Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T et al (2012) Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 149(6):1245–1256
Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, Radtke F (2004) Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med 199(2):221–229
Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J, Kuttler F, Malanchi I, Birchmeier W, Leutz A et al (2008) Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood 111(1):142–149
Staal FJ, Clevers HC (2005) WNT signalling and haematopoiesis: a WNT–WNT situation. Nat Rev Immunol 5(1):21–30
Gandillet A, Park S, Lassailly F, Griessinger E, Vargaftig J, Filby A, Lister TA, Bonnet D (2011) Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia 25(5):770–780
Siapati EK, Papadaki M, Kozaou Z, Rouka E, Michali E, Savvidou I, Gogos D, Kyriakou D, Anagnostopoulos NI, Vassilopoulos G (2011) Proliferation and bone marrow engraftment of AML blasts is dependent on beta-catenin signalling. Br J Haematol 152(2):164–174
Griffiths EA, Gore SD, Hooker C, McDevitt MA, Karp JE, Smith BD, Mohammad HP, Ye Y, Herman JG, Carraway HE (2010) Acute myeloid leukemia is characterized by Wnt pathway inhibitor promoter hypermethylation. Leukemia Lymphoma 51(9):1711–1719
Hu Y, Chen Y, Douglas L, Li S (2009) beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 23(1):109–116
Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, Lagoo A, Reya T (2007) Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 12(6):528–541
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, So CW (2010) beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18(6):606–618
Giampaolo A, Sterpetti P, Bulgarini D, Samoggia P, Pelosi E, Valtieri M, Peschle C (1994) Key functional role and lineage-specific expression of selected HOXB genes in purified hematopoietic progenitor differentiation. Blood 84(11):3637–3647
Kawagoe H, Humphries RK, Blair A, Sutherland HJ, Hogge DE (1999) Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 13(5):687–698
Moretti P, Simmons P, Thomas P, Haylock D, Rathjen P, Vadas M, D’Andrea R (1994) Identification of homeobox genes expressed in human haemopoietic progenitor cells. Gene 144(2):213–219
Pineault N, Helgason CD, Lawrence HJ, Humphries RK (2002) Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp Hematol 30(1):49–57
Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, Humphries RK (1994) Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci USA 91(25):12223–12227
Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26(47):6766–6776
Buske C, Humphries RK (2000) Homeobox genes in leukemogenesis. Int J Hematol 71(4):301–308
Tedeschi FA, Zalazar FE (2006) HOXA9 gene expression in the chronic myeloid leukemia progression. Leuk Res 30(11):1453–1456
Krumlauf R (1994) Hox genes in vertebrate development. Cell 78(2):191–201
Lengerke C, Schmitt S, Bowman TV, Jang IH, Maouche-Chretien L, McKinney-Freeman S, Davidson AJ, Hammerschmidt M, Rentzsch F, Green JB et al (2008) BMP and Wnt specify hematopoietic fate by activation of the Cdx-Hox pathway. Cell Stem Cell 2(1):72–82
Perkins A, Kongsuwan K, Visvader J, Adams JM, Cory S (1990) Homeobox gene expression plus autocrine growth factor production elicits myeloid leukemia. Proc Natl Acad Sci USA 87(21):8398–8402
Scholl C, Bansal D, Dohner K, Eiwen K, Huntly BJ, Lee BH, Rucker FG, Schlenk RF, Bullinger L, Dohner H et al (2007) The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Invest 117(4):1037–1048
Borrow J, Shearman AM, Stanton VP Jr, Becher R, Collins T, Williams AJ, Dube I, Katz F, Kwong YL, Morris C et al (1996) The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 12(2):159–167
Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, Chen SJ, Willman CL, Chen IM, Feinberg AP et al (1996) Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet 12(2):154–158
Sauvageau G, Thorsteinsdottir U, Hough MR, Hugo P, Lawrence HJ, Largman C, Humphries RK (1997) Overexpression of HOXB3 in hematopoietic cells causes defective lymphoid development and progressive myeloproliferation. Immunity 6(1):13–22
Antonchuk J, Sauvageau G, Humphries RK (2002) HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell 109(1):39–45
Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, Largman C, Humphries RK (1997) Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol Cell Biol 17(1):495–505
Valent P, Sadovnik I, Racil Z, Herrmann H, Blatt K, Cerny-Reiterer S, Eisenwort G, Lion T, Holyoake T, Mayer J (2014) DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur J Clin Invest 44(12):1239–1245
Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R et al (2004) Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol 24(2):617–628
Abramovich C, Humphries RK (2005) Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol 12(3):210–216
Varjosalo M, Taipale J (2008) Hedgehog: functions and mechanisms. Genes Dev 22(18):2454–2472
Irvine DA, Copland M (2012) Targeting hedgehog in hematologic malignancy. Blood 119(10):2196–2204
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V et al (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425(6960):851–856
Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277(5329):1109–1113
Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr et al (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272(5268):1668–1671
Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB (2003) Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 422(6929):313–317
Corcoran RB, Scott MP (2006) Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci USA 103(22):8408–8413
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D et al (2009) Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458(7239):776–779
Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF et al (2007) Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med 13(8):944–951
Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA et al (2007) Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA 104(10):4048–4053
Sands WA, Copland M, Wheadon H (2013) Targeting self-renewal pathways in myeloid malignancies. Cell Commun Signal 11(1):33
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H et al (2008) Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14(3):238–249
Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, Kharas MG, Miller PG, Cornejo MG, Okabe R et al (2009) Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 4(6):559–567
Amakye D, Jagani Z, Dorsch M (2013) Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med 19(11):1410–1422
Woods JW, Coffey MJ, Brock TG, Singer II, Peters-Golden M (1995) 5-Lipoxygenase is located in the euchromatin of the nucleus in resting human alveolar macrophages and translocates to the nuclear envelope upon cell activation. J Clin Invest 95(5):2035–2046
Evans JF, Ferguson AD, Mosley RT, Hutchinson JH (2008) What’s all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci 29(2):72–78
Radmark O, Werz O, Steinhilber D, Samuelsson B (2007) 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci 32(7):332–341
Werz O, Burkert E, Fischer L, Szellas D, Dishart D, Samuelsson B, Radmark O, Steinhilber D (2002) Extracellular signal-regulated kinases phosphorylate 5-lipoxygenase and stimulate 5-lipoxygenase product formation in leukocytes. FASEB J 16(11):1441–1443
Werz O, Szellas D, Steinhilber D, Radmark O (2002) Arachidonic acid promotes phosphorylation of 5-lipoxygenase at Ser-271 by MAPK-activated protein kinase 2 (MK2). J Biol Chem 277(17):14793–14800
Catalano A, Rodilossi S, Caprari P, Coppola V, Procopio A (2005) 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J 24(1):170–179
Chen X, Sood S, Yang CS, Li N, Sun Z (2006) Five-lipoxygenase pathway of arachidonic acid metabolism in carcino-genesis and cancer chemoprevention. Curr Cancer Drug Targets 6(7):613–622
Graham SM, Vass JK, Holyoake TL, Graham GJ (2007) Transcriptional analysis of quiescent and proliferating CD34+ human hemopoietic cells from normal and chronic myeloid leukemia sources. Stem Cells 25(12):3111–3120
Chen Y, Hu Y, Zhang H, Peng C, Li S (2009) Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet 41(7):783–792
DeKelver RC, Lewin B, Lam K, Komeno Y, Yan M, Rundle C, Lo MC, Zhang DE (2013) Cooperation between RUNX1-ETO9a and novel transcriptional partner KLF6 in upregulation of Alox5 in acute myeloid leukemia. PLoS Genet 9(10):e1003765
Undi RB, Kandi R, Gutti RK (2013) MicroRNAs as haematopoiesis regulators. Adv Hematol 2013:695754
Machova Polakova K, Lopotova T, Klamova H, Burda P, Trneny M, Stopka T, Moravcova J (2011) Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer 10:41
Croce CM (2013) MicroRNA dysregulation in acute myeloid leukemia. J Clin Oncol 31(17):2065–2066
Han YC, Park CY, Bhagat G, Zhang J, Wang Y, Fan JB, Liu M, Zou Y, Weissman IL, Gu H (2010) microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med 207(3):475–489
Morris VA, Zhang A, Yang T, Stirewalt DL, Ramamurthy R, Meshinchi S, Oehler VG (2013) MicroRNA-150 expression induces myeloid differentiation of human acute leukemia cells and normal hematopoietic progenitors. PLoS One 8(9):e75815
Sackstein R (2009) Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: engineering a roadmap for cell migration. Immunol Rev 230(1):51–74
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR et al (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425(6960):841–846
Naveiras O, Daley GQ (2006) Stem cells and their niche: a matter of fate. Cell Mol Life Sci 63(7–8):760–766
Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, Bertoncello I, Bendall LJ, Simmons PJ, Haylock DN (2005) Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106(4):1232–1239
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ et al (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425(6960):836–841
Ninomiya M, Abe A, Katsumi A, Xu J, Ito M, Arai F, Suda T, Ito M, Kiyoi H, Kinoshita T et al (2007) Homing, proliferation and survival sites of human leukemia cells in vivo in immunodeficient mice. Leukemia 21(1):136–142
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA (2008) Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 322(5909):1861–1865
Konoplev S, Rassidakis GZ, Estey E, Kantarjian H, Liakou CI, Huang X, Xiao L, Andreeff M, Konopleva M, Medeiros LJ (2007) Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 109(6):1152–1156
Mohle R, Schittenhelm M, Failenschmid C, Bautz F, Kratz-Albers K, Serve H, Brugger W, Kanz L (2000) Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br J Haematol 110(3):563–572
Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE (2004) Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 104(2):550–557
Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT, Lin CP (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435(7044):969–973
Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, Shemtov N, Deutsch V, Naparstek E, Nagler A et al (2004) CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res 64(8):2817–2824
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, Bhatia R (2013) Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 121(10):1824–1838
Sison EA, Rau RE, McIntyre E, Li L, Small D, Brown P (2013) MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br J Haematol 160(6):785–797
Agarwal A, Fleischman AG, Petersen CL, MacKenzie R, Luty S, Loriaux M, Druker BJ, Woltjer RL, Deininger MW (2012) Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 120(13):2658–2668
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 12(10):1167–1174
Krause DS, Lazarides K, von Andrian UH, Van Etten RA (2006) Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 12(10):1175–1180
Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M et al (2003) Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med 9(9):1158–1165
Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W, Mills GB, Ohsaka A, Nagaoka I, Andreeff M et al (2007) Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res 67(2):684–694
Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441(7092):475–482
Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH (2007) Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129(6):1081–1095
Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129(6):1097–1110
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO et al (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464(7290):852–857
Anderson KM, Seed T, Plate JM, Jajeh A, Meng J, Harris JE (1995) Selective inhibitors of 5-lipoxygenase reduce CML blast cell proliferation and induce limited differentiation and apoptosis. Leuk Res 19(11):789–801
Cadigan KM, Liu YI (2006) Wnt signaling: complexity at the surface. J Cell Sci 119(Pt 3):395–402
Ring A, Kim YM, Kahn M (2014) Wnt/catenin signaling in adult stem cell physiology and disease. Stem cell reviews 10(4):512–525
McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R et al (2014) Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28(1):15–33
Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, Gardner D et al (2010) Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 17(4):388–399
Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Kim J, Chen B, King EM, Borodovsky A et al (2013) Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23(1):23–34
Nakamura M, Kubo M, Yanai K, Mikami Y, Ikebe M, Nagai S, Yamaguchi K, Tanaka M, Katano M (2007) Anti-patched-1 antibodies suppress hedgehog signaling pathway and pancreatic cancer proliferation. Anticancer Res 27(6A):3743–3747
Hunt R, Bragina O, Drews M, Kasak L, Timmusk S, Valkna A, Kogerman P, Jarvekulg L (2007) Generation and characterization of mouse monoclonal antibody 5E1 against human transcription factor GLI3. Hybridoma 26(4):231–240
Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ et al (2011) Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest 121(1):148–160
Kim J, Lee JJ, Kim J, Gardner D, Beachy PA (2010) Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA 107(30):13432–13437
Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A et al (2010) Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 28(3):275–280
Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M (2003) Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 102(3):972–980
Xu Q, Thompson JE, Carroll M (2005) mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 106(13):4261–4268
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T et al (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10(5):375–388
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676
Suzuki N, Yamazaki S, Yamaguchi T, Okabe M, Masaki H, Takaki S, Otsu M, Nakauchi H (2013) Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther 21(7):1424–1431
Acknowledgments
We apologize to the authors whose works are not cited here due to limitations in space and timing. YH would like to thank State Key Laboratory of Biotherapy, West China Hospital, and Sichuan University, for generous support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors indicate no potential conflicts of interest.
Rights and permissions
About this article

Cite this article
Hu, Y., Li, S. Survival regulation of leukemia stem cells. Cell. Mol. Life Sci. 73, 1039–1050 (2016). https://doi.org/10.1007/s00018-015-2108-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2108-7