
Abstract
Hematopoietic stem cells (HSCs) and leukemic stem cells (LSCs) utilize many of the same signaling pathways for their maintenance and survival. In this review, we will focus on several signaling pathways whose roles have been extensively studied in both HSCs and LSCs. Our main focus will be on the PI3K/AKT/mTOR pathway and several of its regulators and downstream effectors. We will also discuss several other signaling pathways of particular importance in LSCs, including the WNT/β-catenin pathway, the NOTCH pathway, and the TGFβ pathway. For each of these pathways, we will emphasize differences in how these pathways operate in LSCs, compared to their function in HSCs, to highlight opportunities for the specific therapeutic targeting of LSCs. We will also highlight areas of crosstalk between multiple signaling pathways that may affect LSC function.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
Hematopoiesis is a hierarchical process, with hematopoietic stem cells (HSCs) residing at the top of the hierarchy. These cells have the ability to both self-renew and differentiate to generate all mature blood cells. To retain homeostasis under normal conditions, as well as to initiate the required responses under stress conditions, hematopoiesis needs to be a tightly regulated process. To maintain homeostasis, HSCs need to respond to extracellular cues, such as growth factors and chemokines within the bone marrow niche, in order to make the decision whether to maintain self-renewal or to enter the cell cycle and differentiate. These growth factors and chemokines are the ligands for key signaling networks that either instruct or facilitate these critical decisions that orchestrate HSC function. Examples of signaling pathways required for HSC regulation include the PI3K, WNT/β-catenin, NOTCH, and TGFβ pathways.
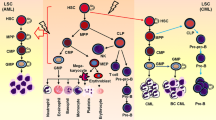
Without proper regulation by such signaling pathways, HSCs and downstream progenitors can acquire unlimited self-renewal potential at the expense of differentiation, as well as increased proliferation and survival. The acquisition of the first genetic or epigenetic alteration in an HSC can give that cell a clonal advantage over its counterparts and may lead to the generation of preleukemic stem cells (pre-LSCs) (reviewed in Corces-Zimmerman et al. [1]) (Fig. 1.1). Pre-LSCs are still capable of multilineage differentiation but have increased self-renewal capacity. They can then acquire additional genetic alterations that will promote full transformation to leukemic stem cells (LSCs) (Fig. 1.1). Unlike pre-LSCs, LSCs cannot contribute to normal adult blood cells. Instead, they differentiate only into leukemic blasts, which form the bulk of the leukemia. Some studies have shown that LSCs can also arise directly from more committed myeloid progenitors, through genetic alterations that reprogram these progenitors to express a self-renewal program usually associated with HSCs [2,3,4] (Fig. 1.1). Phenotypically, LSCs may share some of the same cell surface markers as HSCs, but they are defined functionally by their ability to generate leukemia in the bone marrow transplantation setting. As mutations in components of signaling pathways are prominent in leukemias, it is important to understand how these signaling pathways regulate LSC function.

Normal and leukemic hematopoiesis
In this review, we will focus on several signaling pathways whose roles have been extensively studied in both HSCs and LSCs and their effects on myeloid LSC self-renewal, cycling, quiescence, and differentiation capacity. Our main focus will be on the PI3K/AKT/mTOR pathway and some of its regulators and downstream effectors, whose roles in HSC and LSC function have been explored. We will also discuss other signaling pathways of particular importance in LSCs, including the WNT/β-catenin pathway, the NOTCH pathway, and the TGFβ pathway. For each of these pathways, we will emphasize differences in how these pathways operate in LSCs, compared to their function in HSCs, to highlight potential opportunities for the specific therapeutic targeting of LSCs. We will also highlight areas of crosstalk between multiple signaling pathways that may affect LSC function.
1.2 The PI3K/AKT/mTOR Signaling Pathway
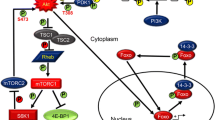
Many hematopoietic growth factors and cytokines signal through the PI3K/AKT pathway. Phosphatidylinositol 3-kinase (PI3K) can be activated by growth factors through receptor tyrosine kinase (RTK) receptors, by chemokines through G-protein-coupled receptors (GPCRs) or, by RAS family proteins (Fig. 1.2). The PI3K family consists of three distinct classes, and the Class I isoforms have been the most extensively studied in hematopoiesis, so we will focus on Class I PI3K for this discussion. Class I PI3Ks are heterodimeric lipid kinases, which are composed of a p110-kDa catalytic subunit and a regulatory subunit (reviewed in Vanhaesebroeck et al. [5]). Depending on the type of regulatory subunit, Class I PI3Ks are classified as either Class 1A or Class1B enzymes. Mammals have three Class 1A p110 isoforms (p110α, p110β, and p110δ), which are encoded by three separate genes (PIK3CA, PIK3CB, and PIK3CD, respectively), and one Class 1B isoform p110ɣ, encoded by the PIK3CG gene. The regulatory subunits are encoded by different genes and diversified by alternative promoter usage as follows: PIK3R1 encodes p85α and the splice variants p55α and p50α, while PIK3R2 encodes the p85β subunit, PIK3R3 encodes p55γ, PIK3R5 gene encodes p101, and PIK3R6 encodes the p84/p87regulatory subunit. Class 1A p85 regulatory subunits (p85, p55, p50) harbor two SRC homology 2 domains (nSH2, cSH2) and an intervening p110-binding region (iSH2) and constitutively interact with the p110α, p110β, and p110δ catalytic subunits [6]. The Class 1A catalytic isoforms can all bind to the same p85 regulatory subunits, so they can functionally compensate for one another (reviewed in [5]). In contrast, the Class1B catalytic subunit p110γ does not have a p85-binding domain and is almost exclusively activated by GPCRs. The Class I PI3Ks p110α, p110δ, and p110γ also harbor a RAS-binding domain and all except p110β are thought to be RAS effectors [5]. In contrast, p110β uses its RBD to bind to RAC and RHO GTPase family members and also interacts with Rab5 GTPase [7,8,9]. Upon stimulation, Class I PI3Ks produce the lipid second messenger phosphatidylinositol (3,4,5)-triphosphate (PIP3) from phosphatidylinositol (3,4)-diphosphate (PIP2), and this process can be antagonized by phosphatase and tensin homolog (PTEN) or Src-homology 2 (SH2)-containing inositol 5′-phosphatase (SHIP), both of which dephosphorylate PIP3 to PIP2. PIP3 recruits the inactive serine/threonine-protein kinase AKT and pyruvate dehydrogenase kinase 1 (PDK1) from the cytosol through their pleckstrin homology (PH) domains, where PDK1 then phosphorylates AKT at Thr308. For complete activation, AKT must also be phosphorylated by the mTOR complex 2 (mTORC2) at Ser473 [10]. Intriguingly, activation of PI3K/AKT in tumors can be frequently accompanied by JNK activation, and this activation seems to be PI3K-dependent, since it is promoted by loss of PTEN, with constitutive activation of JNK and the transcription factor c-JUN [11]. Furthermore, c-JUN promotes cellular survival by negatively regulating the expression of PTEN through direct binding to a variant AP-1 site on the PTEN promoter, thus activating the AKT pathway [12]. AKT has multiple downstream effectors, which regulate diverse cell processes, including cellular metabolism, glucose homeostasis, inflammation, apoptosis, cell cycle regulation, protein synthesis, and autophagy [5]. Here we will focus on those AKT effectors that have been shown to play a role in HSCs and LSCs: mechanistic target of rapamycin (mTOR) and FOXO.

The PI3K/AKT/mTOR and RAS/MEK/ERK signaling cascades and their main downstream effectors
mTOR is the major intracellular component that senses and reacts to dynamic environmental changes in response to nutrient and growth factor fluctuation to coordinate cell metabolism and growth. mTOR is a serine/threonine kinase that forms two distinct functional complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (reviewed in Zoncu et al. [13]). mTORC1 has six known protein components, while mTORC2 has seven components [14]. These two complexes share five proteins: the mTOR catalytic subunit, mammalian lethal with sec-13 protein 8 (mLST8, also known as GbL) [15, 16], DEP domain-containing mTOR-interacting protein (DEPTOR) [17], and the TTI1/TEL2 complex counts as 2 proteins [18]. In addition, mTORC1 contains regulatory-associated protein of mammalian target of rapamycin (RAPTOR) [19, 20] and proline-rich AKT substrate 40 kDa (PRAS40) [21,22,23], while mTORC2 contains the following unique components: rapamycin-insensitive companion of mTOR (RICTOR) [24, 25], mammalian stress-activated map kinase-interacting protein 1 (mSIN1) [24, 26], and protein observed with Rictor 1 and 2 (PROTOR1/2) [27, 28]. mTORC1 integrates signals from nutrients, growth factors, energy, and stress, while mTORC2 is primarily regulated by growth factors [13]. The small GTPase RAS homolog enriched in brain (RHEB) stimulates mTORC1 activity when it is GTP-bound. AKT activates mTORC1 indirectly, through phosphorylation and subsequent inactivation of tuberous sclerosis 2 (TSC2) in the tuberous sclerosis 1 (TSC1)/TSC2 complex. TSC1-TSC2 acts as a GTPase-activating protein (GAP) for RHEB, thereby inactivating RHEB. Therefore, AKT activation ultimately leads to the stimulation of mTORC1. AKT also increases mTORC1 activity through phosphorylation of proline-rich AKT substrate PRAS40 [13].
The FOXO transcription factors are evolutionarily conserved, and their roles have been implicated in a multitude of fundamental biological processes, including metabolism, stem cell maintenance, lifespan regulation, and tumorigenesis [29, 30]. The mammalian FOXOs include FOXO1, FOXO3, FOXO4, and FOXO6. The FOXOs are potent transcriptional activators that bind to the consensus motif TTGTTTAC (termed DBE for DAF-16a family member binding element) to regulate the transcription of target genes [31,32,33]. Multiple studies have shown complex regulation of FOXOs by phosphorylation, which determines their subcellular localization and governs their transcriptional activity (reviewed in Tothova et al. [34)]). In HSC and progenitor cells, upon growth factor stimulation, AKT phosphorylates FOXO 1, FOXO 3, and FOXO 4 at three conserved residues (in FOXO3 – Thr32, Ser253, Ser315), which inactivates FOXOs by creating a binding motif for the 14-3-3 chaperone proteins. This then facilitates translocation of the FOXOs from the nucleus and to the cytoplasm while preventing nuclear reentry by masking the nuclear localization sequence [31, 35, 36]. It is interesting to note that FOXO phosphorylation by AKT can be overridden by stress stimuli that activate Jun N-terminal kinase (JNK), which phosphorylates FOXOs at distinct sites from that of AKT to facilitate their nuclear import [37,38,39,40]. Interestingly, Yang et al. showed that ERK is also able to downregulate FOXO3a activity by phosphorylation of FOXO3a at Ser 294, Ser 344, and Ser 425, consequently promoting cell proliferation and tumorigenesis in a breast cancer model [41]. In addition to phosphorylation, FOXOs are posttranslationally regulated by acetylation/deacetylation, methylation, ubiquitination, and redox modulation. This fine-tuned regulation allows FOXOs to induce specific gene-expression programs in response to many environmental stimuli, such as insulin, growth factors, nutrients, and oxidative stress (reviewed in [42, 43]).
1.3 The PI3K Isoforms in HSCs and in Leukemia
Of the Class I catalytic isoforms, Pik3ca and Pik3cb are ubiquitously expressed, and their germline deletion in mice results in embryonic lethality (reviewed in Vanhaesebroeck et al. [5]). In contrast, Pik3cd and Pik3cg are enriched in leukocytes and are not required for development. In fact, mice with germline deletion of Pik3cd or Pik3cg are viable and have normal blood counts. While p110α and p110δ isoforms primarily transduce RTK signals, p110β and p110ɣ play a less prominent role in RTK signaling and are functionally redundant in signaling downstream of GPCRs [9]. P110α is essential for growth factor signaling and oncogenic signaling in fibroblasts [44]. However, functional redundancy between p110α and p110β in transducing RTK signals has also been described in some contexts [45, 46]. Surprisingly, postnatal conditional deletion of p110α in the hematopoietic system results only in mild anemia, without affecting HSC self-renewal, while conditional deletion of p110β in hematopoietic cells does not affect blood counts (Table 1.1) [47, 48]. In fetal liver HSCs, compound deletion of the regulatory subunits p85α and p85β decreases HSC frequency and multilineage repopulating capacity [49]. This suggests that there is increased redundancy between the Class IA isoforms in transducing growth factor signals in HSCs.
However, there is evidence that the same level of redundancy may not be present in the context of hematologic malignancies. For example, a requirement for p85α, but not of p85β, regulatory subunit was shown to be necessary for myeloproliferative disease driven by an activation loop mutant of proto-oncogene receptor tyrosine kinase (KIT), which encodes the receptor for stem cell factor [50] and has been described in AML [51,52,53]. An isoform-selective role for the catalytic isoform p110α was also demonstrated in RAS-mutated myeloid leukemia, in a mouse model of juvenile myelomonocytic leukemia (JMML) driven by oncogenic Kras, and in a xenograft model of human RAS-mutated AML [47]. Furthermore, this study showed that combined pharmacologic inhibition of p110α and mitogen-activated protein kinase (MEK) could be an effective therapeutic strategy for RAS-mutated myeloid malignancies [47]. In contrast, in a mouse leukemia model driven by loss of Pten in HSCs, of all the Class I PI3K isoforms, p110β was the most important in promoting myeloid leukemogenesis, though a Rac-dependent mechanism [48]. Importantly, deletion of p110β also was able to restore normal HSC function, while depleting leukemia-initiating cells, as was demonstrated in transplantation assays. This is consistent with studies in other tumor models driven by Pten loss, such as prostate cancer, which also showed preferential dependency upon p110β [54]. Furthermore, this study showed that a p110β-selective inhibitor could prolong survival in leukemia driven by Pten loss [48]. All of these studies demonstrate the concept of isoform-selective PI3K dependencies in myeloid leukemia, suggesting that it should be possible to achieve an antitumor effect with a good therapeutic window using selective PI3K isoform inhibition. In fact, several isoform-selective PI3K inhibitors, such as idelalisib, or dual-isoform inhibitors, such as duvelisib, are already in clinical use for indolent lymphoid malignancies, with reasonable toxicity profiles. However, these studies also illustrate that the specific PI3K isoform dependencies may vary depending on the oncogenic driver, as in other tumor types.
1.4 AKT in HSCs and Leukemia
Constitutive activation of PI3K/AKT signaling is observed in many cancers, including AML. Interestingly, unlike in solid tumors, mutations in members of the PI3K pathway are rarely detected in AML. However, increased AKT phosphorylation has been observed in up to 72% of AML cases [55]. In many cases, AKT activation in AML is thought to be secondary to mutations or chromosomal rearrangements causing constitutive activation of upstream RTKs, such as FLT3 or c-KIT. Inactivation of PTEN, either through genetic or epigenetic mechanisms, may also play a role in AKT activation. However, the prognostic significance of AKT phosphorylation is controversial. Phosphorylation of the AKT at Thr308 was shown to confer a poor prognosis in AML [56], whereas phosphorylation of AKT at Ser473 correlates with a favorable response to chemotherapy [57]. To determine whether AKT activation is a true mechanism of transformation, or simply a marker of the transformed state, Kharas et al. generated a murine retroviral bone marrow transplantation model in which they expressed myristoylated AKT in mouse bone marrow cells [58]. The transplant recipients developed a myeloproliferative phenotype, as well as transplantable AML or T-ALL, demonstrating that AKT activation is an important driver of transformation in leukemia. Interestingly, this study also showed that PI3K/AKT pathway activation is deleterious to normal HSC function, due to increased HSC cycling and depletion of the stem cell pool. Both the T-ALL disease phenotype and the HSC maintenance phenotype could be rescued with the mTORC1 inhibitor rapamycin, highlighting the important role of mTORC1 in HSCs and in T-ALL, but not in AML. Interestingly, this study did not show any change in ROS levels in HSCs expressing myristoylated AKT [58].
In a different study, Juntilla et al. have shown that AKT1 and AKT2 play essential redundant roles in fetal HSC maintenance. Akt1/2 double knockout fetal HSCs are unable to enter cell cycle and persist in the G0 phase, causing long-term functional defects due to increased quiescence. This increased quiescence of AKT1/2-deficient HSCs was associated with decreased ROS levels, suggesting that AKT1 and AKT2 are important for regulating ROS levels in hematopoiesis [59]. This work confirmed the important roles of AKT in the regulation of HSC homeostasis.
1.5 The Phosphatases PTEN and SHIP in HSCs and LSCs
PTEN is commonly deleted or otherwise inactivated in diverse cancers (reviewed in [60]), including hematologic malignancies [61, 62]. The phenotype observed with conditional deletion of Pten in the hematopoietic system using Mx1-Cre resembles the phenotype observed with activation of AKT in hematopoietic cells [63, 64], also reviewed in [65]). Pten deletion in hematopoietic cells led to the development of MPN, AML, and T-ALL and resulted in enhanced LSC function while compromising normal HSC function [63]. Zhang and colleagues also demonstrated that Pten inactivation in murine bone marrow causes short-term expansion, followed by the subsequent long-term decline of HSCs, primarily due to enhanced HSC activation, followed by exhaustion of the HSC pool [64]. Although Pten-deficient HSCs engraft normally in recipient mice, their ability to sustain hematopoietic reconstitution is impaired due to cell cycle dysregulation and decreased bone marrow retention [64]. Both phenotypes, the HSC exhaustion and leukemia initiation, are mediated by mTORC1, because they can be rescued by rapamycin [63]. However, it was later shown by Guo et al. that in established T-ALL, the effects of Pten deletion in LSCs couldn’t be completely reversed by rapamycin [66]. This likely reflects the different roles of mTORC1 in leukemia initiation, as opposed to leukemia maintenance. Interestingly, Lee et al. showed that the effects of rapamycin on Pten-deficient HSCs and LSCs can be partially rescued by increased expression of cyclin-dependent kinase inhibitor 2A, p16(Ink4a) and p53 in HSCs. However, while both p53 and p16(Ink4a) promoted the depletion in Pten−/− HSCs, only p53 could suppress leukemogenesis [67]. In a CML model driven by BCR-ABL expression, Peng et al. demonstrated that Pten deletion accelerates disease development, while Pten overexpression delayed disease progression by suppressing LSC function [68]. This study further supports the role of PTEN as a tumor suppressor in myeloid LSCs. All of these studies highlight the distinct effects of PTEN deletion in HSCs and in LSCs, again suggesting that there is a therapeutic window for targeting elevated AKT signaling in LSCs.
In another study, Li et al. showed that PTEN protein levels are similar among different HSC populations and multipotent progenitors (MPPs), but that there is increased PTEN phosphorylation (p-PTEN) during the transition from long-term to short-term HSCs, and further to MPPs, where p-PTEN levels are inversely correlated with the self-renewal capacity and long-term repopulation capacity of HSPCs [69]. Serial transplantation experiments demonstrated that unphosphorylated Pten augments the self-renewal of wild-type long-term repopulating cells and promotes myeloid over lymphoid differentiation. Moreover, unphosphorylated Pten restricts HSCs to a quiescent state in the bone marrow niche by dual inhibition of PI3K/AKT and SRC signaling. In contrast, p-PTEN represses PI3K/AKT, but promotes the niche contact-stimulated signaling that regulates the proliferation and lodging of hematopoietic stem and progenitor cells (HSPCs) in the bone marrow niche, whereas unphosphorylated PTEN represses both types of signaling [69]. Thus, this study highlights the dual cell-autonomous and non-cell-autonomous regulation of HSCs by PTEN.
Conditional deletion of another PIP3 phosphatase, Ship, also led to the development of a myeloproliferative disease, and bone marrow progenitor cells from Ship−/− mice were hypersensitive to hematopoietic growth factors [70, 71]. Luo et al. evaluated the potential tumor suppressor role of SHIP in myeloid leukemogenesis, using AML patient samples and myeloid leukemia cell lines [72]. Strikingly, they discovered in one AML patient a dominant negative mutation in the SHIP gene, SHIPV684E, which replaces Val with Glu at the phosphatase active site, reducing its catalytic activity.
Leukemia cells bearing this mutation have enhanced AKT phosphorylation following IL-3 stimulation. These cells also have a growth advantage, and are resistant to apoptosis upon serum deprivation, and in response to the chemotherapy drug etoposide [72]. These results suggest that mutated SHIP is a potential mechanism for the development of chemotherapy resistance through the PI3K/AKT signaling pathway.
Magee et al. addressed the effects of patient age on activation of the PI3K/AKT pathway in HSCs [73]. They asked why PTEN is sometimes mutated in adult, but rarely in early childhood leukemias, and showed that there is a different effect of Pten deletion on the neonatal vs. adult HSCs. They revealed that PTEN is required by adult HSCs to dampen AKT signaling and prevents leukemia development in adults. However, neonatal HSCs do not activate AKT signaling as a result of Pten deletion. Moreover, they showed that in adult mice, Rictor deletion in Pten−/− HSCs prevents leukemogenesis and HSC depletion, which for the first time implicates mTORC2 in HSC maintenance in the context of Pten inactivation. On the contrary, in normal HSCs, deletion of Rictor has little effect. Together with the fact that in neonatal HSCs, Pten deletion does not activate PI3K/AKT pathway and does not promote HSC proliferation, depletion, or leukemogenesis, this demonstrates the difference in critical tumor suppressor mechanisms and in the roles of PI3K/AKT signaling between adult and neonatal HSCs [73]. This study highlights the importance of the developmental changes in key signaling pathways that confer temporal alterations upon stem cell self-renewal and tumor suppressor mechanisms.
Kalaitizidis et al. confirmed the observation of the importance of RICTOR in the context of Pten loss in adult mice in their study of Raptor and Rictor deletion in the hematopoietic system (74). They found that deletion of Raptor in Pten-lox-lox;Mx1-Cre; mice significantly improved survival, but without much improvement in HSC function in transplantation assays. This is consistent with prior studies showing that rapamycin can prevent leukemia initiation in the context of hematopoietic Pten deletion [63]. However, in contrast to Raptor deletion, rapamycin was also able to rescue HSC function in the context of Pten loss, suggesting that RAPTOR may have some rapamycin-insensitive functions that are required for HSC maintenance [74]. Interestingly, conditional deletion of only Rictor, but not Raptor, after disease onset could significantly improve HSC function in the context of Pten loss [74]. Overall, these studies underscore the importance of both mTORC1 and mTORC2 in leukemic transformation induced by Pten loss, while only mTORC1 appears to play an important role in the maintenance of normal HSCs. Therefore, components of mTORC2 should be explored further as potential therapeutic targets for myeloid malignancies.
1.6 PDK1 in HSCs and Leukemia
It has been reported that pyruvate dehydrogenase kinase 1 (PDK1), which contributes to AKT activation by phosphorylating AKT on Thr308, also plays important roles in the hematopoietic system. Wang et al. revealed an indispensable role for PDK1 in fetal liver HSCs. Using the Vav-iCre system, they showed that Pdk1 is crucial for HSC expansion, and Pdk1 deletion severely impairs the repopulating potential and differentiation capacity of fetal liver HSCs [75]. Interestingly, another study showed that conditional deletion of Pdk1 in the MLL-AF9 murine AML model prolonged survival due to increased apoptosis of LSCs, which was accompanied by reduced expression of Stat5, which is often activated in leukemia [76]. These data highlight the essential role of PDK1 during fetal hematopoiesis and in LSC maintenance. It would be interesting to further delineate the differences between the function of PDK1 in adult HSCs and in LSCs, given the differences in PI3K/AKT signaling previously reported between neonatal and adult HSCs [77].
1.7 mTOR in HSCs and Leukemia
mTOR has been shown to play a central role in both embryonic and adult hematopoiesis. Guo et al. showed that conditional mTOR deletion using Mx1-Cre results in loss of HSC quiescence, with the consequent exhaustion of HSCs, as well as impaired HSC engraftment and repopulation (78). Interestingly, conditional deletion of Tsc1, and consequent mTOR activation, also causes HSC cycling and premature depletion, due to increased mitochondrial biogenesis followed by elevated levels of reactive oxygen species (ROS) [79]. This suggests that TSC1 is an essential regulator of HSC quiescence. In a follow-up study, Chen et al. also showed that mTOR activity in HSCs increases with age, and that Tsc1 deletion mimics the characteristics of aged HSCs in young mice [80]. Furthermore, they showed that decreasing mTOR signaling with rapamycin restores HSC function in old mice, thus highlighting a fundamental function of mTOR signaling in HSC aging [80]. However, activation of mTOR signaling by conditional deletion of Tsc1 is insufficient to cause hematologic malignancies in mice [79]. Interestingly, Kalaitzidis et al. also showed that Raptor deficiency leads to HSC expansion in Pten-wild-type HSCs and promotes the transition of short-term HSCs from the G0 to G1 phase [74]. Furthermore, Raptor-deficient BM cells cannot reconstitute hematopoiesis in lethally irradiated recipient mice [74]. Although under homeostatic conditions mTORC1 is not required for HSC maintenance, its loss leads to HSC and progenitor mobilization. A similar phenotype was reported by Hoshii et al. for Raptor deletion in the hematopoietic system; they observed myeloid expansion, but no effects on progenitor proliferation or survival [81]. Both groups also showed that deletion of Raptor, but not of Rictor, caused an increase in AKT activation in hematopoietic stem and progenitor cells (HSPCs). This is consistent with the previously described effects of prolonged rapamycin treatment on AKT activation, which can occur through the unopposed activity of mTORC2, leading to phosphorylation of AKT on Ser473 [82]. Given the known effects of AKT activation on HSC function, this could potentially explain the HSC cycling phenotype in Raptor−/− mice [58, 74].
Surprisingly, the GTP-binding protein RHEB, which is known to activate mTORC1 activity in other cell types, was found to only moderately activate mTORC1 in HSCs and progenitors [83]. Peng et al. showed that Rheb deletion did not significantly affect HSC maintenance in mice, in contrast to Raptor deletion, signifying that under steady-state conditions, RHEB activity is expendable for mTORC1 activity and HSC self-renewal [83].
Dysregulation of the mTORC1-S6K1 signaling pathway occurs frequently in AML patients. Primary AML cells derived from patients show increased activation of S6K1 [84]. The role of mTORC1-S6K1 signaling and its role in regulation of HSCs and LSCs were recently reviewed in detail by Ghosh and Kapur, where they highlight recent progress in identifying the roles of different components of the pathway and pharmacological targeting of mTORC1 and S6K1 [85]. Ghosh et al. showed that S6K1 deficiency in hematopoietic cells results in the reduced quiescence of L-HSCs, and their impaired self-renewal, concomitant with the reduced expression of cyclin-dependent kinase inhibitor 1A (Cdkn1a) [86]. They showed that loss of S6K1 increases the susceptibility of mice to repeated stress. Moreover, the authors reported that the leukemic potential of cells driven by the mixed lineage leukemia (MLL) fusion oncogene MLL-AF9 is similarly dependent on S6K1. Inhibition of S6K1 activity reduced the proliferation of MLL-rearranged human AML cells, and loss of S6K1 impaired in vivo LSC function in MLL-AF9 AML model, by modulating AKT and 4E-BP1 phosphorylation [86]. This data demonstrated that S6K1 loss decreases mTOR activation, consequently leading to reduced phosphorylation of AKT and 4E-BP1 in HSCs. Thus, S6K1 is a potential therapeutic target in AML LSCs. Hoshii reported similar findings with Raptor deletion in MLL-AF9 LSCs and also showed that loss of Raptor led to differentiation of LSCs [81]. Also, consistent with the results of Kalaitzidis et al. in the Pten−/− leukemia model, they reported that loss of Raptor failed to rescue normal hematopoiesis in the MLL-AF9 AML model. However, Hoshii et al. also reported the intriguing finding that Raptor deletion compromised the leukemia-initiating cells in MLL-AF9 AML mice, without compromising the self-renewal of LSCs, arguing that these are two different cell populations with differential dependence on RAPTOR [81]. This finding brings into question the usefulness of targeting mTORC1 alone in AML.
1.8 The FOXO Transcription Factors in HSCs and Leukemia
In the hematopoietic system, FOXO1 and FOXO3 are thought to be the main players, with FOXO3 being more active than FOXO1, and they have been shown to be key regulators of both HSC and LSC maintenance. It was shown that FOXOs regulate the intracellular levels of reactive oxygen species (ROS), where loss of FOXO activity elevates ROS levels, leading to increased HSC cycling and apoptosis [87, 88]. Yalcin et al. also showed that loss of FOXO3 leads to an increased number of hematopoietic progenitors with hypersensitivity to cytokines and led to a myeloproliferative phenotype. These progenitors had increased ROS levels, increased AKT/mTOR signaling, and a relative deficiency of LNK, a negative regulator of cytokine receptor signaling [89]. More recently, it was shown by Rimmele et al. that in Foxo3(−/−) HSCs and HSPCs, mitochondrial metabolism is suppressed, affecting HSC long-term repopulation activity [90]. This repression was associated with altered expression of mitochondrial and metabolic genes, which was independent of ROS levels or mTOR signaling [90].
Remarkably, accumulating evidence in the field raises the possibility that in stem cells, including in HSCs, AKT is not the only regulator of FOXO. It has been demonstrated that nuclear FOXO localization and activity in HSPCs are dependent on other upstream regulators, such as P38 mitogen-activated protein kinases (p38MAPK) or NAD-dependent deacetylase sirtuin-1 (SIRT1) [87, 91]. In fact, at steady state, FOXO seems to be primarily regulated independently from PI3K/AKT signaling. As Miyamoto et al. have shown using their Foxo3a conditional knockout mouse model, Foxo3a−/− HSCs have normal proliferation and differentiation in the bone marrow, but lose their self-renewal capacity due to loss of HSC quiescence, associated with increased p38-MAPK phosphorylation [87]. However, during myelosuppressive stress and aging, FOXO3a in HSCs is regulated via AKT and ERK signaling [92].
It has been reported that high levels of FOXO3a mRNA and phosphorylation of FOXO3a are adverse prognostic factors in AML [93, 94]. Interestingly, Chapuis et al. showed that in AML samples, the localization of FOXO3a is able to escape AKT control and is also independent of the ERK/MAPK signaling pathway, but it is dependent on FOXO3a Ser644 phosphorylation by IκB kinase (IKK) [95]. They proposed an interesting possibility that IKK activity retains FOXO3a in the cytoplasm, thereby inactivating FOXO3a and affecting the proliferation and survival of AML cells. Although FOXOs are primarily known as tumor suppressors, the roles of FOXOs in myeloid malignancies are controversial. High level of FOXO3 expression is associated with an adverse prognosis in AML [93], and genetic ablation of Foxo3 has been shown to reduce disease burden in a murine model of CML [96]. Strikingly, Sykes et al. demonstrated the opposite roles for AKT/FOXOs in AML [97]. There is a generally accepted concept that activated AKT signaling, which should lead to FOXO inhibition, is associated with hematopoietic transformation, but they observed that in 40% of AML patient samples, regardless of genetic subtype, FOXOs are active. In these patients, instead of decreased FOXO activity, FOXO is active and inversely correlated with c-Jun N-terminal kinase of (c-JUN)/JNK signaling. Moreover, leukemic cells resistant to FOXO inhibition responded to JNK inhibition, which revealed a molecular role for AKT/FOXO and JNK/c-JUN in maintaining the differentiation blockade, opening an exciting avenue for the inhibition of leukemias with a range of genetic lesions [97].
1.9 Integration of PI3K/AKT with the MAPK Pathway in HSCs and LSCs
Baumgartner et al. recently showed that during emergency hematopoiesis, the MEK/ERK and PI3K pathways are synchronously activated in HSCs [98]. Moreover, MEK1 phosphorylation by activated ERK provides a feedback mechanism to counterbalance AKT/mTORC1 activation, presenting HSCs with the rate-limiting feedback mechanism that controls their fitness and reentry into quiescence (Fig. 1.2) [98]. In myeloid leukemogenesis, oncogenic NRASG12D activates both MAPK and PI3K signaling and can promote the expansion of preleukemic clones in some contexts [99,100,101,102]. It is interesting to consider that disruption of this feedback loop that controls the duration of PI3K signaling and the production of PIP3 could exhaust the pool of fast-proliferating HSCs induced by chemotherapy, while at the same time this could benefit the expansion of a pool of self-renewing preleukemic clones.
Many of the growth factors and cytokines that orchestrate hematopoiesis and HSC maintenance signal through the PI3K/AKT canonical signaling pathway, and dysregulation of this pathway can lead to hematopoietic transformation. It is becoming clear that tight regulation of PI3K/AKT pathway activation is critical for the maintenance of HSC homeostasis, at least in adults. There is also sufficient evidence that activation of AKT in HSCs, either directly or indirectly through inactivation of PTEN or SHIP, can lead to hematopoietic transformation. However, modulation of the downstream components of AKT generally is not sufficient to promote full transformation, though it can cause significant alterations in HSC homeostasis. This highlights the importance of understanding the roles of the individual components of the PI3K/AKT pathway in hematopoietic transformation, as well as their crosstalk with parallel pathways, such as with the MAPK pathway. Exciting studies have already provided evidence that the FOXOs can act as a “crosstalk hub” between several signaling pathways, including the PI3K/AKT, MEK/ERK, and JNK/c-JUN pathways, suggesting that any mutation that alters FOXO3 function in HSCs might contribute to malignant transformation. Further research in this area should focus not only on further delineating the roles of PI3K/AKT pathway members in LSCs but also on the interplay between PI3K and other signaling pathways.
1.10 The WNT/β-Catenin Signaling Pathway in HSCs and LSCs
The WNT signaling pathway has been well characterized for its stimulatory role in adult HSC maintenance and self-renewal [103, 104] (Fig. 1.3, Table 1.2). The canonical WNT signaling pathway is activated when WNT ligands within the bone marrow stroma interact with the FRIZZLED-LRP5/6 receptor complex, which is expressed on the HSC surface (reviewed in Staal et al. [105]). This leads to inhibition of the multi-protein destruction complex composed of GSK-3β, AXIN1 or AXIN2, and APC, which normally phosphorylates β-catenin, leading to its proteasomal degradation. β-Catenin is then able to accumulate in cytoplasm, leading to its translocation into the nucleus, where it activates the TCF/LEF transcription factor family to activate the expression of genes essential for expanding the HSC pool [105].

Summary of WNT/β-catenin pathway activation in HSCs and LSCs
Despite the conserved role of WNT signaling in regulating HSC self-renewal, there has been conflicting evidence regarding the effects of β-catenin signaling on the HSC population. Knockout studies have demonstrated that WNT signaling is dispensable for HSC function. For example, deletion of β-catenin in bone marrow progenitors did not impair self-renewal or differentiation [106]. In support of this, deletion of Porcupine (Porcn), an O-acyltransferase essential for all WNT ligands, in HSCs did not impair their ability to self-renew, proliferate, or differentiate [107]. A recent review has covered the concept that the proper dosage of WNT signaling must be achieved to moderate HSC self-renewal versus differentiation decisions [108]. Using transgenic mouse lines expressing different doses of the negative regulator APC, they elegantly demonstrated that slightly enhanced levels of WNT signaling can enhance HSC reconstitution, while much higher levels of WNT signaling can result in the failure to engraft and reconstitute lethally irradiated mice [109]. This supports earlier work, which showed through conditional expression of a stable constitutively active form of β-catenin that HSCs lose their ability to repopulate and also lose the capacity for myeloid lineage commitment [110]. Scheller et al. also observed a similar HSC exhaustion phenotype when they expressed a different constitutively active mutant of β-catenin in hematopoietic cells [111]. Notably, using the SCL-Cre-ER mouse model, β-catenin activation more specifically in HSCs resulted in HSC apoptosis [112]. However, when β-catenin activation was coupled with Pten deletion, there was an expansion of LT-HSCs at the expense of differentiation, suggesting that the WNT and PI3K/AKT pathways cooperate in regulating LT-HSC proliferation, apoptosis, and differentiation [112]. Altogether, it is clear that the HSCs require some level of activation of the WNT signaling pathway for self-renewal, survival, and proliferation and that this level must be tightly regulated.
The WNT signaling pathway has also been shown to play important roles in LSCs, as this population also requires the WNT signaling pathway for self-renewal and survival. In human CML LSCs, it has been shown that β-catenin is localized in the nucleus and that in vitro LSC activity could be reduced by expression of AXIN [113]. Transplantation of BCR-ABL LSCs depleted of β-catenin into mice results in a decreased disease burden and impaired serial transplantation [114, 115]. Importantly, it has been shown that progression of CML from the chronic phase to blast crisis occurs through the stimulation of β-catenin activity through activation of the PI3K pathway by BCR-ABL [116]. Hu et al. also showed that inhibiting BCR-ABL or the PI3K pathway results in decreased levels of β-catenin. Subsequently, in vivo inhibition of this pathway delays CML development and impairs the tumor-initiating ability of CML LSCs in secondary transplantation, implying that the crosstalk between the PI3K pathway and WNT pathway is important for LSC maintenance in CML [116].
Importantly, β-catenin also plays a role in BCR-ABL tyrosine kinase inhibitor (TKI) resistance, as it has been reported that β-catenin is stabilized in CML cells to promote TKI resistance through a BCR-ABL kinase-independent resistance mechanism [117]. It has been reported that CML cells treated with imatinib maintain their β-catenin expression and have subsequent activation of downstream targets, promoting their survival and colony formation ability [117]. In addition, it has been shown that CML LSCs require the transcription factors TCF1 and LEF1 to propagate CML in secondary recipients, and combining imatinib with induced deletion of Tcf1 and Lef1 in vivo diminishes the LSC population [118]. Importantly, HSCs are less sensitive to loss of Tcf1 and Lef1, suggesting that targeting TCF1 and LEF1 in combination with a TKI could be a novel therapeutic strategy to eliminate CML LSCs, without much toxicity to HSCs [118].
In AML LSCs, inhibiting β-catenin in either the HOXA9/MEIS1 or MLL-AF9 mouse models prevents AML initiation from either the HSC or GMP populations, suggesting that WNT signaling is essential for LSC function [119]. In addition, conditional deletion of Ctnnb1, the gene that encodes β-catenin, in MLL-rearranged AML mouse models reduces the number of LSCs and overall incidence of leukemia development [120]. In both CML and AML LSC populations, β-catenin- and WNT pathway-associated genes are increased in comparison to normal HSCs [113, 120], further supporting the important roles of the WNT signaling pathway in LSCs. More recently, it has been shown that GPR84, a positive regulator of β-catenin, is also upregulated in LSCs in comparison with normal HSCs, and suppression of GPR84 reduced MLL-rearranged LSC functionality in vitro and in vivo [121]. WNT signaling has also been studied in FLT3-mutant AML models, as FLT3 signaling can increase nuclear β-catenin levels [122]. High levels of β-catenin were found in bone marrow resident leukemic cells in patients with FLT3-mutated AML. Notably, the combination of a β-catenin antagonist and a FLT3 TKI impaired the engraftment of FLT3-mutated leukemic cells into immunodeficient mice, suggesting that β-catenin inhibitors can synergize with FLT3 inhibitors in FLT3-mutated AML, due to the effects of this drug combination on LSC function [122].
Noncanonical WNT signaling has also been shown to play a role in myeloid LSCs. The noncanonical WNT ligand WNT5a can signal through both the planar cell polarity and WNT/calcium noncanonical pathways, both of which are extensively reviewed in Komiya and Habas [123]. In short, WNT5a signaling through the WNT/calcium pathway results in increased intracellular calcium levels and subsequent inhibition of β-catenin [123]. This has been shown in HSCs, as WNT5a can inhibit the activation of canonical WNT signaling, and this inhibition results in an increase of quiescent HSCs [124]. WNT5a has implications in AML, as WNT5a levels are low or absent in primary AML samples, and WNT5a hemizygous mice develop myeloid leukemias [125]. In addition, high levels of methylation of the WNT5a gene locus correlate with lower expression of WNT5a, and this high methylation status correlates with poor prognosis of a subset of AML patients [126]. More recently, the role of WNT5a in the niche has been explored. It was shown that both HSCs and BCR-ABL LSCs are impaired in engrafting into a WNT5a-deficient niche [127]. Furthermore, transplanting LSCs exposed to a WNT5a-deficient niche reduces the incidence of leukemia in mice, as well as the ability to propagate leukemia to secondary recipient mice [127]. A more recent study has confirmed a role for both canonical and noncanonical WNT signaling in leukemia, showing in vivo that the canonical WNT signaling pathway is upregulated in the leukemic HSPC compartment at the expense of a downregulated WNT5a signaling axis [128]. Altogether, it is evident that myeloid LSCs, like HSCs, rely on WNT signaling for functionality, but that different levels of WNT/β-catenin signaling may be required to promote HSC and LSC function.
1.11 The NOTCH Pathway in HSCs and LSCs
The NOTCH pathway is best characterized for its crucial role in embryonic development and in HSC emergence during embryogenesis. However, the efforts to elucidate the roles of NOTCH signaling in adult hematopoiesis and HSC maintenance have expanded in an effort to understand how dysregulated NOTCH signaling affects the leukemic state (Fig. 1.4, Table 1.3). Within this signaling network is a family of four NOTCH receptors expressed on the cell surface that can interact with one of five ligands (Jagged 1, Jagged 2, Delta 1, Delta 2, and Delta 3), which are presented on the surface of a neighboring cell (reviewed in Pajcini et al. [129]) . Ligand binding results in cleavage of the intracellular domain of the NOTCH receptor by a γ-secretase complex to allow this portion of the receptor to translocate into the nucleus. In the nucleus, the intracellular domain of NOTCH (NICD) can interact with co-activator proteins such as CBF-1 and MAM1, resulting in transcriptional activation of NOTCH target genes, such as the HES family [129].

NOTCH signaling in HSCs and LSCs
NOTCH signaling has been shown to play a stimulatory role in adult HSCs, and promotes HSC expansion. Notably, constitutive activation of NOTCH1 signaling via expression of NICD in hematopoietic cells results in immortalized, cytokine-dependent HSCs capable of differentiating into both myeloid and lymphoid progeny [130]. This result was confirmed by additional work showing that expression of NICD in vivo resulted in HSC expansion and self-renewal at the expense of differentiation [131]. Several groups have shown that NOTCH pathway activation also leads to expansion of the hematopoietic progenitor compartment. For example, Karanu et al. showed that adding soluble forms of the human ligands Delta 1 and Delta 4 results in increased proliferation of hematopoietic progenitors in vivo, and transplantation of Jagged 1 or Delta 1 into immunodeficient mice expands HSPCs that are capable of pluripotent hematopoietic reconstitution [132, 133]. Supporting this work, it was shown in transgenic Notch reporter mice that NOTCH signaling is upregulated in HSCs, but that this signaling is lost upon lineage commitment [134]. Moreover, inhibiting NOTCH signaling was able to accelerate the differentiation of HSCs in vitro, and this inhibition resulted in the depletion of HSCs in vivo [134]. Thus, activation of the NOTCH pathway, either through overexpression of NICD or by exogenous ligands, promotes HSC expansion and self-renewal.
Surprisingly, examination of the role of NOTCH through genetic deletion has suggested that NOTCH signaling is not required for adult HSC function. It has been shown that genetic deletion of Notch1 does not affect the reconstitution capacity of HSCs, though it does lead to a block in T-cell development [135]. Studying the effects of Notch2 deletion in the hematopoietic context has also shown that HSC function is NOTCH-independent [136]. However, Notch2-deficient mice have impaired hematopoietic recovery after 5-fluorouracil (5-FU), suggesting a requirement for NOTCH signaling for HSC function during stress hematopoiesis [136]. In addition, NOTCH may regulate HSC function through the niche. For example, osteoblasts within the niche express high levels of JAGGED 1, correlating with an increase in HSC number in vivo [137]. In addition, endothelial cells within the niche promote the proliferation and survival of LT-HSCs via expression of NOTCH ligands in vivo, further suggesting a role for NOTCH signaling dependence for HSC function [138].
The roles of the NOTCH pathway in LSCs have been controversial, as NOTCH signaling has been reported to play both a tumor suppressor and oncogenic role in LSCs – depending on the lineage context. The roles of NOTCH signaling in LSCs have been extensively studied in the context of T-ALL and CLL, in which NOTCH signaling is considered oncogenic (reviewed in Lobry et al. [139]). Work is still being done to fully elucidate the role of the NOTCH pathway in myeloid LSCs. However, several groups have begun to unravel that NOTCH signaling may play a tumor suppressor role in the myeloid context [140,141,142,143]. NOTCH signaling has an accepted role in promoting myeloid differentiation by upregulation of the transcription factor PU.1 [140]. It was shown that Notch 1 expression is downregulated in AML cell lines and in primary patient blasts, and this downregulation correlates with a decrease in PU.1-mediated differentiation, suggesting that NOTCH signaling may be responsible for maintaining the immature myeloid compartment seen in leukemia [140]. Furthermore, data suggests that induced activation of the NOTCH signaling axis in AML in vivo results in cell growth arrest, differentiation, and apoptosis [142]. Consistent with this, activation of NOTCH signaling in AML by NOTCH ICN1 expression in vivo decreases AML cell proliferation, and this phenotype is reversed through the inhibition of NOTCH signaling using a dominant-negative MAML construct [143]. Additionally, inactivation of NOTCH signaling in HSCs in vivo can promote a chronic myelomonocytic leukemia (CMML)-like phenotype, further supporting a tumor-suppressive function [141]. Additionally, recent work has shown that the NOTCH target HES1 binds to the FLT3 promoter to suppress its transcription, and loss of HES1 results in AML development in vivo [144]. Importantly, in MLL-AF9 leukemic cells, FLT3 was significantly upregulated upon HES1 deletion [144]. However, the role of NOTCH signaling as a tumor suppressor pathway in AML LSCs was recently challenged by results revealing that crosstalk between the WNT and NOTCH signaling pathways in osteoblasts of the bone marrow results in activation of NOTCH signaling between osteoblasts and HSPCs, actually promoting leukemic transformation [145]. Thus, more work is still needed to fully elucidate the roles of NOTCH signaling in AML LSCs.
There is also evidence to suggest that the NOTCH signaling pathway plays a role in CML LSCs, although the specific effects of NOTCH in this context also require further exploration. For example, induced expression of Hes1 was shown to cooperate with BCR-ABL to induce blast crisis CML in vivo [146]. Importantly, HES1 is highly expressed in CML patients in blast crisis, suggesting its potential role in the progression from chronic phase to blast phase in CML [146]. In addition, genetic deletion of the transcription factor Twist1 in the microenvironment activates the NOTCH signaling pathway via upregulation of Jagged 2, and this activation is required for MLL-AF9-induced AML [147]. In contrast, it was shown that overexpression of NICD was sufficient to inhibit the proliferation of the CML blast crisis cell line K562 [148]. A tumor-suppressive role for NOTCH has also been demonstrated within the CML niche. Specifically, ablation of osteoblasts in a CML transgenic mouse model results in accelerated development of CML [149]. Since it was shown that JAGGED 1 levels were increased in CML osteoblasts, this group went on to show that the addition of JAGGED 1 to CML LSCs reduced their cycling, thus supporting the hypothesis that NOTCH signaling promotes an antileukemic environment [149]. However, while it is clear that NOTCH signaling is important in normal HSC and myeloid LSC function, understanding and deciphering the contexts in which it promotes or suppresses the leukemic state still remains an active area of research.
1.12 The TGFβ Pathway in HSCs and LSCs
The TGFβ pathway is known to play an important role in controlling HSC function, but its potential role in myeloid LSCs has only been explored within the last decade (Fig. 1.5, Table 1.4). Recently reviewed by Vaidya and Kale [150], the TGFβ family consists of three ligands – TGFβ1, TGFβ2, and TGFβ3 – all of which signal through type I and type II serine/threonine kinase membrane receptors [150]. The primary type I receptor for TGFβ is ALK5/TβRI and the sole type II receptor for TGFβ is TβRII. In short, TGFβ is found in its dormant form within the extracellular matrix, but upon cleavage of its pro-domain, it is able to bind to a TβRII homodimer. TβRII is then able to multimerize with a TβRI homodimer, resulting in subsequent phosphorylation of TβRI. After the required receptor transphosphorylation, the canonical downstream TGFβ pathway involving the SMAD transcription factors is activated. The SMAD family consists of five receptor-mediated SMADs (R-SMADs 1, 2, 3, 5, and 8), one common-partner SMAD (Co-SMAD 4), and two inhibitory SMADs (I-SMADs 6 and 7). The R-SMADs contain MH2 domains that can bind via adaptor proteins to activated TβRI receptors. This interaction results in the phosphorylation of the R-SMAD, which is now able to dissociate and bind to SMAD 4. This heterodimeric complex is able to translocate into the nucleus to regulate the transcription of TGFβ target genes. The inhibitory SMADs turn off TGFβ signaling by either dephosphorylating or recruiting E3 ligases to TβRI or by competitively binding to SMAD 4. TGFβ can also signal through a SMAD-independent pathway, specifically by activating the MAPK pathway and subsequent ERK, JNK, and/or p38 activity [150]. TGFβ is expressed by both hematopoietic and bone marrow stromal cells [151], as well as by other niche components, such as megakaryocytes [152]. It has also been shown that both TβRI and TβRII are expressed on hematopoietic cells, and they can induce SMAD activity [153]. Thus, HSCs can both express TGFβ and respond to TGFβ signaling.

Summary of TGFβ pathway activation in HSCs and LSCs
It has been well documented that TGFβ is an inhibitor of HSC proliferation and an inducer of HSC quiescence [154,155,156,157,158]. In addition, it was reported that Tgfβ1-null mice have enhanced myelopoiesis [159]. Due to this profound regulatory role of the TGFβ pathway, many groups have examined the effect of deleting its components in HSCs. It is evident that TGFβ plays a crucial role in embryonic development, as both Tgfβ1 and Tgfβ2 knockout mice have an embryonic lethal phenotype [160, 161]. While conditional deletion of TβRI in vivo does not impact HSC self-renewal or long-term repopulating capacity [162], deletion of TβRII results in impaired HSC long-term repopulation [163]. Consistent with this finding, conditional deletion of Smad 4 also results in impaired HSC self-renewal and reconstitution capacity [153]. In an attempt to remove the major source of TGFβ from HSCs, Zhao et al. depleted megakaryocytes, which resulted in the loss of HSC quiescence and subsequent HSC proliferation [152]. Importantly, these HSCs had reduced SMAD 2 and SMAD 3 phosphorylation, supporting the hypothesis that megakaryocyte ablation is sufficient to reduce TGFβ signaling in HSCs. This loss of HSC quiescence phenotype could be rescued by injecting TGFβ1 into megakaryocyte-depleted mice. Furthermore, conditional deletion of TGFβ1 in megakaryocytes induced the same HSC proliferation phenotype as megakaryocyte ablation [152].
Several groups have investigated the mechanisms by which TGFβ signaling controls HSC quiescence and self-renewal. Most evidence suggests a SMAD-dependent mechanism, as seen in one study in which the inhibitory SMAD 7 was overexpressed in murine HSCs, leading to an increase in their self-renewal capacity [164]. Furthermore, the TGFβ-SMAD-p57Kip2 axis has been shown to be essential for inducing HSC quiescence. Expression of p57Kip2 increases upon TGFβ stimulation in primary human hematopoietic cells, and this upregulation is essential to drive cell cycle arrest [165]. It was later shown that quiescent murine CD34- HSCs exhibit both increased SMAD 2/SMAD 3 and p57Kip2 activity as opposed to their cycling CD34+ counterparts [163], further suggesting that SMAD-dependent TGFβ signaling is required to maintain a dormant HSC population. In support of this, conditional deletion of p57 was shown to impair HSC self-renewal and reduce the size of the LSK population [166]. Additionally, Musashi 2 (MSI2), an RNA-binding protein that is important for HSC function and myeloid malignancies, was shown to maintain HSC function and regulate HSC self-renewal fate decisions by regulating TGFβ-SMAD 2/3-p57Kip2 [167]. Msi2-deficient HSCs have reduced reconstitution capability due to enhanced myeloid differentiation at the expense of HSC maintenance, and this is accompanied by decreased TGFβ-SMAD 2/3-p57Kip2 signaling [167]. The TGFβ-SMAD-p57 axis has also been implemented in restoring HSC quiescence after stress-induced cycling. After 5-FU treatment, murine HSCs exhibit a transient increase in TGFβ expression that correlates with the amount of time required for HSCs to reenter a quiescent state [168]. In summary, there is clear evidence that the TGFβ-SMAD-p57Kip2 signaling axis is required for HSC quiescence.
Furthermore, recent evidence suggests that the tyrosine phosphatase SHP-1 is essential for activating the TGFβ pathway in HSCs to induce quiescence, raising the possibility that the TGFβ-SMAD-p57Kip2 axis requires upstream SHP-1 activity. Specifically, it was shown that SHP-1 physically interacts with TβRI to potentiate downstream signaling, and SHP-1-depleted HSCs lose their quiescence and subsequently enter cycling until exhaustion [169]. As expected, these Shp-1-depleted HSCs have impaired long-term self-renewal, and, importantly, they are incapable of becoming quiescent upon TGFβ stimulation [169]. While more work is required to delineate all the possible mechanisms by which TGFβ controls HSC function, it is evident that a TGFβ-induced SMAD-dependent mechanism mediates HSC quiescence and cycling.
Despite the apparent role that TGFβ plays in regulating HSC function, very little information is known about how the TGFβ pathway regulates LSCs. As expected by its accepted role in limiting HSC proliferation, there is evidence that TGFβ acts as a tumor suppressor. The cyclin-dependent kinase p27 is critical for regulating the cell cycle of hematopoietic cells, and one growth factor that regulates p27 expression is TGFβ [170]. It was shown that BCR-ABL-expressing cells have decreased expression of p27 and are incapable of responding to TGFβ stimulation, suggesting that the BCR-ABL fusion protein functions to turn off TGFβ signaling to maintain a proliferative advantage [170]. It was previously reported that loss of the transcription factor JunB in HSCs is adequate to transform HSCs and induce myeloid malignancies in vivo [171, 172]. One possible mechanism for this phenotype is that loss of JunB renders HSCs incapable of responding to TGFβ stimulation due to dysregulation of the JunB downstream target gene Hes1, suggesting a role for the antiproliferative effects induced by the TGFβ pathway in HSCs in protection against myeloid malignancies [173].
The SMADs have also been shown to play tumor suppressor roles. For example, it was shown that HOXA9, a key transcriptional regulator of HSC self-renewal that is commonly dysregulated in AML, can be stabilized in the cytoplasm by SMAD 4, thereby losing its ability to translocate into the nucleus and mediate events required for leukemic transformation [174]. Importantly, transplanting SMAD 4-deficient HSCs transduced with the oncogenic NUP98-HOXA9 fusion into mice resulted in rapid onset of a more pronounced myeloproliferative disease, and secondary transplantation of these cells displayed a competitive advantage compared to wild-type cells, with faster development of leukemia. Notably, reintroducing the SMAD 4 domain required for HOXA9 cytoplasmic stabilization induced apoptosis of the HOXA9-transduced cells in vitro and limited their ability to initiate leukemia in vivo [174]. In summary, it is plausible that TGFβ plays a protective role in myeloid LSCs by regulating downstream parallel pathways that are essential for regulating proliferation.
However, other studies have suggested that TGFβ plays an oncogenic role in LSCs. In one study, TGFβ was shown to be a critical regulator of AKT activity, and AKT-dependent suppression of active nuclear FOXO3a is required for eliminating CML LSCs [96]. Importantly, the combination of inhibiting TGFβ signaling and imatinib was sufficient to impair CML transformation, and this was shown to be due to decreased nuclear FOXO levels [96]. It has also been suggested that TGFβ can regulate the activity of LYN kinase to protect CML cells from imatinib, and inhibiting the TGFβ pathway can suppress LYN kinase activity and subsequently sensitize CML cells to imatinib treatment [175]. In AML, examining the role of TGFβ in the microenvironment has shown that the hypoxic bone marrow microenvironment reinforced by leukemic cells stimulates the TGFβ pathway, which in turn induces the expression of CXCR4 – a critical factor involved in promoting the survival of resident chemoresistant LSCs [176]. Furthermore, the inhibition of both TGFβ and CXCR4 in a FLT3-mutated AML model was sufficient to decrease the leukemic burden and prolong survival [176]. Lastly, it has recently been suggested that several different isoforms of TβRII play a role in AML. The TβRII splice variant TβRII-B is expressed in normal hematopoietic cells and functions to promote apoptosis and differentiation of AML cells [177]. However, the canonical TβRII receptor, which is the isoform primarily expressed in AML cells, inhibits the ability of TβRII-B to mediate its tumor suppressor role in vivo, overall blocking the differentiation of the leukemic cells [177]. This further complicates the potential role that the TGFβ pathway is playing in LSC function, displaying the necessity for further exploring the TGFβ pathway in the leukemic context.
1.13 Concluding Remarks
It is evident that proper signaling through the PI3K, WNT/β-catenin, NOTCH, and TGFβ pathways is fundamental for regulating the fate decisions and proliferation of HSCs. With the frontline treatment of AML still being chemotherapy and stem cell transplantation for most patients, and given the resistance observed in CML patients to targeted therapies like imatinib, it is critical that new treatment options become available to fully eradicate LSCs. Key advances in considering new therapeutic avenues include the emerging field of studying the characteristics of the LSC, as well as the acceptance of a pre-LSC that has the ability to contribute to normal hematopoiesis, while accumulating key mutations that eventually push it toward the threshold of transforming to a LSC. Mutations in components of each of these signaling pathways are common in myeloid leukemia, further validating studying these signaling pathways in the context of regulating the function of the LSC.
A classic “two-hit” model for the accumulation of mutations in AML to generate a fully functional LSC posits that two distinct types of mutations are required for full transformation (Fig. 1.1) [178]. First, a mutation in a gene critical for regulating HSC differentiation and self-renewal decisions is required. Traditionally, genes within this class were thought to be transcription factors, but it is now understood that alterations in multiple other types of genes, including splicing factors and epigenetic regulators, could also serve a similar function. This type of mutation will confer properties of self-renewal to the LSC at the expense of differentiation. Second, the LSC also requires an activating mutation in a signaling pathway essential for regulating HSC proliferation. This mutation will confer a proliferative and survival advantage to the LSC. This requirement for second mutations, the so-called “two-hit hypothesis”, has been supported by multiple studies. For example, it was shown that signaling mutations alone, such as BCR-ABL and FLT3-ITD, are not sufficient to impair LSC differentiation and confer self-renewal, suggesting that the proliferative advantage alone is not adequate to generate a fully functional LSC [179]. On the other hand, mutations and/or rearrangements in transcription factors, such as AML1-ETO or CBFB-MYH11, are sufficient to confer self-renewal properties to LSCs, but insufficient to trigger leukemogenesis without additional mutations [177, 180, 181]. Given this, since the PI3K, WNT/β-catenin, NOTCH, and TGFβ signaling pathways play essential roles in regulating HSC cycling and self-renewal decisions, mutations in components of either of these pathways can directly impact the functionality of the LSC.
Epigenetic mutations also play an essential role in leukemia development at the stem cell level. However, their potential cooperation with signaling pathway mutations is often overlooked. Mutations in enzymes that affect DNA methylation or chromatin modifications, including TET2, DNMT3a, IDH1/2, and ASXL1/2, are considered to be “initiating” mutations, which are acquired at or before the pre-LSC stage (reviewed in Corces-Zimmerman et al. [1]). It is clear how such mutations may cooperate with mutations in transcription factors in leukemogenesis, since these epigenetic modifiers function in establishing the genomic landscape required for the recruitment of transcription factors. However, little has been explored regarding how epigenetic mutations cooperate with signaling mutations in LSCs. The “two-hit” model of leukemogenesis still helps us to understand the requirements for leukemic transformation. However, perhaps it would be helpful to think of these two, or more, classes of mutations not as simply additive, but as potentially synergistic in promoting hematopoietic transformation. The specific nonrandom patterns of co-occurrence of genetic alterations in AML, such as AML1-ETO with C-KIT mutations, or MLL rearrangements with RAS mutations [182], could provide clues about such potential synergistic relationships that should be further explored. For example, in other tumors, evidence for the direct regulation of epigenetic modifying enzymes by PI3K/AKT signaling is emerging (reviewed in Spangle et al. [183]). Thus, it is necessary to explore the interactions between signaling pathways and epigenetics in the context of LSCs, ultimately with the goal of refining the standard of care for myeloid leukemia patients to prevent relapse.
Lastly, given that the PI3K, WNT/β-catenin, NOTCH, and TGFβ pathways all play important roles in regulating HSC self-renewal and proliferation, it is not surprising that there would be crosstalk between these pathways. One example of crosstalk in other cell types includes the NOTCH and PI3K pathways, in which NOTCH signaling requires activation of the PI3K/AKT pathway to induce megakaryocyte differentiation [184]. A second example is between the PI3K and WNT pathways, as it has been shown that β-catenin is a direct substrate of AKT [185]. Lastly, it has been demonstrated extensively that PI3K/AKT signaling is a SMAD-independent pathway that can be activated by TGFβ. Exploring this crosstalk in the context of LSCs may help to elucidate the contradictory role of TGFβ in LSCs. Although such crosstalk between signaling pathways has been studied in hematopoietic cells and in other tissues, it may be different in LSCs, and it is crucial to decipher these targetable differences. In summary, the PI3K, WNT/β-catenin, NOTCH, and TGFβ pathways are all important signaling pathways in HSCs, but with distinct roles in LSCs in some contexts. Further characterization of these pathways in the context of LSCs is important for understanding leukemogenesis and for developing new treatment paradigms with a safe therapeutic window.
Abbreviations
- AML:
-
Acute myeloid leukemia
- APC:
-
Adenomatous polyposis coli
- BCR-ABL:
-
Fusion of breakpoint cluster region (BCR) and Abelson murine leukemia viral oncogene homolog 1 (ABL)
- Cbf-1:
-
Centromere-binding protein 1
- CLL:
-
Chronic lymphoblastic leukemia
- CML:
-
Chronic myeloid leukemia
- Co-Smad:
-
Common-partner Smad
- DEPTOR:
-
DEP domain-containing mTOR-interacting protein
- GPCRs:
-
G-protein-coupled receptors
- GSK-3β:
-
Glycogen synthase kinase-3β
- Hes-1:
-
Hairy and enhancer of split-1
- HSC:
-
Hematopoietic stem cell
- HSPC:
-
Hematopoietic stem and progenitor cell
- IKK:
-
IκB kinase
- I-Smad:
-
Inhibitory Smad
- JNK:
-
Jun N-terminal kinase
- LEF:
-
Lymphoid enhancer factor
- Lrp5/6:
-
Lipoprotein receptor-related protein 5 or 6
- LSC:
-
Leukemic stem cell
- LSK:
-
Lineage negative, Sca1+, cKit-
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Mitogen-activated protein kinase
- MLL:
-
Mixed lineage leukemia
- mLST8:
-
Mammalian lethal with sec-13 protein 8
- mSin1:
-
Mammalian stress-activated map kinase-interacting protein 1
- mTOR:
-
Mechanistic target of rapamycin
- mTORC1:
-
mTOR complex 1
- mTORC2:
-
mTOR complex 2
- NICD:
-
Notch intracellular domain
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PH:
-
Pleckstrin homology
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2:
-
Phosphatidylinositol (3,4)-diphosphate
- PIP3:
-
Phosphatidylinositol (3,4,5)-triphosphate
- PORCN:
-
Porcupine
- PRAS40:
-
Proline-rich Akt substrate 40 kDa
- Protor1/2:
-
Protein observed with Rictor 1 and 2
- PTEN:
-
Phosphatase and tensin homolog
- Raptor:
-
Regulatory-associated protein of mammalian target of rapamycin
- R-Smad:
-
Receptor-mediated Smad
- RTK:
-
Receptor tyrosine kinase
- SH2:
-
Src-homology 2
- SHIP:
-
SH2-containing inositol 5′-phosphatase
- SIRT1:
-
NAD-dependent deacetylase sirtuin-1
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TCF:
-
T-cell factor
- TGFβ:
-
Transforming growth factor-beta
- TKI:
-
Tyrosine kinase inhibitor
- TSC1:
-
Tuberous sclerosis 1
- TSC2:
-
Tuberous sclerosis 2
- TβRI:
-
TGFβ-type I receptor
- TβRII:
-
TGFβ-type II receptor
References
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R (2014) Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 111(7):2548–2553
Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N et al (2004) MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6(6):587–596
Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S et al (2011) Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 19(1):138–152
Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J et al (2006) Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442(7104):818–822
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B (2010) The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11(5):329–341
Vadas O, Burke JE, Zhang X, Berndt A, Williams RL (2011) Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal 4(195):re2
Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC et al (1999) Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol 1(4):249–252
Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS et al (2013) RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153(5):1050–1063
Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A et al (2008) The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A 105(24):8292–8297
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P et al (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15(23):6541–6551
Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ et al (2007) Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 11(6):555–569
Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT et al (2007) c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ 14(2):218–229
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12(1):21–35
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A et al (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6(11):1122–1128
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10(8):935–945
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137(5):873–886
Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K et al (2010) Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem 285(26):20109–20116
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S et al (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110(2):177–189
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H et al (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110(2):163–175
Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E et al (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25(6):903–915
Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P et al (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2(11):e1217
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9(3):316–323
Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY et al (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127(1):125–137
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H et al (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol CB 14(14):1296–1302
Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA et al (2006) mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol CB 16(18):1865–1870
Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M et al (2007) Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J 405:513–522
Pearce LR, Sommer EM, Sakamoto K, Wullschleger S, Alessi DR (2011) Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem J 436:169–179
Greer EL, Brunet A (2008) FOXO transcription factors in ageing and cancer. Acta Physiol 192(1):19–28
Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE, Burgering BM (2013) FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep 5(6):1664–1678
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6):857–868
Kops GJ, Burgering BM (1999) Forkhead transcription factors: new insights into protein kinase B (c-akt) signaling. J Mol Med 77(9):656–665
Furuyama T, Nakazawa T, Nakano I, Mori N (2000) Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J 349(Pt 2):629–634
Tothova Z, Gilliland DG (2007) FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1(2):140–152
Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV et al (2002) 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 156(5):817–828
Rena G, Prescott AR, Guo SD, Cohen P, Unterman TG (2001) Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targeting. Biochem J 354:605–612
Wang MC, Bohmann D, Jasper H (2005) JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 121(1):115–125
Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB et al (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125(5):987–1001
Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL et al (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23(24):4802–4812
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303(5666):2011–2015
Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X et al (2008) ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol 10(2):138–148
Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27(16):2276–2288
Salih DA, Brunet A (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 20(2):126–136
Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A et al (2006) The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A 103(44):16296–16300
Marques M, Kumar A, Cortes I, Gonzalez-Garcia A, Hernandez C, Moreno-Ortiz MC et al (2008) Phosphoinositide 3-kinases p110 alpha and p110 beta regulate cell cycle entry, exhibiting distinct activation kinetics in G(1) phase. Mol Cell Biol 28(8):2803–2814
Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC et al (2012) The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev 26(14):1573–1586
Gritsman K, Yuzugullu H, Von T, Yan H, Clayton L, Fritsch C et al (2014) Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J Clin Invest 124(4):1794–1809
Yuzugullu H, Baitsch L, Von T, Steiner A, Tong H, Ni J et al (2015) A PI3K p110beta-Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat Commun 6:8501
Haneline LS, White H, Yang FC, Chen S, Orschell C, Kapur R et al (2006) Genetic reduction of class IA PI-3 kinase activity alters fetal hematopoiesis and competitive repopulating ability of hematopoietic stem cells in vivo. Blood 107(4):1375–1382
Munugalavadla V, Sims EC, Chan RJ, Lenz SD, Kapur R (2008) Requirement for p85alpha regulatory subunit of class IA PI3K in myeloproliferative disease driven by an activation loop mutant of KIT. Exp Hematol 36(3):301–308
Cairoli R, Beghini A, Grillo G, Nadali G, Elice F, Ripamonti CB et al (2006) Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 107(9):3463–3468
Cairoli R, Grillo G, Beghini A, Tedeschi A, Ripamonti CB, Larizza L et al (2003) C-Kit point mutations in core binding factor leukemias: correlation with white blood cell count and the white blood cell index. Leukemia 17(2):471–472
Beghini A, Peterlongo P, Ripamonti CB, Larizza L, Cairoli R, Morra E et al (2000) C-kit mutations in core binding factor leukemias. Blood 95(2):726–727
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH et al (2008) Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454(7205):776–779
Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK et al (2003) Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 17(5):995–997
Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C et al (2009) The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23(6):1029–1038
Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broet P et al (2007) Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 110(3):1025–1028
Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M et al (2010) Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115(7):1406–1415
Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA (2010) AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 115(20):4030–4038
Di Cristofano A, Pandolfi PP (2000) The multiple roles of PTEN in tumor suppression. Cell 100(4):387–390
Aggerholm A, Gronbaek K, Guldberg P, Hokland P (2000) Mutational analysis of the tumour suppressor gene MMAC1/PTEN in malignant myeloid disorders. Eur J Haematol 65(2):109–113
Dahia PLM, Aguiar RCT, Alberta J, Kum JB, Caron S, Sill H et al (1999) PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Hum Mol Genet 8(2):185–193
Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H et al (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441(7092):475–482
Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT et al (2006) PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441(7092):518–522
Fragoso R, Barata JT (2014) PTEN and leukemia stem cells. Adv Biol Regul 56:22–29
Guo W, Schubbert S, Chen JY, Valamehr B, Mosessian S, Shi H et al (2011) Suppression of leukemia development caused by PTEN loss. Proc Natl Acad Sci U S A 108(4):1409–1414
Lee JY, Nakada D, Yilmaz OH, Tothova Z, Joseph NM, Lim MS et al (2010) mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 7(5):593–605
Peng C, Chen Y, Yang Z, Zhang H, Osterby L, Rosmarin AG et al (2010) PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 115(3):626–635
Li J, Zhang J, Tang MH, Xin JP, Xu Y, Volk A et al (2016) Hematopoietic stem cell activity is regulated by Pten phosphorylation through a niche-dependent mechanism. Stem Cells 34(8):2130–2144
Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM et al (1998) Targeted disruption of SHIP leads to hemopoietic perturbations lung pathology, and a shortened life span. Genes Dev 12(11):1610–1620
Sattler M, Verma S, Byrne CH, Shrikhande G, Winkler T, Algate PA et al (1999) BCR/ABL directly inhibits expression of SHIP, an SH2-containing polyinositol-5-phosphatase involved in the regulation of hematopoiesis. Mol Cell Biol 19(11):7473–7480
Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4(4):257–262
Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ (2012) Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 11(3):415–428
Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C et al (2012) mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked Leukemogenesis. Cell Stem Cell 11(3):429–439
Wang W, Sun X, Hu T, Wang L, Dong S, Gu J et al (2018) PDK1 regulates definitive HSCs via the FOXO pathway during murine fetal liver hematopoiesis. Stem Cell Res 30:192–200
Hu T, Li C, Zhang Y, Wang L, Peng L, Cheng H et al (2015) Phosphoinositide-dependent kinase 1 regulates leukemia stem cell maintenance in MLL-AF9-induced murine acute myeloid leukemia. Biochem Biophys Res Commun 459(4):692–698
Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ (2012) Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 11(3):415–428
Guo F, Zhang S, Grogg M, Cancelas JA, Varney ME, Starczynowski DT et al (2013) Mouse gene targeting reveals an essential role of mTOR in hematopoietic stem cell engraftment and hematopoiesis. Haematologica 98(9):1353–1358
Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y et al (2008) TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205(10):2397–2408
Chen C, Liu Y, Liu Y, Zheng P (2009) mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2(98):ra75
Hoshii T, Tadokoro Y, Naka K, Ooshio T, Muraguchi T, Sugiyama N et al (2012) mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J Clin Invest 122(6):2114–2129
Carracedo A, Pandolfi PP (2008) The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27(41):5527–5541
Peng H, Kasada A, Ueno M, Hoshii T, Tadokoro Y, Nomura N et al (2018) Distinct roles of Rheb and raptor in activating mTOR complex 1 for the self-renewal of hematopoietic stem cells. Biochem Biophys Res Commun 495(1):1129–1135
Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ et al (2011) Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res 17(13):4378–4388
Ghosh J, Kapur R (2017) Role of mTORC1-S6K1 signaling pathway in regulation of hematopoietic stem cell and acute myeloid leukemia. Exp Hematol 50:13–21
Ghosh J, Kobayashi M, Ramdas B, Chatterjee A, Ma P, Mali RS et al (2016) S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J Clin Invest 126(7):2621–2625
Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S et al (2007) Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1(1):101–112
Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE et al (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128(2):325–339
Yalcin S, Marinkovic D, Mungamuri SK, Zhang X, Tong W, Sellers R et al (2010) ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3(−/−) mice. EMBO J 29(24):4118–4131
Rimmele P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN et al (2015) Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep 16(9):1164–1176
Liang R, Rimmele P, Bigarella CL, Yalcin S, Ghaffari S (2016) Evidence for AKT-independent regulation of FOXO1 and FOXO3 in haematopoietic stem and progenitor cells. Cell Cycle 15(6):861–867
Miyamoto K, Miyamoto T, Kato R, Yoshimura A, Motoyama N, Suda T (2008) FoxO3a regulates hematopoietic homeostasis through a negative feedback pathway in conditions of stress or aging. Blood 112(12):4485–4493
Santamaria CM, Chillon MC, Garcia-Sanz R, Perez C, Caballero MD, Ramos F et al (2009) High FOXO3a expression is associated with a poorer prognosis in AML with normal cytogenetics. Leuk Res 33(12):1706–1709
Kornblau SM, Singh N, Qiu YH, Chen WJ, Zhang NX, Coombes KR (2010) Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin Cancer Res 16(6):1865–1874
Chapuis N, Park S, Leotoing L, Tamburini J, Verdier F, Bardet V et al (2010) I kappa B kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood 116(20):4240–4250
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y et al (2010) TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463(7281):676–U111
Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B et al (2011) AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 146(5):697–708
Baumgartner C, Toifl S, Farlik M, Halbritter F, Scheicher R, Fischer I et al (2018) An ERK-dependent feedback mechanism prevents hematopoietic stem cell exhaustion. Cell Stem Cell 22(6):879–92 e6
Xue L, Pulikkan JA, Valk PJM, Castilla LH (2014) NrasG12D oncoprotein inhibits apoptosis of preleukemic cells expressing Cbfbeta-SMMHC via activation of MEK/ERK axis. Blood 124(3):426–436
Wang J, Kong G, Liu Y, Du J, Chang Y-I, Tey SR et al (2013) Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood 121(26):5203–5207
Nowacka JD, Baumgartner C, Pelorosso C, Roth M, Zuber J, Baccarini M (2016) MEK1 is required for the development of NRAS-driven leukemia. Oncotarget 7(49):80113–80130
Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC et al (2013) Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature 504(7478):143–147
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K et al (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423(6938):409–414
Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM et al (2008) Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2(3):274–283
Staal FJ, Famili F, Garcia Perez L, Pike-Overzet K (2016) Aberrant Wnt signaling in leukemia. Cancers (Basel) 8(9):78
Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R et al (2004) Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med 199(2):221–229
Kabiri Z, Numata A, Kawasaki A, Edison TDG, Virshup DM (2015) Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood 126(9):1086–1094
Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJ (2012) Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 26(3):414–421
Luis TC, Naber BA, Roozen PP, Brugman MH, de Haas EF, Ghazvini M et al (2011) Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 9(4):345–356
Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C (2006) Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol 7(10):1048–1056
Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG et al (2006) Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol 7(10):1037–1047
Perry JM, He XC, Sugimura R, Grindley JC, Haug JS, Ding S et al (2011) Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev 25(18):1928–1942
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL et al (2004) Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351(7):657–667
Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM et al (2007) Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 12(6):528–541
Hu Y, Chen Y, Douglas L, Li S (2009) Beta-catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 23(1):109–116
Hu J, Feng M, Liu ZL, Liu Y, Huang ZL, Li H et al (2016) Potential role of Wnt/beta-catenin signaling in blastic transformation of chronic myeloid leukemia: cross talk between beta-catenin and BCR-ABL. Tumour Biol 37:15859–15872
Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC et al (2015) Beta-catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia 29(12):2328–2337
Yu S, Li F, Xing S, Zhao T, Peng W, Xue HH (2016) Hematopoietic and leukemic stem cells have distinct dependence on Tcf1 and Lef1 transcription factors. J Biol Chem 291(21):11148–11160
Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z et al (2010) The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327(5973):1650–1653
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D et al (2010) Beta-catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18(6):606–618
Dietrich PA, Yang C, Leung HH, Lynch JR, Gonzales E, Liu B et al (2014) GPR84 sustains aberrant beta-catenin signaling in leukemic stem cells for maintenance of MLL leukemogenesis. Blood 124(22):3284–3294
Jiang X, Mak PY, Mu H, Tao W, Mak DH, Kornblau S et al (2018) Disruption of Wnt/beta-catenin exerts Antileukemia activity and synergizes with FLT3 inhibition in FLT3-mutant acute myeloid leukemia. Clin Cancer Res 24(10):2417–2429
Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organogenesis 4(2):68–75
Nemeth MJ, Topol L, Anderson SM, Yang Y, Bodine DM (2007) Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc Natl Acad Sci U S A 104(39):15436–15441
Liang H, Chen Q, Coles AH, Anderson SJ, Pihan G, Bradley A et al (2003) Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 4(5):349–360
Martin V, Valencia A, Agirre X, Cervera J, San Jose-Eneriz E, Vilas-Zornoza A, Rodriguez-Otero P, Sanz MA, Herrera C, Torres A, Prosper F, Roman-Gomez J (2010) Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci 101(2):425–32
Schreck C, Istvanffy R, Ziegenhain C, Sippenauer T, Ruf F, Henkel L et al (2017) Niche WNT5A regulates the actin cytoskeleton during regeneration of hematopoietic stem cells. J Exp Med 214(1):165–181
Chattopadhyay S, Chaklader M, Law S (2018) Aberrant Wnt signaling pathway in the hematopoietic stem/progenitor compartment in experimental leukemic animal. J Cell Commun Signal 13(1):39–52
Pajcini KV, Speck NA, Pear WS (2011) Notch signaling in mammalian hematopoietic stem cells. Leukemia 25(10):1525–1532
Varnum-Finney B, Xu L, Brashem-Stein C, Nourigat C, Flowers D, Bakkour S et al (2000) Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat Med 6(11):1278–1281
Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT (2002) Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 99(7):2369–2378
Karanu FN, Murdoch B, Gallacher L, Wu DM, Koremoto M, Sakano S et al (2000) The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J Exp Med 192(9):1365–1372
Karanu FN, Murdoch B, Miyabayashi T, Ohno M, Koremoto M, Gallacher L et al (2001) Human homologues of Delta-1 and Delta-4 function as mitogenic regulators of primitive human hematopoietic cells. Blood 97(7):1960–1967
Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C et al (2005) Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol 6(3):314–322
Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR et al (1999) Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10(5):547–558
Varnum-Finney B, Halasz LM, Sun M, Gridley T, Radtke F, Bernstein ID (2011) Notch2 governs the rate of generation of mouse long- and short-term repopulating stem cells. J Clin Invest 121(3):1207–1216
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC et al (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425(6960):841–846
Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT et al (2010) Endothelial cells are essential for the self-renewal and repopulation of notch-dependent hematopoietic stem cells. Cell Stem Cell 6(3):251–264
Lobry C, Oh P, Mansour MR, Look AT, Aifantis I (2014) Notch signaling: switching an oncogene to a tumor suppressor. Blood 123(16):2451–2459
Chen PM, Yen CC, Wang WS, Lin YJ, Chu CJ, Chiou TJ et al (2008) Down-regulation of Notch-1 expression decreases PU.1-mediated myeloid differentiation signaling in acute myeloid leukemia. Int J Oncol 32(6):1335–1341
Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S et al (2011) A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 473(7346):230–233
Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N et al (2013) Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med 210(2):301–319
Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM et al (2013) Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med 210(2):321–337
Kato T, Sakata-Yanagimoto M, Nishikii H, Ueno M, Miyake Y, Yokoyama Y et al (2015) Hes1 suppresses acute myeloid leukemia development through FLT3 repression. Leukemia 29(3):576–585
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N et al (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506(7487):240–244
Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haraguchi K et al (2010) Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood 115(14):2872–2881
Liu X, Ma Y, Li R, Guo D, Wang N, Zhao Y et al (2018) Niche TWIST1 is critical for maintaining normal hematopoiesis and impeding leukemia progression. Haematologica 103(12):1969–1979
Yin DD, Fan FY, Hu XB, Hou LH, Zhang XP, Liu L et al (2009) Notch signaling inhibits the growth of the human chronic myeloid leukemia cell line K562. Leuk Res 33(1):109–114
Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R (2015) Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 125(17):2678–2688
Vaidya A, Kale VP (2015) TGF-beta signaling and its role in the regulation of hematopoietic stem cells. Syst Synth Biol 9(1–2):1–10
Ruscetti FW, Akel S, Bartelmez SH (2005) Autocrine transforming growth factor-beta regulation of hematopoiesis: many outcomes that depend on the context. Oncogene 24(37):5751–5763
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC et al (2014) Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med 20(11):1321–1326
Karlsson G, Blank U, Moody JL, Ehinger M, Singbrant S, Deng CX et al (2007) Smad4 is critical for self-renewal of hematopoietic stem cells. J Exp Med 204(3):467–474
Sing GK, Keller JR, Ellingsworth LR, Ruscetti FW (1988) Transforming growth factor beta selectively inhibits normal and leukemic human bone marrow cell growth in vitro. Blood 72(5):1504–1511
Keller JR, McNiece IK, Sill KT, Ellingsworth LR, Quesenberry PJ, Sing GK et al (1990) Transforming growth factor beta directly regulates primitive murine hematopoietic cell proliferation. Blood 75(3):596–602
Sitnicka E, Ruscetti FW, Priestley GV, Wolf NS, Bartelmez SH (1996) Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood 88(1):82–88
Soma T, Yu JM, Dunbar CE (1996) Maintenance of murine long-term repopulating stem cells in ex vivo culture is affected by modulation of transforming growth factor-beta but not macrophage inflammatory protein-1 alpha activities. Blood 87(11):4561–4567
Fortunel N, Batard P, Hatzfeld A, Monier MN, Panterne B, Lebkowski J et al (1998) High proliferative potential-quiescent cells: a working model to study primitive quiescent hematopoietic cells. J Cell Sci 111(Pt 13):1867–1875
Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L, Nakabayashi T et al (1996) Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest 98(9):2109–2119
Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC et al (1993) Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A 90(2):770–774
Oshima M, Oshima H, Taketo MM (1996) TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol 179(1):297–302
Larsson J, Blank U, Helgadottir H, Bjornsson JM, Ehinger M, Goumans MJ et al (2003) TGF-beta signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood 102(9):3129–3135
Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H (2009) TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 113(6):1250–1256
Blank U, Karlsson G, Moody JL, Utsugisawa T, Magnusson M, Singbrant S et al (2006) Smad7 promotes self-renewal of hematopoietic stem cells. Blood 108(13):4246–4254
Scandura JM, Boccuni P, Massague J, Nimer SD (2004) Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc Natl Acad Sci U S A 101(42):15231–15236
Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y et al (2011) p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 9(3):262–271
Park SM, Deering RP, Lu Y, Tivnan P, Lianoglou S, Al-Shahrour F et al (2014) Musashi-2 controls cell fate, lineage bias, and TGF-beta signaling in HSCs. J Exp Med 211(1):71–87
Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM (2013) TGFbeta restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med 210(3):623–639
Jiang L, Han X, Wang J, Wang C, Sun X, Xie J et al (2018) SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-beta signaling. J Exp Med 215(5):1337–1347
Jonuleit T, van der Kuip H, Miething C, Michels H, Hallek M, Duyster J et al (2000) Bcr-Abl kinase down-regulates cyclin-dependent kinase inhibitor p27 in human and murine cell lines. Blood 96(5):1933–1939
Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF (2001) Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell 104(1):21–32
Passegue E, Wagner EF, Weissman IL (2004) JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119(3):431–443
Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL et al (2009) JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 15(4):341–352
Quere R, Karlsson G, Hertwig F, Rissler M, Lindqvist B, Fioretos T et al (2011) Smad4 binds Hoxa9 in the cytoplasm and protects primitive hematopoietic cells against nuclear activation by Hoxa9 and leukemia transformation. Blood 117(22):5918–5930
Smith PG, Tanaka H, Chantry A (2012) A novel co-operative mechanism linking TGFbeta and Lyn kinase activation to imatinib resistance in chronic myeloid leukaemia cells. Oncotarget 3(5):518–524
Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y et al (2013) TGF-beta-neutralizing antibody 1D11 enhances cytarabine-induced apoptosis in AML cells in the bone marrow microenvironment. PLoS One 8(6):e62785
Wu Y, Su M, Zhang S, Cheng Y, Liao XY, Lin BY et al (2017) Abnormal expression of TGF-beta type II receptor isoforms contributes to acute myeloid leukemia. Oncotarget 8(6):10037–10049
Gilliland DG (2002) Molecular genetics of human leukemias: new insights into therapy. Semin Hematol 39(4 Suppl 3):6–11
Huntly BJ, Gilliland DG (2004) Blasts from the past: new lessons in stem cell biology from chronic myelogenous leukemia. Cancer Cell 6(3):199–201
Cabezas-Wallscheid N, Eichwald V, de Graaf J, Lower M, Lehr HA, Kreft A et al (2013) Instruction of haematopoietic lineage choices, evolution of transcriptional landscapes and cancer stem cell hierarchies derived from an AML1-ETO mouse model. EMBO Mol Med 5(12):1804–1820
Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S et al (1999) The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat Genet 23(2):144–146
Chen SJ, Shen Y, Chen Z (2013) A panoramic view of acute myeloid leukemia. Nat Genet 45(6):586–587
Spangle JM, Roberts TM, Zhao JJ (2017) The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim Biophys Acta Rev Cancer 1868(1):123–131
Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK et al (2011) Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood 118(5):1264–1273
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H et al (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282(15):11221–11229
Acknowledgments
We thank all members of the Gritsman lab for helpful discussions. We apologize to any authors whose work was not cited due to space limitations. This work was supported by the NIH/NCI R01 #R01CA196973 (KG), the American Society of Hematology (KG), the V Foundation for Cancer Research (KG), the Sinsheimer Foundation (KG), the Training Program in Cellular and Molecular Biology and Genetics at Albert Einstein College of Medicine #5T32GM007491-44 (LG), and the IRACDA-BETTR Postdoctoral Training Grant at Albert Einstein College of Medicine #2K12GM102779 (KA).
Conflicts of Interest
The authors declare that no conflict of interest exists.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gurska, L.M., Ames, K., Gritsman, K. (2019). Signaling Pathways in Leukemic Stem Cells. In: Zhang, H., Li, S. (eds) Leukemia Stem Cells in Hematologic Malignancies. Advances in Experimental Medicine and Biology, vol 1143. Springer, Singapore. https://doi.org/10.1007/978-981-13-7342-8_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-7342-8_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7341-1
Online ISBN: 978-981-13-7342-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)