
Abstract
Phenylacetone monooxygenase from Thermobifida fusca (EC 1.14.13.92, PAMO) belongs to the Baeyer–Villiger family of monooxygenases and catalyzes the oxidation of various aromatic ketones to the corresponding esters using NADPH as a cofactor. In this study we analyzed the structure of the cofactor binding site, selected the most important amino acid residues for recognition of the phosphate group of the cofactor, and simulated enzyme structures with amino acid substitutions that could potentially lead to a change in the coenzyme specificity of the enzyme from NADPH to NADH. Based on the modeling, the most promising amino acid substitutions, T218D, T218E, K336A, and K336R, were proposed. Using site-directed mutagenesis we obtained genetic constructs containing genes encoding PAMOs with the corresponding amino acid substitutions, and the enzymes were expressed and purified. The resulting mutant PAMOs are able to bind NADH, but lack the ability to catalyze the oxidation of benzylacetone in the presence of NADH and show a deterioration of the Michaelis constants for NADPH. The catalytic constants of the mutant enzymes studied decrease slightly, and remain within the allowed experimental error.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Large quantities of costly reduced forms of nicotinamide cofactors NADH and NADPH are used as an energy source for biotechnological processes involving oxidoreductases. Although the only structural difference between NADPH and NADH is the phosphate group, most enzymes have a very high (often absolute) coenzyme specificity to one of them. Investigating the mechanism of such specificity is in itself an important and interesting fundamental task. However, the solution of such a problem is also of practical importance, because when considering the possibility of implementing industrial processes, NADH is the preferable form, as it is much cheaper and more stable than NADPH.
A crucial step in the implementation of chiral synthesis processes using NAD(P)+-dependent dehydrogenases was the introduction of a second reaction, in which the oxidized form of the coenzyme was again converted into the reduced one [1]. Formate dehydrogenase (FDH) from methanol utilizing microorganisms has been shown to be the optimal enzyme for NADH regeneration [2, 3]. At the same time, due to the physiological role of NADPH in in vivo biosynthesis reactions, the processes involving it were of greater interest. However, these processes were not implemented for a long time, because NADP+-specific FDH was not known at that time. In fact, the first processes of chiral synthesis using oxidoreductases and NADPH regeneration were described only in the mid-1990s [4, 5] after the first variant of mutant FDH from the bacterium Pseudomonas sp. 101 (PseFDH) with altered specificity from NAD+ to NADP+ was obtained in our laboratory [6]. Later, mutant NADP+-specific PseFDH with improved catalytic properties and stability were obtained [7–11]. These enzymes have been used in a number of chiral synthesis processes, including those catalyzed by Baeyer–Villiger monooxygenases [12]. Currently, to reduce the cost of biocatalytic synthesis processes, the main trend is to change the coenzyme specificity of practically important oxidoreductases from NADP+ to NAD+. Successful examples of such a change by directed mutagenesis can be found in the literature. For example, the introduction of two amino acid substitutions K21A/N272D made it possible to obtain an NADH-specific xylose reductase, while the wild-type enzyme uses exclusively NADPH as an electron transporter [13].
Phenylacetone monooxygenase from Thermobifida fusca (EC 1.14.13.92, PAMO) is a thermostable Baeyer–Villiger type I monooxygenase. It contains FAD in its active center and catalyzes the oxidation reactions of aromatic ketones to esters [14]. PAMO, like most Baeyer–Villiger type I monooxygenases, uses NADPH as a cofactor and shows significantly less activity with NADH. An exception to this pattern is the MekA enzyme from Pseudomonas veronii MEK700, which has comparable Michaelis constants for NADPH (29 µM) and NADH (11 µM) [15]. Work on changes in coenzyme specificity has been performed on several enzymes of this family. In the work devoted to 4-hydroxyacetophenone monooxygenase (HAPMO), it was shown that replacement of the lysine amino acid residue at the 439 position (corresponding to K336 in PAMO), which is close to the NADPH phosphate group in the coenzyme binding center, with phenylalanine and alanine residues leads to a decrease in the catalytic properties of the enzyme in reaction with NADPH, and to improvement with NADH. In addition, it was shown that any substitutions of the canonical arginine residue (R339 in HAPMO, R217 in PAMO) coordinating the phosphate group of NADPH lead to a significant deterioration of all enzyme properties [16].
Amino acid substitutions S208D/E, Q210N/S, K326H/N, K349R, L55R, S186P, T187L, and W490Y were studied [17] and resulted in improved properties of cyclohexanon monooxygenase from Acinetobacter sp. NCIMB 9871 (CHMOAcineto). Many of these residues are present in the PAMO structure and are also in close proximity to the NADPH binding center.
Analysis of the structure of the coenzyme-binding domain of the PAMO active center revealed the involvement of the T218 residue in the binding of the NADPH phosphate group. Its replacement with an alanine residue resulted in an increase in the catalytic constant of the enzyme in the reaction with NADH, but the Michaelis constant also increased [18]. In the same work, it was shown that H220N/Q substitutions led to a significant decrease in the Michaelis constant for NADH, and the catalytic constant also increased in the case of H220N change. The K336N mutation did not result in positive effects, although a similar substitution resulted in a sixfold increase in the HAPMO efficiency with NADH.
In this study, we continued the experiments to study the mechanism of coenzyme specificity of PAMO. The structure of the enzyme’s active center was analyzed and new promising substitutions were proposed based on the results of previous work. The possible effect of the selected substitutions was modelled, and genetic constructs containing genes encoding mutant PAMOs with these amino acid changes were obtained. The corresponding enzymes were obtained, purified, and studied.
EXPERIMENTAL
Computer Modelling
Analysis of the structure and modeling of mutant PAMO variants were performed with the UCSF Chimera program [19] using structures of an enzyme with a bound coenzyme (PDBID 2YLR, 2YM1, 2YM2). Side replacements of amino acid radicals were carried out using a library of various side conformations of radicals (rotamers). Rotamers were selected automatically. The main selection criteria were the following: minimization of steric stress, increase in the energy of hydrogen bonds, and compliance with conformations of side radicals from the library. To optimize the structural mutant models, the procedure of minimizing energy using the UCSF Chimera minimization routine was carried out without fixing any atoms in the system.
Site-Directed Mutagenesis
The pamo gene from T. fusca with the additional nucleotide sequence encoding 6 histidine residues at the N-terminus [20] was used as the template for obtaining genetic constructs. All constructs are based on the pET28a(+) vector. The required nucleotide substitutions were introduced with a two-step polymerase chain reaction using the primers presented in Table 1.
Polymerase chain reaction (PCR) was carried out on the T 100 Thermal Cycler amplifier (Bio-Rad). A 2.5 µL 10× PCR buffer supplied by the manufacturer together with the enzyme, 1 µL MgCl2 solution (25 mM), 2 µL dNTP solution (2.5 mM each), 1 µL solution of genomic DNA (50 ng/µL), 0.5 µL Phusion DNA polymerases (ThermoFisher Scientific, 5 U/µL), and 17 µL of deionized water was added to the primer mixture solution (20 pmol each) up to a total volume of 25 µL. PCR was performed according to the following scheme: 30 cycles (10 s at 98°С; 10 s at 72°C; 1.5 min at 72°C), then elongation for 10 min at 72°C.
The PCR products were purified by electrophoresis in 1% agarose gel and digested with restrictases NcoI and HindIII. Then the DNA was purified by electrophoresis in 1% agarose gel and ligated with the pET28a(+) plasmid treated with the same restriction endonucleases. After ligation the reaction mixture was transformed in E. coli DH5α cells and they were grown on Petri dishes with an agar medium containing kanamycin (30 μg/mL) and incubated for 16 h at 37°C. For every construct three colonies were taken from each dish. Plasmids were isolated from each colony. To control the prepared constructs, the sequencing of plasmid DNA at the Genome Shared Use Center (Engelhardt Institute of Molecular Biology, Russian Academy of Sciences) was carried out.
Expression of Mutant PAMOs in E. coli Cells
For the expression of enzymes, the plasmid DNA was transformed into E. coli BL21(DE3). Cell colonies of the producer strain were placed in 4 mL of an LB medium containing 30 μg/mL of kanamycin and cultured overnight at 37°C and 180 rpm. The cells from the overnight culture were subcultured into 200 mL of a fresh TB medium (yeast extract 24 g/L; bactotripton 24 g/L; glycerol 4 mL/L; 0.017 M KH2PO4; 0.072 M K2HPO4; pH 7.0) with a similar concentration of kanamycin before reaching the absorption A600 = 0.08–0.10. The cells were grown at 120 rpm and 37°C. Upon reaching absorption A600 = 0.8, 200 μM of IPTG was added. Expression was carried out for 24 or 7 h at 120 rpm and different temperatures (30 or 37°C). The cells were pelleted by centrifugation for 20 min at 6000 rpm and 4°C. The pellet was resuspended in a chilled buffer containing 50 mM Tris HCl, 500 mM NaCl, and 20 mM imidazole; pH 7.5.
Isolation and Purification
An aqueous solution of FAD was added to the cell suspension to a final concentration of 10 µM. Cells were sonicated during 4–5 cycles of 90 s each in ice with breaks for cooling. Next, the solution was incubated at 55°С for 10 min, followed by sedimentation of the debris in a centrifuge (40 min, 10 000 rpm, 4°C). The cell-free extract was applied to a 1-mL HisTrap HP column filled with a buffer solution containing 50 mM Tris-HCl, 500 mM NaCl, and 20 mM imidazole (pH 7.5), and washed with an increasing linear gradient of the imidazole concentration from 20 to 500 mM. Fractions, containing the target protein were transferred into a 50 mM sodium phosphate buffer solution (pH 7.5) by gel filtration through Sephadex G-25. The efficiency of the purification was monitored by protein electrophoresis in denaturing conditions.
Determination of Catalytic Parameters
The PAMO solution was incubated at 55°С for 10 min before measurements. The progress of the reaction was monitored at 30°С by the NADPH consumption, by measuring the decrease in absorption at 340 nm. The volume of the reaction medium was 1 mL. For measurements, we used a 50 mM sodium phosphate buffer (pH 7.5) containing 50 μM NADPH and 10 mM benzylacetone. For determination of the Michaelis constants, the concentrations of NADPH and benzylacetone were varied in the range of 5 to 500 μM and 0.25 to 15 mM, respectively. The KM values were calculated from the experimental dependences of the reaction rate on the concentration of the varied substrate with nonlinear regression analysis using the Origin Pro 8.5 program. The concentration of the active sites of the enzyme were determined spectrophotometrically by the absorption of FAD on 441 nm (ε = 12.4 mM–1 cm–1).
RESULTS AND DISCUSSION
Modeling Amino Acid Substitutions
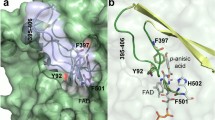
Based on the results of the published analysis and a detailed study of the phenylacetone monooxygenase active center structure, the amino acid residues, H220, T218, T194, K336, and R217, were selected based on the enzyme structures in complex with NADPH (PDBID 2YLR, PDBID 2YM2, PDBID 2YM1) available in the PDB database (Fig. 1). Substitutions for residue H220 have already been described in the literature. The R217 residue plays an important role in catalysis [21]; thus, its replacement is not appropriate. The amino acid residues T194 and S196 are farther away from the NADPH phosphate group. Therefore, the effect of amino acid substitutions of residues T218 and K336 was studied first.

The structure of the coenzyme-binding domain of the PAMO active center and the position of a number of amino acid residues in the region of phosphate group binding.
The simulations showed that in the case of the amino acid residue threonine, substitutions T218D and T218E should have the greatest effect on the coenzyme binding center (Fig. 2a). The choice of these amino acid substitutions is due to steric hindrance near the NADPH ribose phosphate group, which can prevent the binding of the phosphorylated form of the cofactor, and facilitate the NADH binding.

Structures of fragment of the phenylacetone monooxygenase active center near the NADPH molecule for the wild-type enzyme (T218, K336) and model structures for enzymes with amino acid substitutions T218E and T218D (a) and K336R and K336A (b).
Similarly, the amino acid K336R substitution was chosen based on the assumption of the emerging steric hindrance in the NADPH binding (Fig. 2b). Nevertheless, the introduced arginine residue is positively charged, as is the lysine residue. Since this could favorably influence the binding of the coenzyme phosphate group, it was interesting to obtain a variant of the enzyme with a short uncharged tail. The K336A substitution was made according to this logic.
To verify the correct choice of the key amino acid residues, we compared the results of our analysis with the prediction performed by the CSR-Salad server (Cofactor Specificity Reversal Structural Analysis & Library Design, http://www.che.caltech.edu/groups/ fha/CSRSALAD/index.html) using the PAMO PDB2YLR structure. This server also indicated that residues R217, T218, H220, and K336 are key for coenzyme specificity. The same server suggested possible substitutions. In the case of the H220 residue, no substitutions were suggested, although a positive effect of H220N and H220Q substitutions on the cationic activity of PAMO with NADH was shown in [18]. Residue R217 stands alone—the earlier experiments [20] showed that this residue cannot be mutated because it is canonical for monooxygenases.
Preparation and Characterization of Mutant Variants of PAMO
The obtained genetic constructs were expressed according to the standard technique. The phenylacetone monooxygenase expression conditions were optimized earlier in our laboratory at the stage of studying the influence of the histidine tag [13]; therefore, it was not performed in this study. The presence of the His-tag sequence at the N-terminus of all the studied PAMO variants allows us to use metal-chelate chromatography for purification. The purity of the obtained preparations was controlled by analytical gel electrophoresis under denaturing conditions. As can be seen from Fig. 3, all enzymes were obtained in a nearly homogeneous form.

Results of purification of wild-type PAMO and its mutant forms. (1) PAMO; (2) PAMO T218D; (3) PAMO T128E; (4) PAMO K336R; (5) PAMO K336A; (6–8) PAMO T218D fractions at different purification steps ((6) two hours after induction; (7) end of cultivation before affinity chromatography; (8) after affinity chromatography); M, molecular weight marker.
Table 2 shows the catalytic parameters of the obtained enzymes in comparison with the wild-type enzyme. The data in the table show that the introduction of selected amino acid substitutions leads to a slight deterioration of the Michaelis constants for NADPH, indicating that the binding of this coenzyme is impaired. When studying the conversion of benzylacetone by mutant forms of PAMO in the reaction with NADH, a decrease in the concentration of the reduced form of the coenzyme is detected, indicating the appearance of the ability to bind NADH. However, a similar rate of NADH consumption is detected at any starting concentration of benzylacetone as well as without the addition of benzylacetone to the reaction medium, indicating no conversion of benzylacetone. This idle coenzyme consumption effect is a fairly common problem for monooxygenases [21, 22].
The results of our experiments, together with the data from other authors, indicate that a dramatic change in the coenzyme specificity of PAMOs from NADP+ to NAD+ cannot be achieved by single amino acid substitutions. The problem can most likely be solved by several simultaneous substitutions. Before that it is necessary to carry out a complex simulation of the effect of several substitutions on the structure of the active center, which is planned to be done further.
The results obtained in this work with PAMOs are entirely consistent with the general pattern that changing the coenzyme specificity of the enzyme from NADP+ to NAD+ is a much more difficult problem than the reverse procedure. A striking example is the first successful paper on changing the coenzyme specificity of glutathione reductase (GTR) from NADPH to NADH [23]. The coenzyme-binding domain of this enzyme has a very high structural homology to the NAD+-specific dihydrolipoamide dehydrogenase (DHLDH). Only because of this homology, the authors were able to identify a number of key residues, which were subjected to directed mutagenesis—the third residue of alanine in the canonical sequence GxGxxG(A)—Ala179 was replaced by the Gly residue, the Arg198 residue that interacts with the 2'-phosphate coenzyme was replaced by the Met residue, and the mutation Val197Glu was introduced in the 197 residue to create hydrogen bonds between the side carboxyl group of the Glu residue and the OH-groups of ribose. However, there was no noticeable effect on improving the catalytic properties with NADH. As a result, the authors simply inserted additional parts of the amino acid sequence from the DHLDH active center into the coenzyme-binding domain of the GTR. Only after such complex (dotted and linear) substitutions it was possible to obtain a mutant enzyme that was 71.8 times more specific to NADPH than to NADPH, but its catalytic efficiency (kcat/KM) with the GTR mutant with NADH was worse by a factor of more than 30 than that of the wild-type enzyme with NADPH.
REFERENCES
Wichmann, R., Wandrey, C., Buckmann, A.F., and Kula, M.R., Biotechnol. Bioeng., 1982, vol. 23, no. 12, p. 2789.
van der Donk, W.A. and Zhao, H., Curr. Opin. Biotechnol., 2003, vol. 14, no. 4, p. 421.
Tishkov, V.I., Vestn. Mosk. Univ., Ser. 2: Khim., 2002, vol. 43. no. 6, p. 380.
Seelbach, K., Riebel, B., Hummel, W., Kula, M.R., Tishkov, V.I., Egorov, A.M., Wandrey, C., and Kragl, U., Tetrahedron Lett., 1996, vol. 37, no. 9, p. 1377.
Rissom, S., Schwarz-Linek, U., Vogel, M., Tishkov, V.I., and Kragl, U., Tetrahedron: Asymmetry, 1997, vol. 8, no. 15, p. 2523.
Tishkov, V.I., Galkin, A.G., and Egorov, A.M., Proc. Int. Conf. “Enzyme Engineering XII,” Deu Ville, France, 1993.
Tishkov, V.I., Galkin, A.G., Fedorchuk, V.V., Savitsky, P.A., Rojkova, A.M., Gieren, H., and Kula, M., Biotechnol. Bioeng., 1999, vol. 64, no. 2, p. 187.
Serov, A.E., Popova, A.S., Fedorchuk, V.V., and Tishkov, V.I., Biochem. J., 2002, vol. 367, p. 841.
Tishkov, V.I. and Popov, V.O., Biomol. Eng., 2006, vol. 23, nos. 2–3, p. 89.
Alekseeva, A.A., Fedorchuk, V.V., Zarubina, S.A., Sadykhov, E.G., Matorin, A.D., Savin, S.S., and Tishkov, V.I., Acta Nat., 2015, vol. 7, no. 1, p. 60.
Pometun, A.A., Parshin, P.D., Galanicheva, N.P., Uporov, I.V., Atroshenko, D.L., Savin, S.S., and Tishkov, V.I., Moscow Univ. Chem. Bull., 2020, vol. 75, p. 250.
Schwarz-Linek, U., Krödel, A., Ludwig, F.-A., Schulze, A., Rissom, S., Kragl, U., Tishkov, V.I., and Vogel, M., Synthesis, 2001, vol. 33, no. 6, p. 947.
Zeng, Q.K., Du, H.L., Wang, J.F., Wei, D.Q., Wang, X.N., Li, Y.X., and Lin, Y., Biotechnol. Lett., 2009, vol. 31, no. 7, p. 1025.
Fraaije, M.W., Wu, J., Heuts, D.P.H.M., van Hellemond, E.W., Spelberg, J.H.L., and Janssen, D.B., Appl. Microbiol. Biotechnol., 2005, vol. 66, no. 4, p. 393.
Völker, A., Kirschner, A., Bornscheuer, U.T., and Altenbuchner, J., Appl. Microbiol. Biotechnol., 2008, vol. 77, no. 6, p. 1251–1260.
Kamerbeek, N.M., Fraaije, M.W., and Janssen, D.B., Eur. J. Biochem., 2004, vol. 271, no. 11, p. 2107.
Beier, A., Bordewick, S., Maika, G., Schmidt, S., Bergh, T., van den Peters, C., Joosten, H., and Bornscheuer, U.T., ChemBioChem, 2016, vol. 17, no. 24, p. 2312.
Dudek, H.M., Torres Pazmiño, D.E., Rodríguez, C., de Gonzalo, G., Gotor, V., and Fraaije, M.W., Appl. Microbiol. Biotechnol., 2010, vol. 88, no. 5, p. 1135.
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., and Ferrin, T.E., J. Comput. Chem., 2004, vol. 25, no. 13, p. 1605.
Parshin, P.D., Pometun, A.A., Martysuk, U.A., Kleymenov, S.Y., Atroshenko, D.L., Pometun, E.V., Savin, S.S., and Tishkov, V.I., Biochemistry (Moscow), 2020, vol. 85, no. 5, p. 575.
Balke, K., Beier, A., and Bornscheuer, U.T., Biotechnol. Adv., 2018, vol. 36, p. 247.
Kokorin, A., Parshin, P.D., Bakkes, P.J., Pometun, A.A., Tishkov, V.I., and Urlacher, V.B., Sci. Rep., 2021, vol. 11, p. 21706.
Scrutton, N.S., Berry, A., and Perham, R.N., Nature, 1990, vol. 34, p. 38.
ACKNOWLEDGMENTS
The equipment of the Center for Collective Use “Genom” of the Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, and the Center for Collective Use “Industrial Biotechnologies” of the Federal Research Center “Fundamentals of Biotechnology” of the Russian Academy of Sciences” was used.
Funding
This study was partially supported by the Russian Foundation for Basic Research (project no. 20-34-90120).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Abbreviation: FDH, formate dehydrogenaseа; PAMO, phenylacetone monooxygenase from Thermobifida fusca.
About this article

Cite this article
Parshin, P.D., Martysuk, U.A., Atroshenko, D.L. et al. Study of the Mechanism of Coenzyme Specificity of Phenylacetone Monooxygenase from Thermobifida fusca by Site-Directed Mutagenesis. Moscow Univ. Chem. Bull. 77, 242–248 (2022). https://doi.org/10.3103/S0027131422050091
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0027131422050091