
Abstract
A major problem when xylose is used for ethanol production is the intercellular redox imbalance arising from different coenzyme specificities of xylose reductase (XR) and xylitol dehydrogenase. The residue Lys21 in XR from Pichia stipitis was subjected to site-directed mutagenesis to alter its coenzyme specificity. The N272D mutant exhibited improved catalytic efficiency when NADH was the coenzyme. Both K21A and K21A/N272D preferred NADH to NADPH, their catalytic efficiencies for NADPH were almost zero. The catalytic efficiency of K21A/N272D for NADH was almost 9-fold and 2-fold that of K21A and the wild-type enzyme, respectively. Complete reversal of coenzyme specificity toward NADH and improved catalytic efficiency were achieved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most important functions of biomass energy systems is to produce ethanol by fermentation of a biomass such as lignocellulose, which is abundant and inexpensive (Lin and Tanaka 2006). D-Xylose is the main constituent of the lignocellulose hydrolyzate. Unfortunately, Saccharomyces cerevisiae, a prominent ethanol-producing organism, is unable to utilize d-xylose. Therefore, processes that efficiently utilize the xylose component of lignocellulose could significantly decrease the cost of bioethanol production (Nigam 2001).
S. cerevisiae can grow on xylulose, an isomer of xylose that is produced by two key enzymes: xylose reductase (XR; EC 1.1.1.21) and xylitol dehydrogenase (XDH; EC 1.1.1.9). Therefore, the cloning of XR and XDH into S. cerevisiae would allow xylose to be used as a substrate (Katahira et al. 2006; Hahn-Hagerdal et al. 2007). However, one of the key problems in utilizing xylose for ethanol production is the impairment of the redox balance that arises from the different coenzyme specificities of XR (NADPH) and XDH (NADH) in the xylose metabolic pathway (Jeppsson et al. 2003). Generation of an NADH-dependent XR by protein engineering could resolve this problem. Decreasing the NADPH-preferring activity of Pichia stipitis XR (PsXR) might therefore increase ethanol production in S. cerevisiae (Watanabe et al. 2007b).
Efforts have been made to alter the coenzyme specificity of XR by site-directed mutagenesis, but there has been limited success so far (Petschacher et al. 2005; Klimacek et al. 2001; Jeppsson et al. 2006; Jeong et al. 2001; Chu and Lee 2006). The main reason is that there is no experimental data on the structure of XR. In our previous study, we determined the three-dimensional (3D) structures of PsXR and some key amino acids that affect the coenzyme preference of PsXR (Wang et al. 2007). In the present study, Lys21, which is the only amino acid in PsXR that hydrogen bonds with NADPH but not with NADH in the binding pocket (Wang et al. 2007), was specifically targeted for site-directed mutagenesis in order to alter the coenzyme specificity of PsXR from NADPH and NADH to NADH alone. Analysis of the data of Petschacher et al. (2005) prompted us to create the N272D mutant in order to improve the catalytic efficiency toward NADH.
Materials and methods
Simulations for rational site-directed mutagenesis by amino acid substitution
Lys21 in xylose reductase from Pichia stipitis was specifically targeted for site-directed mutagenesis. It was substituted by each of the 19 other protein amino acids so that 19 PsXR mutants were produced. The 3D structure modeling, ligand docking, and molecular dynamics (MD) simulations for each mutant were carried out based on previously reported methods (Wang et al. 2007).
Cloning of the PsXR gene
Genomic DNA was extracted from the P. stipitis strain (JCM 10742), which was purchased from RIKEN Company, Japan. Two primers (forward: 5′-ACTGCTAGCCCTTCTATTAAGTTGAACTCTG-3′ and reverse: 5′-GATAAGCTTTTAGACGAAGATAGGAATCTTG-3′) were designed based on the published sequence of PsXR (GenBank accession number: X59465). The polymerase chain reaction (PCR) was performed in a final volume of 50 μl with 30 cycles at 94°C for 30 s, 57°C for 30 s, and 68°C for 120 s. The PCR product was purified, digested, and inserted into the pET-28b(+) plasmid. E. coli BL21 (DE3) cells were used throughout for the cloning and expression of PsXR.
Site-directed mutagenesis
The mutants were constructed by PCR-based site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The primers for mutagenesis are described in Supplementary Table 1. Each mutant was verified by DNA sequencing (Sangon Biotechnology Company, Shanghai, China) and amino acid sequence alignment.
Expression and purification of PsXRs
E. coli BL21 (DE3) cells harboring the expression plasmid with His-tagged wild-type (WT) or mutated PsXRs were grown overnight at 37°C in LB medium supplemented with 50 μg kanamycin/ml. Saturated overnight cultures were diluted 100-fold into fresh LB media containing 50 μg kanamycin/ml and grown at 37°C for 2 h. After adding 1 mM IPTG, the cultures were grown at 25°C for 4 h to induce the expression of PsXR. These were subsequently stored overnight at 4°C. The cells were then harvested by centrifugation and suspended in 50 ml Buffer A (20 mM sodium phosphate (pH 7.4) containing 500 mM NaCl and 20 mM imidazole) and subjected to 99 pulses of sonication. The supernatant was centrifuged again and purified on an ÄKTA purification system using a Ni2+-charged HisTrap FF crude column. Enzymes were eluted with Buffer B (Buffer A plus 500 mM imidazole). Purified enzymes were stored at −20°C in 15% (v/v) glycerol until further use. SDS-PAGE was carried out on 10% (v/v) slab gels.
PsXR enzyme activity assays
PsXR activity was determined by measuring the oxidation of NAD(P)H at 340 nm and 35°C. The standard assay mixture contained 80 mM xylose and 100 mM NAD(P)H in 50 mM sodium phosphate buffer (pH 6.5). All reactions were initiated by adding 100 μl enzyme to the standard assay mixture in a final volume of 3 ml. Enzyme units were defined as the decrease in the number of μmol of NAD(P)H per min. The kinetic parameters K m and k cat were calculated from the Lineweaver-Burk plot. Protein concentrations were estimated by measuring the absorbance at 280 nm.
Results and discussion
Rational design to reverse coenzyme specificity
The potential energy of each mutant with NAD or NADP was calculated by the formula E = E str + E ang + E stb + E tor + E oop +E ele + E vdw + E sol , in which str represents bond stretch energies; ang, angle bend energies; stb, stretch-bend cross term energies; tor, dihedral rotation energies; oop, out-of-plane energies; ele, electrostatic interactions; vdw, van der Waals interactions; and sol, implicit solvent electrostatic correction. Amber force field parameters were used for all computations.
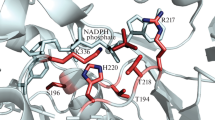
The potential energy of the K21A mutant with NAD (10897.6091 kcal/mol) was slightly different from that of the WT enzyme (10906.6419 kcal/mol). Lys21 has strong hydrogen bond interactions with NADP but not with NAD in the binding pocket (Wang et al. 2007). The interaction of PsXR with NADP can be weakened by mutating Lys21 to Ala21, which has little influence on the interaction of PsXR with NAD, as shown in Fig. 1.

An illustration of the interactions between Lys21 in wild-type Pichia stipitis xylose reductase and NAD (a) or NADP (b). The interactions between Ala21 in the K21A mutant and NAD or NADP are also shown in (c) and (d), respectively. Lys21 has strong hydrogen bond interactions with NADP (b) but has no such interactions with NAD (a). Ala21 has no hydrogen bond interactions with NAD (c) or NADP (d)
Expression and purification of PsXRs
All mutant PsXRs had His6 tags at their N terminals. They were expressed and purified, and SDS-PAGE analysis revealed that the molecular mass of the purified PsXRs was approximately 36,000 Da, which is consistent with that of the wild-type enzyme (Zeng et al. 2008).
Characterization of PsXRs
The specific activities and kinetic parameters of all PsXRs are summarized in Table 1. The specific activities and catalytic efficiencies of K21A and K21A/N272D could not be determined in the presence of NADPH; however, both values could be estimated in the presence of NADH, suggesting that complete reversal of PsXR coenzyme specificity was achieved. These results are in good agreement with the theoretical predictions made on the basis of the simulation studies. The catalytic efficiency of the K21A/N272D mutant for NADH was almost 9-fold that of the K21A mutant and 2-fold that of the WT enzyme. These data suggest that the N272D mutation could improve the catalytic efficiency of XR for NADH, which is consistent with our assumptions made on the basis of previously published data (Petschacher et al. 2005).
Two new NADH-dependent XR mutants, i.e., K21A and K21A/N272D, are reported in this study. Complete reversal of the PsXR coenzyme specificity with improved catalytic efficiency was achieved by rational site-directed mutagenesis. Considering that the redox imbalance caused by the different coenzyme specificities of XR and XDH results in low ethanol yields, attempts have been made to increase the affinity of XDH for NADP. A PsXDH mutant with completely reversed coenzyme specificity toward NADP was generated (Watanabe et al. 2005) and introduced into S. cerevisiae together with WT PsXR. It resulted in a 41% increase in ethanol production in comparison with the reference strain expressing WT PsXDH and WT PsXR (Watanabe et al. 2007a). PsXR mutants with higher affinity for NADH than NADPH have also been introduced in S. cerevisiae. Increased ethanol yield was reported in recombinant S. cerevisiae that harbored mutated K270 M PsXR and WT PsXDH (Jeppsson et al. 2006). Moreover, recombinant S. cerevisiae with mutated K270R PsXR showed a 5.1% increase in ethanol production in comparison with the control expressing WT XR (Watanabe et al. 2007b). Since the K21A/N272D mutant exhibited complete reversal of coenzyme specificity and improved catalytic efficiency, it is expected that the efficiency of ethanol fermentation would be improved by expressing the K21A/N272D mutant and WT PsXDH in S. cerevisiae.
References
Chu BCH, Lee H (2006) Investigation of the role of a conserved glycine motif in the Saccharomyces cerevisiae xylose reductase. Curr Microbiol 53:118–123
Hahn-Hagerdal B, Karhumaa K, Fonseca C et al (2007) Towards industrial pentose-fermenting yeast strains. Appl Microbiol Biotechnol 74:937–953
Jeong EY, Sopher C, Kim IS et al (2001) Mutational study of the role of tyrosine-49 in the Saccharomyces cerevisiae xylose reductase. Yeast 18:1081–1089
Jeppsson M, Traff K, Johansson B et al (2003) Effect of enhanced xylose reductase activity on xylose consumption and product distribution in xylose-fermenting recombinant Saccharomyces cerevisiae. FEMS Yeast Res 3:167–175
Jeppsson M, Bengtsson O, Franke K et al (2006) The expression of a Pichia stipitis xylose reductase mutant with higher Km for NADPH increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Biotechnol Bioeng 93:665–673
Katahira S, Mizuike A, Fukuda H et al (2006) Ethanol fermentation from lignocellulosic hydrolysate by a recombinant xylose- and cellooligosaccharide-assimilating yeast strain. Appl Microbiol Biotechnol 72:1136–1143
Klimacek M, Szekely M, Griessler R et al (2001) Exploring the active site of yeast xylose reductase by site-directed mutagenesis of sequence motifs characteristic of two dehydrogenase/reductase family types. FEBS Lett 500:149–152
Lin Y, Tanaka S (2006) Ethanol fermentation from biomass resources: current state and prospects. Appl Microbiol Biotechnol 69:627–642
Nigam JN (2001) Ethanol production from wheat straw hemicellulose hydrolysate by Pichia stipitis. J Biotech 87:17–27
Petschacher B, Leitgeb S, Kavanagh KL et al (2005) The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem J 385:75–83
Wang JF, Wei DQ, Lin Y et al (2007) Insights from modeling the 3D structure of NAD(P)H-dependent D-xylose reductase of Pichia stipitis and its binding interactions with NAD and NADP. Biochem Biophys Res Commun 359:323–329
Watanabe S, Kodaki T, Makino K (2005) Complete reversal of coenzyme specificity of xylitol dehydrogenase and increase of thermostability by the introduction of structural zinc. J Biol Chem 280:10340–10349
Watanabe S, Abu Saleh A, Pack SP et al (2007a) Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein engineered NADP(+)-dependent xylitol dehydrogenase. J Biotech 130:316–319
Watanabe S, Pack SP, Abu Saleh A et al (2007b) The positive effect of the decreased NADPH-preferring activity of xylose reductase from Pichia stipitis on ethanol production using xylose-fermenting recombinant Saccharomyces cerevisiae. Biosci Biotechnol Biochem 71:1365–1369
Zeng QK, Du HL, Zhai ZC et al (2008) Mutational research on the role of lysine 21 in the Pichia stipitis xylose reductase. Chin J Biotech 24:1108–1111
Acknowledgements
This study was supported by grants from the National 863 Bioinformatics projects under contract no. 2007AA02Z333, the Chinese National Science Foundation under contract nos. 20773085 and 30870476, and the Guangdong Natural Science Foundation under contract no. 06300199. It was also supported by the Virtual Laboratory for Computational Chemistry of CNIC and the Supercomputing Center of CNIC, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Qi-Kai Zeng, Hong-Li Du, Jing-Fang Wang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zeng, QK., Du, HL., Wang, JF. et al. Reversal of coenzyme specificity and improvement of catalytic efficiency of Pichia stipitis xylose reductase by rational site-directed mutagenesis. Biotechnol Lett 31, 1025–1029 (2009). https://doi.org/10.1007/s10529-009-9980-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-009-9980-x