
Abstract
For the investigation of the NADPH-dependent Baeyer-Villiger monooxygenase MekA from Pseudomonas veronii MEK700, the encoding gene mekA with a C-terminal strep-tag was cloned and expressed under the control of a l-rhamnose inducible promoter from Escherichia coli. The mekA gene was found by analyzing the methylethylketone (MEK) degradation pathway by Onaca et al. J Bacteriol 189:3759–3767, 2007. Sequence analysis of the corresponding protein, which catalyzes the Baeyer-Villiger oxidation of MEK to ethyl acetate, showed two binding sites (Rossman-fold motifs) for cofactors NAD(P)H and FAD. Although expression of mekA resulted in large amounts of inclusion bodies compared to soluble protein, high amounts of purified and active MekA were obtained by affinity chromatography. The substrate spectrum of MekA was investigated with purified enzyme and whole cells using a variety of aliphatic, aromatic, and cyclic ketones including four chiral substrates. The specific activity of MekA with MEK as substrate was determined to be 1.1 U/mg protein. K M values were determined for MEK and the cofactors NADPH and NADH to be 6, 11, and 29 μM, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Baeyer-Villiger monooxygenases (BVMOs) belong to an interesting enzyme family of flavin-dependent monooxygenases, which are able to catalyze oxidations of ketones to their corresponding esters by introducing one oxygen atom next to a keto group (Mihovilovic et al. 2006). BVMOs are divided into two main groups according to their coenzyme usage: type I BVMOs use NADPH and FAD, whereas type II BVMOs need NADH and FMN, which is indicated by two dinucleotide-binding sites as typical features. Both BVMO types also differ in their subunit organization. Type I BVMOs consist of monomers, homodimers, or homotetramers, whereas type II BVMOs consist of two subunits in α2β organization (van der Werf 2000). Most BVMOs belong to type I; only two type II BVMOs have been purified and characterized from strain Pseudomonas putida PpG1 (Jones et al. 1993).
A further significant BVMO-identifying sequence motif was characterized by studying NADPH-dependent 4-hydroxyacetophenone monooxygenase (HAPMO) from Pseudomonas fluorescens ACB in detail (Fraaije et al. 2002). In addition to BVMOs, also mechanistically related flavin-containing monooxygenases (FMOs) such as p-hydroxybenzoate hydroxylase and N-hydroxylating monooxygenases (NMOs) belong to the same enzyme family, but both of them only contain one dinucleotide-binding site (Fraaije et al. 2002).
Some of the BVMOs have been investigated for their enantio- and regioselectivity, which is important for industrial application of BVMOs in organic synthesis. An example is the type I HAPMO, which was isolated from P. fluorescens ACB growing on 4-hydroxyacetophenone (Higson and Focht 1990). It converts a broad range of acetophenones by Baeyer-Villiger oxidation to the corresponding phenyl acetates, but not cyclopentanone and cyclohexanone. The activity of the purified enzyme was determined with 5.5 U/mg with 4-hydroxyacetophenone as substrate. Enantioselectivity has been investigated with various aromatic ketones, aldehydes, and sulfides showing high selectivity in the asymmetric conversion of sulfides, but only moderate selectivity in the formation of regioisomeric lactones (Kamerbeek et al. 2001, 2003a, b and 2004).
A second HAPMO from Pseudomonas putida JD1 was described by Tanner and Hopper (2000). This enzyme seems to be involved in the catabolism of 4-ethylphenol. One of the best characterized BVMOs is cyclohexanone monooxygenase (CHMO) from Acinetobacter sp. NCIMB 9871. CHMO was screened by random mutagenesis for its substrate acceptance, stereopreference, and catalytic mechanism. Wild-type CHMO and its mutants convert a broad variety of substituted cycloketones to their corresponding lactones with more or less significant changes in stereoselectivity (Mihovilovic et al. 2006). All newly identified CHMOs from other microorganisms showed significant sequence identity with CHMO from Acinetobacter sp. NCIMB 9871.
Two CHMOs were discovered in cyclohexanone-induced Brevibacterium sp. strain HCU1, and the recombinant enzymes were purified. Both monooxygenases convert a broad variety of substituted cyclic ketones into the corresponding lactones (Brzostowicz et al. 2000). Enantioselectivity was also tested for NADPH-dependent phenylacetone monooxygenase (PAMO) from Thermobifida fusca. The enzyme is thermostable with highest activity at 70°C and can also perform sulfide oxidations of substrates like 4-tolylsulfide and ethionamide, which provides the ability to produce enantiomerically pure and biologically active compounds (Fraaije et al. 2005). Another BVMO from Pseudomonas fluorescens DSM 50106 has been characterized by Kirschner et al. (2007). The gene is part of an operon containing in addition adhF1, an alcohol dehydrogenase, and estF1, a lactone specific esterase (Khalameyzer et al. 1999). Solubility of this recombinant BVMO was improved by coexpressing a variety of chaperones.
It could be shown that with whole cells a range of short-chain aliphatic ketones and cyclopentanone are converted. Geitner et al. (2007) compared BVMO from P. fluorescens DSM 50106 with CHMO from Acinetobacter sp. NCIMB 9871, cyclopentanone monooxygenase from Comamonas sp. NCIMB 9872 and a BVMO from Pseudomonas putida KT2440 for their enantioselectivity in the conversion of 3-phenyl-2-ketones. Whole-cell biocatalysts with the BVMO from P. putida KT2440 have been observed to be (R)-selective while all other BVMOs have been found to be (S)-selective.
Because of the low stability and solubility of some wild-type and recombinant BVMOs biotransformations with whole cells are often preferred. Saccharomyces cerevisiae and E. coli have been used to overexpress CHMO and to produce a wide range of optically pure δ- and ɛ-lactones (Chen et al. 1999). Also Kirschner et al. (2007) used whole-cell biocatalysis to determine the substrate acceptance of recombinant P. fluorescens DSM 50106 BVMO in E. coli.
The mekA gene was found by investigating the degradation of methylketones by P. veronii MEK700, which was isolated from a biotrickling filter cleaning waste air (Onaca et al. 2007). It forms an operon with mekB, which has strong esterase activity and high similarity to a homoserine acetyltransferase. Both genes are positively regulated by the transcriptional regulator MekR. So far, only mekB/MekB was studied in detail. The mekB gene was functionally expressed and MekB was purified.
The enzyme hydrolyzes a variety of aliphatic and aromatic esters such as ethyl acetate or p-nitrophenyl acetate, which could be products from a Baeyer-Villiger oxidation by MekA—a protein of 549 amino acids and two typical dinucleotide binding motifs. MekA shares 27% amino acid sequence identity with HAPMO and 28% identity with BVMO from P. fluorescens DSM 50106, respectively. MekA has not yet been studied because of accumulating inclusion bodies during heterologous expression (Onaca et al. 2007). The sequence of the MekA protein presents the two dinucleotide-binding motifs (GXGXXG) for its cofactors NADPH and FAD and also possesses the highly conserved type I BVMO-identifying sequence motif FXGXXXHXXXW(P/D), which was identified by Fraaije et al. (2002). This motif comprises amino acids 170 to 180 (FQGQIYHTGLW) of MekA directly ahead of the second dinucleotide-binding site. In this study, we present the results of heterologous expression, purification, and functional characterization of P. veronii MEK700 Baeyer-Villiger monooxygenase MekA and demonstrate its use in whole-cell biocatalysis.
Materials and methods
Enzymes, chemicals, and media
High-fidelity polymerase was obtained from Fermentas, restriction enzymes from New England Biolabs or Roche, and NADPH and NADH from Biomol. For SDS-PAGEs, the standard protein marker from ROTH (Germany) was used as well as dNTPs for polymerase chain reaction (PCR) and l-rhamnose as inducer for gene expression. All other chemicals were obtained from Sigma Aldrich and Fluka (Germany). For DNA purification from PCR and agarose gels, the GE DNA Purification Kit (General Electrics Healthcare) was used. Plasmid DNA was isolated with the Qiagen Miniprep Kit.
For purification of recombinant MekA streptactin-sepharose columns from IBA were used. For cultivation of bacteria Luria Bertani (LB) medium pH 7.2 was used (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl). If necessary, LB medium was supplemented with 100 μg/ml ampicillin.
Amplification and cloning
PCR for amplification of mekA was done with chromosomal DNA from P. veronii MEK700 using oligonucleotides s4211 (5′-AAA AAA CAT ATG AGT GCT CAA TCT AAG C-3′) and s4725 (5′-AAA AAA GGA TCC AGC CAT TTC AAA GCC TGG-3′) with sites for restriction enzymes NdeI and BamHI, respectively. After initial denaturation for 5 min at 94°C, the cycling program was as follows for 30 cycles: 1 min 94°C denaturation, 1 min 50°C annealing and 1:30 min 72°C elongation. A final elongation step was performed over 10 min at 72°C. The resulting 1,668 bp DNA fragment was digested with NdeI and BamHI and ligated into pJOE4042 digested with the same enzymes. The resulting plasmid with a C-terminal strep-tag fusion of mekA was called pAM262 (see Fig. 1).

Vector pAM262 for expression of recombinant mekA from P. veronii MEK700 under control of rhaP in E. coli JM109 and E. coli BL21. The mekA gene was introduced without stop codon using the sites of restriction endonucleases NdeI and BamHI for cloning
The vector pJOE4042 originally is a pJOE3075 derivative with the cer-site of the ColE1 plasmid of E. coli for stabilizing the plasmid in Rec + strains, l-rhamnose-inducible promoter rhaP followed by lacPOZ with C-terminal strep-tag fusion and the beta-lactamase gene bla for ampicillin resistance (Stumpp et al. 2000). The C-terminal his-tag of pJOE3075 was substituted by strep-tag using the restriction endonucleases HindIII and BamHI.
Bacterial strains and culture conditions
Transformation of E. coli strains JM109 (laboratory strain, genotype: recA supE44 endA1 hsdR17 gyrA96 relA thi Δ(lac-proAB) F’[traD36 proAB+lacIqlacZΔM15]) and BL21 (Novagen, genotype: F−ompT hsdSB [\({\text{r}}^{ - }_{{\text{B}}} {\text{m}}^{ - }_{{\text{B}}} \)] gal dcm) with pAM262 was done by the heat shock method as described by Chung et al. (1989). Expression of recombinant mekA in E. coli JM109 pAM262 was carried out by incubating the culture 2 h at 37°C, then adding l-rhamnose (final concentration 0.2%) and shifting the culture to 28°C for another 8 h of cultivation. Expression of recombinant mekA in E. coli BL21 pAM262 was performed by incubating the culture 2 h at 37°C, then adding l-rhamnose (final concentration 0.2%) and shifting the culture to 22°C for another 16 h of cultivation.
Gene expression analysis and activity assays
Gene expression analysis was performed with cell crude extract. Cells were harvested from cultures in aliquots of 5 × 109 cells and resuspended in 500 μl Tris/HCl (0.1 M, pH 8). Cell disruption was performed by ultrasonification (Ultrasonic Sonicator, 2 × 30 s) in iced water. To separate soluble and insoluble protein fractions, the crude extract was centrifuged at 16,200×g for 15 min at 4°C. The supernatant was transferred to a new tube and the pellet (insoluble fraction) was resuspended in 500 μl Tris/HCl (0.1 M, pH 8). For SDS-PAGE, a 12-μl protein solution was used. SDS-PAGE was carried out on 12.5% gels. Proteins were stained with a Coomassie R250/G250 solution.
For enzyme activity measurements, substrates were used in defined concentrations in Tris/HCl (0.1 M, pH 9). As standard substrate methylethylketone (MEK) was used, the others are listed in Table 4. Substrates were used as provided by manufacturers. The cofactor NADPH (ɛ = 6.3 ml μmol−1 cm−1) was used at a final concentration of 0.3 mM. The reaction was started by the addition of 30-μl cell crude extract and carried out at 30°C.
The kinetics were measured at 340 nm by spectrophotometry. Protein content was determined by Bradford assay using BSA as standard (Bradford 1976). Specific activity is given in unit per milligram (U/mg) protein. One unit is defined as the amount of enzyme that catalyzes the oxidation of 1 μmol NADPH per minute at 30°C.
Enzyme purification
Recombinant MekA was purified by affinity chromatography via C-terminal strep-tag. First, a column with streptactin-sepharose (IBA, filling volume 2.5 ml streptactin-sepharose of a 50% suspension) was equilibrated by gravity flow five times with 1-ml 150 mM NaCl in Tris/HCl (0.1 M, pH 8). Because of its low solubility after expression about 2 g (wet weight, corresponding to 740 OD600) of induced cells were used for purification. Cell disruption by ultrasonification was carried out in 8 ml Tris/HCl buffer (0.1 M, pH 8). After centrifugation at 4°C in a Sorvall SS34 rotor at 20,000 rpm for 15 min, the supernatant with recombinant MekA was added to the column.
After passing through of the crude extract, the column has been washed five times with 1-ml 150 mM NaCl in Tris/HCl (0.1 M, pH 8) to remove unattached protein. Elution was carried out by adding six times 800 μl 2.5 mM desthiobiotin and 150 mM NaCl in Tris/HCl (0.1 M, pH 8). Wash and elution fractions have been collected to control purity by SDS-PAGE. To remove desthiobiotin and other possible interfering compounds of the eluate, the yellow fractions were ultrafiltrated via a Centricon column (Millipore, Ultracel® YM-10) by centrifugation for 2.5 h at 4,300×g and 4°C until a final volume of 50–70 μl was left over the membrane. Elution was then carried out in a final volume of 800 μl using potassium phosphate buffer (0.1 M, pH 8).
Biocatalytic reactions and GC analysis
For biocatalysis streptactin-purified MekA and whole cells of E. coli BL21 pAM262 were used. Reactions were carried out in Tris/HCl (0.1 M, pH 9) with different substrates in concentrations listed in Tables 5 and 6. Purified MekA has been used in concentrations of 20–23 μg/ml per reaction. Cells were used in concentrations of 45 OD600/ml. For GC analysis of biocatalytic reactions, the samples were intensively mixed with 500 μl ethyl acetate for extraction of substrates and products.
After a short centrifugation step (1 min, table centrifuge, room temperature, 16,100×g) the organic phase was transferred to a new tube and anhydrous sodium sulfate was added to bind residual water. After another centrifugation step the ethyl acetate phase was analyzed by GC. For chiral substrates extraction was done twice. Samples were concentrated by a vacuum centrifuge. The optical purity of remaining substrate (%eeS) and product (%eeP), conversion and enantioselectivity (E value) were determined by GC analysis as described by Kirschner and Bornscheuer (2006) and Geitner et al. (2007). For GC programs and measurement conditions, see Tables 1 and 2.
Results
Expression of mekA in E. coli JM109 pAM262 and E. coli BL21 pAM262
The mekA gene of P. veronii MEK700 was isolated by Onaca et al. (2007) and expressed under control of the E. colil-rhamnose inducible promoter rhaP BAD at 30°C using E. coli JM109 pJOE5302.3. Expression led to nearly 100% inclusion bodies. For our study, we newly amplified the mekA gene to fuse it C-terminally with a strep-tag. With the new plasmid pAM262, it was possible to purify MekA by affinity chromatography. Expression of recombinant mekA under the l-rhamnose-inducible rhaP BAD-promoter in E. coli JM109 pAM262 at an induction temperature of 28 and 30°C, respectively, also led to the accumulation of large amounts of inclusion bodies (>95%) compared to the fraction with soluble MekA (data not shown). Thus, specific activity of the soluble fraction of the induced sample toward MEK was quite low, with about 0.009 U/mg compared to 0.005 U/mg of the uninduced control sample.
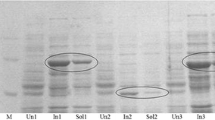
The addition of FAD to the activity assay reaction mix did not increase enzyme activity, which means that the enzyme was either saturated with FAD, or FAD could not bind to the already folded protein. Because of the low formation of active MekA in E. coli JM109 pAM262, the expression of mekA was carried out in E. coli BL21 pAM262 at an induction temperature of 22°C for 16 h. This resulted in less inclusion bodies, formation of more soluble MekA as shown by a sharp protein band at 60 kDa on lane 1 in Fig. 2a, and some increase in the specific activity of the crude cell extract to 0.012 U/mg. The typical SDS polyacrylamide gel for mekA expression by induced and noninduced cells of E. coli BL21 pAM262 is presented in Fig. 2a showing soluble and insoluble cell crude extracts.

a Comparison of crude cell extracts of induced (+) and noninduced (−) cells of E. coli BL21 pAM262. Induction was carried out with 0.2% l-rhamnose for 16 h at 22°C. MekA protein was running in the range of 60 kDa and is indicated by an arrow. Lane description: (M) protein standard marker (Roth, Germany); (1) soluble protein fraction of induced sample; (2) pellet fraction of induced sample; (3) soluble protein fraction of noninduced control sample; (4) pellet fraction of control sample. (b) Purification of recombinant MekA via a streptactin column from IBA, Germany as described. The SDS-PAGE presents the elution fractions 1 to 5. Fractions 3 and 4 have been yellow colored and showed BVMO activity with MEK as substrate
Purification of MekA
Recombinant MekA could be partially purified via streptactin-sepharose from E. coli BL21 pAM262 cells. A representative SDS polyacrylamide gel with the elution fractions is shown in Fig. 2b. During elution, active fractions could be identified by yellow coloration, indicating that the cofactor FAD is bound to the enzyme as shown by Fraaije et al. (2005) for thermostable BVMO from T. fusca. Only fractions E3 and E4 of MekA purification have been yellow colored. Noncolored fractions did not show significant activity.
Because of high activity of purified MekA without any substrate added to the reaction mixture, it was concluded that some compound of the eluate influences the NADPH oxidation. So a buffer exchange purification step was carried out with Centricon columns to remove unwanted low molecular weight substances. Results from MekA purifications are summarized in Table 3. After the Centricon purification step, less than 5% background activity were left. Specific activity of purified MekA with 10 mM MEK as substrate was determined to about 1.1 U/mg protein.
Substrate specificity
A series of several aliphatic, arylaliphatic, and cyclic ketones were tested in a photometric assay as substrates for purified recombinant MekA in concentrations of 10 mM in Tris/HCl (0.1 M, pH 9) at 30°C. The substrates and the corresponding specific activities compared to MEK activity are listed in Table 4. Some of the proposed reaction products as ethyl acetate or propyl acetate were found to be substrates for MekB by Onaca et al. (2007). As P. veronii MEK700 does not grow on cyclohexanone, only low activity was expected for this and other cyclic ketones. In contrast, it was found, that p-chloroacetophenone, cyclohexanone, and cyclopentanone were converted into p-chlorophenyl acetate, ɛ-caprolactone, and δ-valerolactone, respectively, with comparable activity to MEK.
The highest activity toward achiral substrates was determined with cyclopentanone and 4-propylcyclohexanone. With linear aliphatic substrates, we observed a decreasing activity from MEK to 2-hexanone and further on an increasing activity from 2-hexanone to 2-undecanone. Keto groups at the second or third C-atom did not significantly affect the activity. Arylaliphatic ketones were oxidized as well.
Because organic solvents are known to be denaturating, we additionally wanted to investigate if the substrates MEK, 2-pentanone, 2-hexanone, 2-octanone, and 3-octanone cause inactivation of MekA leading to low specific activity and low yields in biotransformations. MEK and 2-octanone seemed to be the only stronger inactivating or denaturating compounds and led within 10 min to activity losses of 36 and 47%, respectively.
In addition to achiral substrates, racemic ketones were also tested for conversion by MekA (20 μg/ml) in concentrations of 10 mM at 30°C. Activity measurements have been carried out only once because the racemates were available at very low amounts. Specific activities have been determined with 1.12 U/mg for β-hydroxyoctanone, 0.46 U/mg for β-hydroxydecanone, and 1.08 U/mg for 3-phenyl-2-butanone, respectively. With β-hydroxydodecanone photometric activity measurement was not possible as this compound led to a clouded suspension, which significantly affected the sample extinction.
With reference to the cofactor regeneration in biocatalyses, we also used NADH as cofactor for MEK oxidation. Here, we could observe a decrease in specific activity of 55% compared to the use of NADPH as cofactor.
pH optimum and temperature stability
The pH optimum of MekA was determined with 0.01% (1.38 mM) MEK as substrate. For measuring a broad range of pH values (pH 6 to 11), three different buffers were used overlapping (potassium phosphate buffer [0.1 M], Tris/HCl buffer [0.1 M], and glycine/NaOH buffer [50 mM]). Depending on the used buffer, MekA is active between pH 7.5 and 11, with a strong optimum between pH 9 and 10 as shown in Fig. 3. The maximum activity was measured with nearly 1.2 U/mg of the total protein in Tris/HCl buffer (0.1 M, pH 9). An activity optimum around pH 9 has also been published for CHMO and monocyclic monoterpene ketone monooxygenase, whereas for HAPMO and PAMO pH 7.5 and 8, respectively, were found to be optimal.

pH-optimum of MekA determined with three different buffers (0.1 M potassium phosphate buffer [♦], 0.1 M Tris/HCl [ ], 50 mM glycine buffer [
], 50 mM glycine buffer [ ]). MEK was used as substrate at a concentration of 1.38 mM (0.01%)
]). MEK was used as substrate at a concentration of 1.38 mM (0.01%)
Optimal temperature for MEK conversion was determined between 25 and 60°C, with 10 mM MEK as substrate in Tris/HCl buffer (0.1 M, pH 9) and was shown to be between 30 and 35°C. The temperature stability of MekA was determined between 0 and 60°C, again with 10 mM MEK as substrate in Tris/HCl buffer (0.1 M, pH 9). Hereby, 60 μg of purified MekA was incubated at a specific temperature for 60 min. Afterward, samples were cooled down on ice and reaction was started by adding 20 μl (according to 22 μg of purified MekA) to the reaction mixture. MekA was obviously not thermostable.
As shown in Fig. 4, incubation at 30°C for 1 h already led to loss of 84% of activity. Higher incubation temperatures nearly inactivated the enzyme. Similar results were obtained for monocyclic monoterpene ketone monooxygenase from R. erythropolis by van der Werf (2000), with an optimal reaction temperature at 36°C and fast enzyme inactivation at 40°C. For cyclopentadecanone monooxygenase CpdB from Pseudomonas sp. strain HI-70, maximum enzyme activity was measured at 40°C (Iwaki et al. 2006). The only thermostable BVMO found to date was discovered by Fraaije et al. (2005) in T. fusca with a half-life time of 1 day at 52°C and maximum activity at 70°C.

Investigation of the temperature stability of MekA. The enzyme was incubated at a specific temperature for 1 h and then used for activity assay at a concentration of 20 μg/ml with MEK as substrate
K M and V max concerning substrate and cofactor
K M values of MekA were determined at 30°C for MEK, NADPH, and NADH (Fig. 5). For the determination of K M-MEK, a NADPH concentration of 0.15 mM was used, whereas the MEK concentration for the determination of K M-NADPH and K M-NADH was 1.38 mM. K M values were determined with 6(±2) μM for MEK and 11(±4) μM for NADPH, which correlate well with the data for CHMO toward cyclohexanone (4 μM) and NADPH (20 μM), and for CPMO toward cyclopentanone (<1 μM) and NADPH (<3 μM). In contrast to all other known BVMOs, MekA can also use NADH as cofactor besides NADPH, resulting in 55% less specific activity compared to the use of NADPH. Thus, K M-NADH was determined with 29(±4) μM. This could be caused by missing conserved amino acids, which were found to be important for cofactor specificity of BVMOs by Kamerbeek et al. (2004).

Lineweaver-Burk plot used to evaluate K M and V max of the substrate MEK and the cofactors NADPH and NADH
While Arg220 was identified as corresponding to Arg339 from HAPMO, the conserved amino acids Lys439 and Arg440 from HAPMO could not be found in MekA. Mutations at these sites led to significant changes in cofactor specificity of HAPMO, making the enzyme able to use NADH and other substituted dinucleotides.
Enzyme stability
Long-time storage stability of purified MekA was investigated using glycerol as additive in different concentrations at 4°C and −20°C. Afterward, activity was measured with MEK as substrate under the conditions described before. Storage of MekA at 4°C led to a fast decrease in activity with and without 10% glycerol after 7 days. With 20 and 40% glycerol, the residual enzyme activity could be maintained at 16 and 44%, respectively. Storage at −20°C is possible at glycerol concentrations of 10, 20, and 40% over a period of 3 weeks and probably longer with only minor loss of specific activity. Without addition of glycerol, MekA kept about 10% of its original activity when stored at −20°C. In Fig. 6, the stability of purified MekA stored under different conditions is shown as relative residual activity for samples with and without 40% glycerol.

Stability of MekA under different storage conditions (⋄ without glycerol, 4°C, ▪ without glycerol −20°C, Δ 40% glycerol, 4°C, • 40% glycerol, −20°C). Activity was measured under standard conditions with MEK as substrate
Analysis of substrates and products from purified MekA by GC analysis
Biocatalyses with purified MekA and different achiral and racemic ketones were investigated using GC analysis for detecting substrates and products. Reactions were usually carried out in 1.5-ml Eppendorf tubes at 30°C with horizontal shaking to increase oxygen insertion into the solution. Reaction times were set to 1 h.
Activity measurements showed broad substrate acceptance of MekA toward aliphatic ketones from C6 to C13 (Table 5). MEK has not been used as substrate in biocatalysis as GC analysis of this highly volatile compound was too complicated. Highest conversions were observed after 1 h for 2-nonanone, 2-decanone, and 2-undecanone. With these substrates, good specific activities have been also determined photometrically. 2-Hexanone, 2-octanone, and the acetophenones were only slightly converted, although for p-chloroacetophenone good specific activity was determined spectrophotometrically.
Cyclopentanone and cyclohexanone were moderately converted by MekA compared to high specific activities in photometric measurements. The differences might be explained by protein denaturation through some of the substrate and products, respectively, as indicated by the results presented in Table 4. In the photometric assay, only the initial reaction is measured, and here a slow denaturation of the enzyme has less consequences compared to the long incubation times for GC analysis. All other results agree with the specific activities (see Table 4). MekA also accepts substrates with keto groups at the third or fourth C-atom of the aliphatic chain as shown for different decanones. Here, the conversion of 3- and 4-decanone led to similar product yields under the same reaction conditions like the conversion of 2-decanone.
Next, activity and enantioselectivity of MekA in the kinetic resolution of four chiral substrates—three racemic β-hydroxyketones and 3-phenyl-2-butanone—were studied. Within 1 h, 5 mM β-hydroxydecanone were converted to nearly 87% followed by β-hydroxyoctanone with 47% (see Table 6). In contrast to the results of bioconversions of achiral substrates, specific activities and conversion rates cannot be compared. With β-hydroxydecanone, MekA has shown moderate activity (specific activity 0.458 U/mg), whereas with 3-phenyl-2-butanone specific activity has been as twice as high (specific activity 1.1 U/mg). GC analysis confirmed that MekA showed acceptable enantioselectivity and (R)-selectivity in the resolution of β-hydroxydecanone, but E values were too low for preparative purposes in case of the other three compounds.
Biocatalysis with whole cells of E. coli BL21 pAM262
For biocatalytic reactions with whole cells of E. coli BL21 pAM262 cell concentrations of 45 OD600/ml of a l-rhamnose-induced culture were used. Better solubility of the substrates was achieved with DMSO (final concentration <1%). Different 2-ketones from C10 to C13, some cyclic and the formerly named chiral compounds were investigated as MekA substrates. Reaction times were set to 1 h for 2-decanone, cyclopentanone, and cyclohexanone, to 4 h for the chiral substrates and to 2 h for the rest. For the regeneration of the cofactor NADPH, glucose was added in all cases in a final concentration of 20 mM as already described by Kataoka et al. (2003) for the production of chiral alcohols.
For Baeyer-Villiger oxidations with non-growing E. coli cells with overexpressed Acinetobacter sp. CHMO also Walton and Stewart (2002) added glucose to avoid requirement of exogenous cofactor. Reaction temperature for MekA whole cell Baeyer-Villiger oxidations was set to 30°C. Reactions were carried out in 1.5-ml tubes and analyzed by GC.
In comparison to conversions with purified MekA, it was not surprising that more than 50% of 2-decanone (54%) were converted to its product octyl acetate in 1 h. Longer reaction times would probably lead to similar conversion rates as with 2-undecanone (98.9%) or 2-dodecanone (61.9%) as substrates. 2-tridecanone (23.9%) does not seem to be accepted as well as shorter aliphatic ketones. As shown with purified MekA, conversion of cyclohexanone (21.9%) and cyclopentanone (17.5%) led to similar results under nearly the same reaction conditions. With acetophenone and p-chloroacetophenone, only low product formation could be observed (14.5 and 0.7%).
In former whole-cell biocatalyses for secondary alcohol production, we found that p-chloroacetophenone denaturates cell protein (Menzel 2006), which might explain the low conversions. With the chiral substrates, no enantioselectivity could be determined because substrates were nearly completely converted to their products after 4 h of reaction (data not shown). Because of using whole cells, the formed esters partially have been hydrolyzed to their corresponding (R)- and (S)-alcohols as observed by GC analysis. In some probes, DMSO was metabolized as well.
Discussion
In the present study, a BVMO involved in methylethylketone (MEK) degradation from P. veronii was investigated. The mekA gene has been found in the genome of P. veronii MEK700 by transposon mutagenesis in an operon together with a gene homologous to homoserine acetyltransferases, which encodes an enzyme with high esterase activity and converts a variety of aliphatic and substituted aromatic esters (Onaca et al. 2007). Expression of the recombinant mekA in the pBR322 derivative pJOE5302.3 led to the formation of nearly 100% inclusion bodies and was not further investigated. Thus, mekA was ligated into a l-rhamnose inducible vector with a C-terminal strep-tag fusion. The expression led to low amounts of soluble recombinant protein and large amounts of inclusion bodies as already described by Onaca et al. (2007) for unmodified MekA.
With the BVMO from P. fluorescens DSM 50106, the same problem occurred and could be partially solved by coexpressing different chaperones (Kirschner et al. 2007). Coexpression of chaperones and foldases also increased solubility of CHMO from Acinetobacter sp. NCIMB 9871 during heterologous expression in E. coli (Lee et al. 2004). As already shown for recombinant expression of the hapE gene under control of the T7 promoter in E. coli by Kamerbeek et al. (2001), the inclusion body formation of MekA decreased significantly at low expression temperatures using E. coli BL21. A combination of low temperature and chaperones could probably minimize inclusion body formation and lead to higher solubility of MekA.
Despite low solubility, recombinantly produced MekA could be purified via streptactin columns. Purification of a recombinant his-tagged MekA via Ni-NTA led to inactive enzyme (data not shown) as already found for BVMO from P. fluorescens DSM 50106 (Kirschner et al. 2007). Successful purification of tagged recombinant BVMOs have only been described for PAMO from T. fusca after recombinant expression in E. coli with N-terminal his-tag (Fraaije et al. 2005). Recombinant CHMO and HAPMO have also been purified from E. coli using several chromatography steps (Kamerbeek et al. 2004; Tanner and Hopper 2000).
The substrate spectrum of MekA was shown to be versatile. This BVMO can be used to catalyze Baeyer-Villiger oxidations of a variety of aliphatic, arylaliphatic, and cyclic chiral or achiral ketones. Substituted derivatives such as 4-propylcyclohexanone or p-chloroacetophenone were also converted to the corresponding esters with high specific activity by the purified enzyme. Compared to BVMO from P. fluorescens DSM 50106, MekA is also reactive with longer aliphatic 2-ketones (C9–C13), whereas 2-decanone has been the best converted aliphatic ketone in activity assays and bioconversions. Keto groups at the third or fourth C-atom are accepted but not as well as at the second position. We did not find a reason why MEK and 2-nonanone are good substrates and 2-hexanone, 2- and 3-octanone are not.
Cyclic and aromatic ketones were moderate to bad substrates, both in reactions with purified enzyme and whole cells. With these substrates, yields may be increased by elongated reaction time, decreased reaction temperature because of MekA temperature instability and (enzymatic) cofactor regeneration. Interestingly, also for PAMO, BVMO from P. fluorescens DSM 50106 and monocyclic monoterpene ketone monooxygenase, it has been published that they all do not accept acetophenone as substrate, although otherwise they have a broad substrate range (Kamerbeek et al. 2003b; Fraaije et al. 2005; van der Werf 2000). Only BVMO from P. fluorescens DSM 50106 was found to be very specific for short aliphatic ketones (Kirschner et al. 2007). As chiral substrates different 4-hydroxy-2-ketones and 3-phenyl-2-butanone were used.
Here, using whole cells in all reactions, 5-mM substrates were nearly completely converted in less than 4 h. A significant (R)-selectivity was observed with purified MekA, which is opposite to the selectivity of BVMO from P. fluorescens DSM 50106. Further improvement of the enantioselectivity might be achieved by site-directed mutagenesis or directed evolution as already shown for CHMO from Acinetobacter sp. NCIMB 9871 by Mihovilovic et al. (2006).
It was interesting to observe that MekA can convert a variety of ketones, which provide the possibility to use the enzyme for industrial applications. Here, the stabilized purified enzyme certainly has the advantage to avoid unwanted side reactions and stable reaction conditions caused by known enzyme concentrations as suitable expression of the mekA gene strongly depends on cultivation conditions. As enzymatic Baeyer-Villiger oxidations play an interesting role for the production of chiral ketoesters, MekA is able to use both NADPH and NADH, which is probably caused by missing conserved amino acids that define the cofactor specificity of other BVMOs as described by Kamerbeek et al. (2004). With NADH and MEK as substrate, still 45% activity were measured providing the possibility to use for example a formate dehydrogenase as cofactor regenerating enzyme in biocatalysis with purified MekA and whole cells. Possibly, further investigations could aim at enzyme mutagenesis to increase activity and enantioselectivity and to improve NADH usage making MekA more interesting for industrial applications.
References
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brzostowicz PC, Gibson KL, Thomas SM, Blasko MS, Rouviere PE (2000) Simultaneous identification of two cyclohexanone oxidation genes from an environmental Brevibacterium isolate using mRNA differential display. J Bacteriol 182:4241–4248
Chen CS, Fujimoto Y, Girdaukas G, Sih CJ (1982) Quantitative analyses of biochemical kinetic resolutions of enantiomers. J Am Chem Soc 104:7294–7299
Chen G, Kayser MM, Mihovilovic MD, Mrstik ME, Martinez CA, Stewart JD (1999) Asymmetric oxidations at sulfur catalyzed by engineered strains that overexpress cyclohexanone monooxygenase. New J Chem 23:827–832
Chung CT, Niemela SL, Miller RH (1989) One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. PNAS USA 86:2172–2175
Fraaije MW, Kamerbeek NM, van Berkel WJH, Janssen DB (2002) Identification of a Baeyer-Villiger monooxygenase sequence motif. FEBS Lett 518:43–47
Fraaije MW, Wu J, Heuts DPHM, van Hellemond EW, Spelberg JHL, Janssen DB (2005) Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol 66:393–400
Geitner K, Kirschner A, Rehdorf J, Schmidt M, Mihovilovic MD, Bornscheuer UT (2007) Enantioselective kinetic resolution of 3-phenyl-2-ketones using Baeyer-Villiger monooxygenases. Tetrahedron: Asymmetry 18:892–895
Higson FK, Focht DD (1990) Bacterial degradation of ring-chlorinated acetophenones. Appl Environ Microbiol 56:3678–3685
Iwaki H, Wang S, Grosse S, Gergeron H, Nagahashi A, Lertvorachon J, Yang J, Konishi Y, Hasegawa Y, Lau PCK (2006) Pseudomonad cyclopentadecanone monooxygenase displaying an uncommon spectrum of Baeyer-Villiger oxidations of cyclic ketones. Appl Environ Microbiol 72:2707–2720
Jones KH, Smith RT, Trudgill PW (1993) Diketocampane enantiomer-specific ‘Baeyer-Villiger’ monooxygenases from camphor-grown Pseudomonas putida ATCC 17453. J Gen Microbiol 139:797–805
Kamerbeek NM, Moonen MJH, van der Ven JGM, van Berkel WJH, Fraaije MW, Janssen DB (2001) 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB. Eur J Biochem 268:2547–2557
Kamerbeek NM, Janssen DB, van Berkel WJH, Fraaije MW (2003a) Baeyer-Villiger monooxygenases, an emerging family of flavin-dependent biocatalysts. Adv Synth Catal 345:667–678
Kamerbeek NM, Olsthoorn AJJ, Fraaije MW, Janssen DB (2003b) Substrate specifity and enantioselectivity of 4-hydroxyacetophenone monooxygenase. Appl Environ Microbiol 69:419–426
Kamerbeek NM, Fraaije MW, Janssen DB (2004) Identifying determinants of NADPH specificity in Baeyer-Villiger monooxygenases. Eur J Biochem 271:2107–2116
Kataoka M, Kita K, Wada M, Yasohara Y, Hasegawa J, Shimizu S (2003) Novel bioreduction system for the production of chiral alcohols. Appl Microbiol Biotechnol 62:437–445
Khalameyzer V, Fischer I, Bornscheuer UT, Altenbuchner J (1999) Screening, nucleotide sequence, and biochemical characterization of an esterase from Pseudomonas fluorescens with high activity towards lactones. Appl Environ Microbiol 65:477–482
Kirschner A, Bornscheuer UT (2006) Kinetic resolution of 4-hydroxy-2-ketones catalyzed by a Baeyer-Villiger monooxygenase. Angew Chem Int Ed 45:7004–7006
Kirschner A, Altenbuchner J, Bornscheuer UT (2007) Cloning, expression, and characterization of a Baeyer-Villiger monooxygenase from Pseudomonas fluorescens DSM 50106 in E. coli. Appl Microbiol Biotechnol 73:1065–1072
Lee DH, Kim MD, Lee WH, Kweon DH, Seo JH (2004) Consortium of fold-catalyzing proteins increases soluble expression of cyclohexanone monooxygenase in recombinant Escherichia coli. Appl Microbiol Biotechnol 63:549–552
Menzel A (2006) Genetically changed Escherichia coli cells for biocatalytic synthesis of enantiomeric pure aminoacids and secondary alcohols. Dissertation of the University of Stuttgart, ISBN: 3-9810745-6-4
Mihovilovic MD, Rudroff F, Winninger A, Schneider T, Schulz F, Reetz MT (2006) Microbial Baeyer-Villiger oxidation: stereopreference and substrate acceptance of cyclohexanone monooxygenase mutants prepared by directed evolution. Org Lett 8:1221–1224
Onaca C, Kieninger M, Engesser KH, Altenbuchner J (2007) Degradation of alkyl methyl ketones by Pseudomonas veronii MEK700. J Bacteriol 189:3759–3767
Stumpp T, Wilms B, Altenbuchner J (2000) Ein neues L-Rhamnose induzierbares Expressionssystem für Escherichia coli. Biospektrum 6:33–36
Tanner A, Hopper DJ (2000) Conversion of 4-hydroxyacetophenone into 4-phenyl acetate by a flavin adenine dinucleotide-containing Baeyer-Villiger-type monooxygenase. J Bacteriol 182:6565–6569
van der Werf MJ (2000) Purification and characterization of a Baeyer-Villiger monooxygenase from Rhodococcus erythropolis DCL14 involved in three different monocyclic monoterpene degradation pathways. Biochem J 347:693–701
Walton AZ, Stewart JD (2002) An efficient enzymatic Baeyer-Villiger Oxidation by engineered Escherichia coli cells under non-growing conditions. Biotechnol Prog 18:262–268
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Völker, A., Kirschner, A., Bornscheuer, U.T. et al. Functional expression, purification, and characterization of the recombinant Baeyer-Villiger monooxygenase MekA from Pseudomonas veronii MEK700. Appl Microbiol Biotechnol 77, 1251–1260 (2008). https://doi.org/10.1007/s00253-007-1264-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1264-6