
Abstract
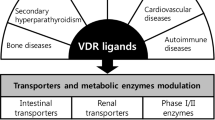
The vitamin D receptor (VDR), in addition to other nuclear receptors, the pregnane X receptor (PXR) and constitutive androstane receptor (CAR), is involved in the regulation of enzymes, transporters and receptors, and therefore intimately affects drug disposition, tissue health, and the handling of endogenous and exogenous compounds. This review examines the role of 1α,25-dihydroxyvitamin D3 or calcitriol, the natural VDR ligand, on activation of the VDR and its crosstalk with other nuclear receptors towards the regulation of enzymes and transporters, notably many of the cytochrome P450s including CYP3A4 and sulfotransferase 2A1 (SULT2A1) as well as cholesterol 7α-hydroxylase (CYP7A1). Moreover, the VDR upregulates the intestinal channel, TRPV6, for calcium absorption, LDL receptor-related protein 1 (LRP1) and receptor for advanced glycation end products (RAGE) in brain for β-amyloid peptide efflux and influx, the sodium phosphate transporters (NaPi), the apical sodium-dependent bile acid transporter (ASBT) and organic solute transporters (OSTα-OSTβ) for bile acid absorption and efflux, respectively, the renal organic anion transporter 3 (OAT3) and several of the ATP-binding cassette protein transporters—the multidrug resistance protein 1 (MDR1) and the multidrug resistance-associated proteins (MRPs). Hence, the role of the VDR is increasingly being recognized for its therapeutic potential and pharmacologic activity, giving rise to drug-drug interactions (DDI). Therapeutically, ligand-activated VDR shows anti-inflammatory effects towards the suppression of inflammatory mediators, improves cognition by upregulating amyloid-beta (Aβ) peptide clearance in brain, and maintains phosphate, calcium, and parathyroid hormone (PTH) balance and kidney function and bone health, demonstrating the crucial roles of the VDR in disease progression and treatment of diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Undeniably, many of the nuclear receptors (NRs) strongly influence the fate of xenobiotics by regulating transporters (1,2,3) and enzymes (4,5,6,7,8), and therefore drug disposition. NRs are multifunctional proteins that transduce signals of their cognate ligands. As a component of the nuclear receptor class 1 (NR1) superfamily, the vitamin D receptor (VDR) is known for its role in the regulation of transporters and enzymes, affecting the balance of not only endogenous molecules such as cholesterol, bile acids, active peptides, and ions but also the disposition of xenobiotics. Like many nuclear receptors, the VDR heterodimerizes with the retinoid X receptor-α (RXRα), and, upon activation by ligand, initiates gene transcription. Common structures of the NRs include an amino terminal ligand-independent activation function domain (AF-1) for interaction with cofactors, a central DNA-binding domain (DBD) consisting of two zinc finger motifs that target the nuclear receptor to highly specific DNA sequences or DNA response elements, a carboxy-terminal ligand-binding domain (LBD) that differs for each NR and allows for specific hormonal and non-hormonal ligand binding, and a less conserved flexible hinge region that connects the DBD and LBD, governing conformational changes, receptor dimerization, and coregulator interactions at the activation function-2 region (AF-2) for biological response (9, 10). The VDR forms a protein complex as a homodimer or heterodimer with RXR when bound to ligands. The active VDR transcription factor complexes by binding to a specific response element sequences (vitamin D response elements (VDREs)) on target genes that are typically comprised of half-sites of direct or inverted hexanucleotide repeats separated by different nucleotide lengths (11).
The Vitamin D Receptor, VDR
Being an important nuclear receptor for calcium, phosphate, and bone homeostasis, the VDR is present ubiquitously in tissues such as the intestine, kidney, and skin, although less in the liver and bone (12,13,14,15,16). The VDR protein is almost undetectable in mouse and human livers and is present at low but perceptible levels in specific regions of the brains of mouse (17), rat (18), and human (19). The protein homology and alignment of rodent and human VDR have been examined (20), revealing that the amino acids in the DNA-binding domain are highly conserved across mouse, rat, and human receptors (100%) (Fig. 1). The mouse VDR ligand-binding domain amino acid sequence is 89% identical to that in human and 96% to that in rat. Although the mouse internal region includes a portion of the ligand-binding domain (aa 159 to 206) that differs from that of man and rat, being only 55% and 78% identical to that in human and rat, respectively (20), the actual hinge region (aa 88 to 125) is highly conserved among mice, rats, and humans (97%) as well as VDR hinge mutations do not affect the ligand-binding affinity (21).

a Amino acid sequences of VDRs in mouse (GenBank: EDL04218.1), rat (GenBank: EDL87095.1), and human (GenBank: BAA83389.1). Identical amino acids are shown as dots. b The homology for the entire amino acid sequences and c for the ligand-binding domain (LBD) of VDRs among mouse, rat, and human
The binding of ligands to the VDR results in a conformational change of the receptor that promotes its translocation from the cytosol to the nucleus and heterodimerization with the RXRα (22). This VDR-RXRα complex then binds to VDREs in the promoter region of VDR-responsive genes, recruiting nuclear proteins/coregulators into the transcriptional pre-invitation complex to initiate gene transcription. There is currently no crystal structure of the unoccupied ligand-binding domain of the VDR. Our understanding of the molecular mechanisms of VDR activation has been advanced with crystal structures of VDR bound to its various ligands, both native and synthetic ones. The crystal structure of VDR, in complex with 1α,25-dihydroxyvitamin D3, active, endogenous VDR ligand (or calcitriol), has revealed the presence of several water molecules near the A-ring linking the ligand C-2 position to the protein surface (23). In rat and human VDR, the 6 amino residues in the ligand-binding pocket form hydrogen bonds with the three hydroxyl groups on the 1α,25-dihydroxyvitamin D3 molecule (23, 24). Ser237 and Arg274 interact with the 1α-hydroxyl group, and Arg274 of the human VDR forms the strongest hydrogen bond with calcitriol. Mutation of this residue is associated with type II rickets (25). Tyr143 and Ser278 interact with the 3-hydroxyl group; His305 and His397 interact with the 25-hydroxyl group. The VDR LBD has a structure of a three-layered, antiparallel α-helical sandwich, and the ligand-binding pocket (LBP) is located in the lower part of the LBD (26, 27). Wan et al. showed that there are 10 and 6 amino acid residues, which stabilize the sandwich structure and form hydrogen bonds with the VDR ligand, respectively, that have identical sequences across mice, rats, and humans (28). Moreover, the amino acid sequences forming LBP are almost the same, except for one amino acid, and this is not important for folding (28, 29). However, there is absence of structural information about the mouse VDR ligand-binding pocket although it is speculated that the amino acids that reside in the ligand-binding pocket would be similar to those for human and rat due to the similarity in the amino acid-binding sequence of the VDR protein (20).
While considerable debate exists regarding the subcellular localization of the VDR and events that transpire after activation (30, 31), there is a general consensus that VDR activation occurs in a similar manner to that for other NRs to regulate channels and transporters, classical targets of the involved in calcium, phosphate, and bone homeostasis (32,33,34). The VDR exerts its effects on its targets both genomically and non-genomically (35), exemplified by the numerous regulatory studies on enzymes and transporters within the last decade. Increasingly, there is documentation on the VDR as a regulator of drug disposition since calcitriol aptly increases CYP3A4 and MDR1 activities, important enzyme and efflux transporter, respectively, which demonstrate many of the functional aspects of the Caco-2 cell line (36,37,38,39,40). Indeed, specific VDREs have been found in the the CYP3A4 and ABCB1 (MDR1) gene promoters (5, 39).
Crosstalk of VDR with Other NRs on Cholesterol and Bile Acid Homeostasis
From in vitro DNA-binding studies, the preferred response elements for PXR, CAR, and VDR are hexanucleotide direct repeats (DR3 or DR4) that are 3- or 4-nucleotide spaced (41,42,43,44,45). Much crosstalk exists between the VDR and other nuclear receptors (NRs). In particular, the VDR shares similar homology with PXR and CAR, and is activated by its cognate ligands such as calcitriol or the alternate VDR ligand, lithocholic acid (LCA) (46). Due to their similar homology, PXR and CAR are able to transactivate CYP24A by binding to VDREs of the CYP24A promoter region (47, 48). The VDR inhibits the farnesoid X receptor (FXR), the bile acid sensor (49), and the liver X receptor-α (LXRα) (50), both important regulators of bile acid and cholesterol homeostasis. The crosstalk between the VDR and FXR in the regulation of cholesterol and bile acid homeostasis in the intestine and liver has been previously examined in vivo in the rat (51). Additionally, VDREs were found in the mouse and human SHP, the small heterodimer partner (17), and the fibroblast growth factor 15 (Fgf15 in rodent or FGF19 in human) (52,53,54). Calcitriol administration to fxr(+/+) and fxr(-/-) mice in vivo increased the activities of liver Cyp7a1, the rate limiting enzyme that metabolizes cholesterol to bile acids in a FXR-independnet manner through inhibition of the repressive nuclear receptor, SHP (17). Consequently, VDR inhibition of Shp/SHP led to induction of Cyp7a1/CYP7A1 in murine and human hepatocytes (17), enhancing cholesterol metabolism to bile acids in mice in vivo (17). However, the VDR was found to stimulate Fgf15 (rodent) and FGF19 (human), hormonal signaling molecules that activate the liver fibroblast growth factor receptor 4 (Fgfr4/FGFR4) (52), which in turn downregulate Cyp7a1/CYP7A1 in liver (54).
Species differences have been found. First, differences exist in the regulation of the apical sodium-dependent transporter (ASBT), an important bile acid uptake transporter in the intestinal ileum (45, 55) that absorbs bile acid in a sodium-dependent fashion (56). Increased bile acid levels trigger FXR activation in the intestine, which in turn represses liver receptor homolog-1 (LRH-1), the competence factor needed for ASBT expression, and activates liver SHP (55, 57). For the rat, there is absence of the cis-element in rat Lrh-1 (45), hence resulting in induction of rat ileal Asbt with the VDR (45) but not mouse Asbt (17). Mouse Asbt expression is aptly repressed by bile acids (45) or remains unchanged with VDR activation (17). Other reports show that the VDR positively impacts the expression of peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ (PPARα and PPARγ) (58, 59). PPARγ binds to VDR and inhibits 1α,25-dihydroxyvitamin D3-mediated transactivation of CYP24A1, the VDR signature gene responsible for the degradation of its ligand (60).
Species difference in VDR activity on rodent Oat3 has been noted. Murine Oat3 expression was upregulated by VDR activation (61) but downregulated in rats (62). We had speculated that the difference could be due to a species difference in protein kinase C (PKC) localization in the kidney: VDR increases PKC activity (63, 64) that could lead to Oat3 inhibition (65). PKC is not expressed in the murine proximal tubules (66), while it is highly expressed in the brush border of the proximal tubes in rats (67). Collectively, the species difference in the regulation of enzymes or transporters by VDR is attributed to differences downstream to VDR, not to VDR itself.
As lithocholic acid, the secondary, toxic bile acid, is another activator of VDR, FXR, and PXR, crosstalk between nuclear receptors in the regulation of bile acid transporters has been investigated. In the intestine, the major bile acid transporters include ASBT and MRP2 at the apical and OSTα-OSTβ and MRP3 at the basolateral enterocyte membranes. VDR activation has resulted in rat Asbt induction (45) but no change or slight inhibition in mouse (68), showing that there are species differences (see later, Table III). Similarly, the observations for OSTα-OSTβ are heterogeneous with respect to the regulation by VDR (see later, Table III). MRP3 induction by VDR occurs by direct regulation of the ABCC3 gene (69). On the other hand, FXR induces OSTα-OSTβ (70) while downregulating ASBT (71) in the intestine. There is some information regarding regulation of intestinal bile acid transporters by PXR or the constitutive androstane receptor (CAR). Indeed, the expression of human OSTβ is induced (72) while mouse Mrp2 and Mrp3 are induced by by CAR activation (73). With respect to the regulation of hepatocyte bile acid transporters, the effects of VDR activation appear modest or inconsequential (Table III). The key hepatocyte bile acid transporters are NTCP, OATP, BSEP, MRP3, and MRP4. Downregulation of BSEP by 1,25(OH)2D3 occurs indirectly through VDR-mediated inhibition of FXR transactivation of the ABCC11 gene (49). However, FXR activation is well known to regulate hepatic bile acid transporter expression including downregulation of NTCP (74) together with induction of BSEP (75) and OSTβ (76). Induction of Mrp3 and Mrp4 in mouse liver by bile acid treatment appears to be independent of FXR activation (77). Morevover, Mrp2, Mrp3, and Mrp4 are upregulated in liver by PXR/CAR activation (78).
1α,25-Dihydroxyvitamin D3 (1,25(OH)2D3 or Calcitriol), Active VDR Ligand
1α,25-Dihydroxyvitamin D3 or calcitriol is the natural, active ligand of the VDR (79) that is formed from inert precursor forms of vitamin D3 or vitamin D2, derived from animals and plants, respectively (80). Vitamin D3 may be obtained from exogenous sources such as milk and cod liver oil, or via exposure to UV rays from sunlight, converting 7-dehydrocholesterol to vitamin D3 (80, 81). Vitamin D3 is lipophilic and is highly bound to the vitamin D-binding protein (DBP) (81, 82) and stored mainly in the adipose tissue rather than circulating in blood (83). The first bioactivation step of vitamin D3 is via hydroxylation of carbon 25 primarily in the liver by 25-hydroxylases, CYP27A1 (84), and CYP2R1 (85) to the relatively inactive metabolite, 25-hydroxyvitamin D3 [25(OH)D3], in higher abundance (nM range) (Fig. 2a). Both 25(OH)D3 and 1,25(OH)2D3 are metabolized to inactive metabolites, 24,25-(OH)2D3 and 1,24,25(OH)3D3, respectively, by the 24-hydroxylase (CYP24A1), the degradation enzyme that is upregulated by the VDR (86, 87). Bioactivation of 25(OH)D3 occurs in the kidney with CYP27B1, the mitochondrial 1α-hydroxylase, to form 1,25(OH)2D3 in a rate-limiting fashion (80). The expression of CYP27B1 is downregulated by the VDR when excess 1,25(OH)2D3 is present. Thus, levels of 1,25(OH)2D3 are tightly regulated, CYP27B1 is inhibited whereas CYP24A1 is induced, at high calcitriol concentrations by the VDR (Fig. 2b) (80, 88). Plasma calcium is a significant contributing factor for concerted regulation of calcitriol levels via CYP27B1 and CYP24A1 controls via the parathyroid hormone (PTH) feedback mechanism, stimulating the calcium-sensing receptor (CaSR) and triggering PTH production in the parathyroid gland that leads to upregulation of renal CYP27B1 and downregulation of CYP24A1, respectively, at low plasma calcium concentrations (80, 89). For vitamin D, it is further known that phase 2 metabolism also occurs (90,91,92), albeit most of the formed metabolites are inactive and the pathways are less important.

a, b Metabolism of vitamin D to 25(OH)D3 with CYP2R1 and CYP27A1 in liver and to 1,25(OH)2D3 via CYP27B1 in kidney. The enzymes CYP27B1 and CYP24A1 are subject to inhibition (red arrow) and induction (blue arrow), respectively, by calcitriol and PTH; the intestinal calcium channel, TRPV6, for calcium absorption is under VDR activation
Over 99% of vitamin D metabolites are bound to plasma proteins, mostly to the vitamin D binding protein (DBP) and to albumin or lipoproteins to lesser extents (93,94,95). In plasma, 25(OH)D3, which exhibits the strongest binding affinity towards DBP and whose concentration is 20 times higher than other vitamin D metabolites, is highly bound (82). The lipophilic nature of vitamin D and the high binding of vitamin D metabolite 25(OH)D3 in plasma contribute to reduced metabolism in tissues and a longer, circulating half-life (28 h). The 25(OH)D3-bound DBP complex is filtered through the glomerulus in the kidney and taken up by endocytic receptor, megalin, present on the brush border of the renal proximal tubule cells (96). Once in tubular cells in the kidney, DBP is degraded by legumain (97), and free 25(OH)D3 is metabolized by the mitochondrial 1α-hydroxylase or CYP27B1 to form the active 1,25(OH)2D3 (98, 99). There is evidence that this rate-limiting enzyme is also present in the brain and skin, albeit playing a much lesser role in the formation of calcitriol (19, 100).
Due to the importance of the VDR, the clinical status of vitamin D sufficiency or deficiency is a health concern. Clinically, the vitamin D status is determined by circulating levels of 25(OH)D3 which is more readily determined as it is present at higher nM range (~75 nM), unlike calcitriol, which is present at much low pM levels (101, 102). It is usually considered that a patient is vitamin D deficient when plasma 25(OH)D3 levels fall below 20 ng/ml or 50 nM (103, 104), and vitamin D insufficient when the 25(OH)D3 plasma level is between 20 and 30 ng/ml (105,106,107,108). Due to the tight regulation of calcitriol on its production and degradation, excess 1,25(OH)2D3 in plasma inhibits renal Cyp27B1 to curtail calcitrol production and enhance CYP24A1-mediated degradation (88). At low doses of calcitriol, when CYP24A1 is not induced, the half-life is the longest, whereas at high levels or with escalating calcitriol doses, CYP24A1 is greatly induced and CYP27B1 completley inhibited, reducing the half-life and increasing calcitriol clearance (88).
Vitamin D Analogs
Many vitamin D analogs have been developed as therapeutic agents to improve the function and activity of the VDR and overcome the well-known hypercalcemic toxicity of calcitriol (see later). These targets include diseases such as secondary hyperparathyroidism (SHPT), psoriasis, cancer, and osteoporosis (109, 110). Structural modification of the vitamin D moiety not only affects ligand-specific conformational change in VDR, influencing pharmacological action by VDR activation, but also affects the binding affinity to DBP in plasma, altering tissue distribution (109). Among the vitamin D analogs, 22-oxa-1,25(OH)2D3 or Oxacalcitriol®, 19-nor-1,25(OH)2D2, 1α(OH)D2, Hecterol® or doxercalciferol, and 1,25(OH)2–26,27-F6-D3 or Falecalcitriol® have been developed to treat SHPT in patients with chronic kidney disease due to their lower hypercalcemic side effects (110). Notably, doxercalciferol (1α(OH)D2), the precursor of 1,25(OH)2D2, requires activation in the liver and is less toxic than 1α(OH)D3, Alfacalcidol®, an active precursor that converts to 1,25(OH)2D3 in the liver (111). Furthermore, several vitamin D analogs have been developed to treat psoriasis and cancers as well as suppress the immune system. Calcipotriol®, possessing a similar binding affinity for VDR as 1,25(OH)2D3 but is 200 times less potent, is designed to treat psoriasis. Tacalcitol® or 1,24(OH)2D3 showed therapeutic potential in psoriasis by inhibiting DNA synthesis of epidermal keratinocytes, having similar activity to that of 1,25(OH)2D3 but less toxic towards hypercalcemia than 1,25(OH)2D3. Not all VDR analogs retain chemical structural similarity to that of 1,25(OH)2D3. As mentioned earlir, lithocholic acid (LCA), the toxic, secondary bile acid, is also a VDR ligand (4). It is structurally dissimilar to 1,25(OH)2D3 but activates the VDR at µM concentrations rather than at nM concentrations (4, 112).
Pharmacologic or Therapeutic Effects of the VDR
The vitamin D receptor, upon binding to its active ligand, acts as biological sensor that is vital to calcium and phosphorus homeostasis and controls many other genes in the regulation of other hormonal actions, immune responses, cell proliferation and differentiation, and tissue health (113). The VDR exerts its pharmacologic action through transcriptional control of various transporters and enzymes. As discussed previously, it tightly regulates plasma calcitriol concentrations via CYP27B1 and CYP24A1. Moreover, the VDR is important for the maintenance of calcium and phosphate homeostasis via the calcium channel, transient receptor potential cation channel, subfamily V, member 6 (TRPV6) in intestine primarily, as well as the sodium-dependent phosphate cotransporter (NaPi) in the intestine and kidney, and osteoblasts and osteoclasts in bone.
Calcium and Phosphate Homeostasis
The presence of the VDR in multiple tissues suggests its definitive and important role in the body (114, 115). Although 1,25(OH)2D3 or calcitriol could be synthesized in many tissues such as skin and brain (19, 100), it is produced specifically for direct cellular use and not purely for mineral requirements. The VDR is best known for its role in calcium and phosphate homeostases and preserving bone health (Fig. 3). The function of the vitamin D endocrine system ensures that calcium and phosphate are kept in a delicate balance for proper bodily functions, requiring adequate communication among tissues such as the kidney, bone, parathyroid gland, and intestine in order to maintain appropriate plasma levels of calcitriol. Calcitriol controls the absorption of calcium from the intestine or reabsorption via the kidney and bone, and regulates PTH in the parathyroid gland (34, 116,117,118,119) as well as phosphate balance via the NaPi in both intestine and kidney. The TRPV6 calcium channel, together with NaPi and PTH, regulates calcium and phosphate levels (Fig. 3) (120,121,122,123,124). In small intestine enterocytes, calcitriol stimulates the VDR to increase calcium absorption from the gut lumen by TRPV6, the epithelial calcium channel (15, 34, 114, 125), increases calcium transport across the cell by inducing calbindin D9K, a cytosolic calcium-binding protein, and elevates basolateral plasma membrane ATPase (PMCA1) that transports calcium ion into the bloodstream (22). Calcitriol upregulates renal TRPV5, though of minor importance in the overall calcium absorption, and calbindin D28K to increase calcium reabsorption (22, 126). VDR activation also increases active phosphate transport through the induction of the apical NaPi cotransporters in the kidney and intestine (127,128,129). In bone, calcitriol activates osteoblasts and stimulates the maturation of osteoclasts to resorb calcium from bone and reverse transport calcium from bone compartment to plasma (80). For control of plasma calcium concentrations, feedback mechanisms exist to curtail calcitriol production and calcium reabsorption. First, the calcium-sensing receptor (CaSR) is present in the parathyroid gland for the detection of plasma calcium (80). When the plasma calcium concenetration is high, calcitonin, a 32-amino acid polypeptide produced in the parafollicular cells of the thyroid gland, increases to inhibit intestinal calcium absorption and osteoclast activity in bones (80). These events act as feedback controls to decrease the plasma levels of calcitriol and calcium (Fig. 3).

Interplay of calcitriol, PTH, and FGF23 on calcium and phosphate homeostasis in kidney, and changes in CKD with alterations in calcium and PTH homeostasis and bone health. Klotho is a membrane protein that enables the function of FGF23, the bone hormone that regulates phosphate homeostasis
Secondary Hyperparathyroidism (SHPT) in Chronic Kidney Disease
Vitamin D analogs are used as therapeutic agents for various diseases such as SHPT in chronic kidney disease (CKD) (117, 127, 130, 131). CKD is a progressive disease characterized by a decline in kidney function that is classified from mild (stage 1) to severe (stage 5) stages according to the glomerular filtration rate (GFR). According to the Kidney Foundation of Canada in 2016 (http://www.cihr-irscgc.ca/e/50872.html), CKD afflicts over one in ten Canadians. In stage 5 CKD, kidney function is minimal or absent, and patients require regular dialysis or kidney transplant for survival. With increasing renal dysfunction, the activity of 1α-hydroxylase or CYP27B1 that rate-limits the formation of calcitriol from 25(OH)D3 is much curtailed. The interaction of calcitriol, calcium, phosphate, PTH, and CYP27B1 is complex (Fig. 3), and the delicate equilibrium is upended by renal diseases. With CKD, the function of the kidney to synthesize the active ligand, 1α,25-dihydroxyvitamin D3, by CYP27B1 is reduced. This leads to a rise in circulating levels of PTH, a condition known as SHPT, which occurs when calcium and calcitriol levels are low. With elevated PTH acting directly on bone cells, the bone hormone, fibroblast growth factor 23 (FGF23) is increased (129) to maintain phosphate balance by increasing urinary phosphate excretion and reducing phosphate reabsorption by NaPi (127) (Fig. 3). FGF23 decreases the abundance of CYP27B1 for calcitriol synthesis (130). When circulating 1,25(OH)2D3 levels are very low, PTH synthesis and secretion are increased (131), and PTH acts on bone to increase efflux of calcium and phosphate and mobilize bone mineralization (132). This condition, when prolonged, causes the softening of bones, calcification of vascular and renal tissues, cardiovascular complications, and death. PTH levels may be restored upon replenishment of calcitriol to normal calcitriol and calcium concentrations in plasma for inhibition of PTH synthesis and secretion (22, 117).
Cholesterol Lowering
In the earlier section on NR crosstalk, we reported that the VDR plays a significant role in the derepression of SHP, thereby removing the negative regulation of SHP on CYP7A1 in a FXR-independent fashion (17) (Fig. 4). Our laboratory has demonstrated unequivocally that the VDR exerts anti-hypercholesterolemic properties via CYP7A1 induction with inhibition of SHP (17). However, the VDR also stimulates intestinal Fgf15/FGF19 (52) that enters the portal vein and binds with liver FGFR4 to result in inhibition of CYP7A1. The stimulation of CYP7A1 by VDR via SHP inhibition overrides that with Fgf15/FGF19 stimulation.

Up-regulation of CYP7A1 in cholesterol lowering by the VDR via inhibition of SHP in liver. In intestine, VDR stimulates Fgf15/FGF19, which travels to the liver and interacts with Fgfr4/FGFR4 to inhibit Cyp7a1/CYP7A1. VDR also exerts an antagonistic effect on FXR in intestine and liver
Cancer
Vitamin D analogs have been used for the treatment of cancer (133, 134). The therapeutic use of 1,25(OH)2D3 for cancer, immune, and endocrine modulation is curtailed due to the hypercalcemic side effects of vitamin D analogs (110). Vitamin D analogs have been used as anticancer agents as these control cell proliferation and differentiation; it has neoplasia-suppressive effects in inhibiting tumor cell proliferation, growth and invasiveness, cell cycle arrest, and inflammatory signaling (135), and regulates cancer development via stem cell proliferation and short non-coding microRNA (miRNAs) expression (135). For these reasons, calcitriol or different formulations are often employed for the treatment or adjunct therapy of prostate, colorectal, and breast cancers (136,137,138,139).
Others: Psoriasis and Possibly Reduction of Brain β-Amyloid Peptides
Vitamin D is an important option for the treatment of psoriasis, a chronic immune-mediated inflammatory skin disease. The epidermis is the natural source of vitamin D synthesis by sunlight due to its role in proliferation and maturation of keratinocytes. The VDR exerts regulation of the immune system (inhibition of T cell proliferation), downregulation of proinflammatory cytokines, and stimulation of anti-microbial peptide expression and regulation of barrier integrity and permeability (140).
Another potential activity is the reduction of pathogenic β-amyloid peptides and plaque in the brains of a mouse model of Alzheimer’s disease due to induction of the multiple drug resistance (MDR) protein and its gene product, P-glycoprotein (P-gp) (141). Moreover, other receptors, for example, RAGE and LRP1, may be subject to VDR regulation. However, the prevalent use of calcitriol therapeutically for these other indications is undermined due to its propensity for hypercalcemia.
VDR Targets Enzymes, Transporters, and Receptors
Levels of calcitriol are tightly regulated by the synthetic mitochondrial enzyme, CYP27B1, in the kidney and the catabolic or degradative enzyme, CYP24A1, which has a more ubiquitous distribution (15). The VDR also targets other noteworthy enzymes and transporters. Western blotting has verified the presence of VDR protein expression in the intestine, liver, kidney, brain, cultured cells in vitro in immortalized Caco2 and HepG2 cells, and intestinal and liver slices. In our laboratory, we identified changes of in the expression of enzymes and transporters upon induction/inhibition via the VDR with four intraperitoneal pharmacologic albeit slightly toxic doses of 2.5 μg/kg of calcitriol, given every day or every other day to rodents. This dose is hypercalcemic and pharmacologically active (15, 17, 51, 142). Pursuant to dose administration, we were able to identify the pharmacokinetics and pharmacodynamics of calcitriol. Upon continued administration of calcitriol to mice, the half-lives of calcitriol become shortened due to induction of Cyp24a1 and inhibition of synthesis via Cyp27b1 in the mouse (15, 17, 51, 88).
Phase I Enzymes
The intestine and liver are both endowed with CYPs for the metabolism of drugs (143, 144). The VDR strongly modulates the expression of Cyp3a isoforms (4, 6, 51, 69, 142, 145,146,147,148,149) in rodents and upregulates CYP3A4 in the Caco-2 cell line (34, 40, 148, 150, 151). Fan et al. (40) further compared the inductive effects of other vitamin D analogs—1α(OH)D2 and 1α(OH)D3—against calcitriol for the intestinal CYP3A4 activity in Caco-2 cells. VDR induction is due to the presence of a VDRE in the human CYP3A4 gene (4, 5). Other studies (Table I) documented that calcitriol treatment of human intestinal cell lines such as Caco-2, LS180, HT-29, and SW480 (5, 36,37,38, 40, 148, 150,151,152,153) as well as human HepG2 (148, 154), HEK293 (4), and HPAC (36) cell lines and human slices induced CYP3A4 expression in vitro. Later studies revealed that the inductive role of VDR is not limited to human CYP3A4 only; the expression of human CYP2B6 and CYP2C9 in primary human hepatocytes (155) is also increased with calcitriol treatment.
In rodents in vivo, rat Cyp3a9 (146, 147) and Cyp3a1/3a23 (6, 51, 147), orthologs of human CYP3A4, are induced in the intestine. Chow et al. (142) showed that calcitriol treatment to rats in vivo induced Cyp3a species differentially in different organs: Cyp3a1 in the intestine and Cyp3a9 in the kidney. Kutuzova and DeLuca (147) demonstrated that calcitriol upregulated genes for detoxification in the rat intestine, such as inducing the expression of the catabolic enzyme, Cyp24a1, as well as Cyp3a1 and Cyp1a1. Rat and human intestinal slices that were incubated with calcitriol led to increased expression of rat Cyp3a1 in jejunum, ileum, and colon and Cyp3a2 in ileum (6, 51, 145) and human CYP3A4 (145).
Phase II Enzymes
In contrast to P450 studies, only a few studies showed that the VDR plays an important role in the regulation of phase II enzymes. 1,25(OH)2D3 treatment elicited an increase in human sulfotransferases SULT1C2 and SULT2A1 in vitro (7, 159) and in mouse (68, 160) as well as mouse Sult2a2 in vivo (69), and rat UDP glucuronosyltransferase, Ugt1a (147) (Table II). However, no significant change in SULT2A1 mRNA levels was observed following 1,25(OH)2D3, 1α(OH)D2, and LCA treatment for 3 consecutive days in Caco-2 cells, despite that other VDR target genes CYP24A1 and TRPV6 were also induced (40). This discrepancy, according to Song et al. (159), may be explained by different cell types and the abundance of SULT2A1 expression among Caco-2 cells (40) vs. liver cells. There is some evidence to suggest that glutathione S-transferase may also be induced by the VDR (147) (Table II).
Regulation of Solute Carrier Family
Several solute carrier (SLC) absorptive transporters may be under the regulation of VDR (Table III). While many of the transporters are tested, only the transporters prone to change with the VDR are reported. Our laboratory noted that a VDRE existed in the rat Asbt gene, and that induction of rat Asbt with calcitriol treatment was due to the absence of the cis-element in rat liver receptor homolog-1, Lrh-1 (45). Hence with calcitriol treatment, Asbt protein was upregulated in the rat intestine (45, 51, 149) and rat kidney (142, 149) (Table III). Without a proper functioning Lrh-1 in rats, VDR-mediated induction of rat Asbt is expected to be absent. Instead, inhibition of mouse Asbt was found with the proper functioning intestinal Lrh-1 (68).
The VDR is involved in the regulation of the folate transporters: there are at least three—the folate receptor-α (FRα), the proton-coupled folate transporter (PCFT), and the reduced folate carrier (RFC). In vitro studies first established that the mRNA expression of the PCFT, which is present abundantly in the small intestine, is increased by calcitriol in a dose-dependent fashion in Caco-2 cell line and rat duodenal segments due to the presence of a VDRE and folate transport function in Caco-2 cells (161). A later study, however, showed that VDR(-/-) mice or vitamin D status (25(OH)D3 or 1,25(OH)2D3 plasma concentrations) in rats are not related to the plasma and liver folate status or intestinal PCFT mRNA expression (163). In brain, where all three folate transporters—PCFT, FRα, and RFC—are expressed (162), FRα is suggested to play a critical role in the maintenance of CNS folate transport across the choroid plexus epithelium. The co-existence of the FRα and PCFT, in HeLa sublines, traps folate into vesicles and enhances folate transport into the cytosol (168). Despite that Eloranta et al. (161) failed to observe changes in RFC mRNA expression, Alam et al. demonstrated that RFC is increased by calcitriol treatment and is capable towards folate uptake in the brain microvessel endothelial cell line, hCMEC/D3, and isolated mouse brain capillaries, and increased protein and mRNA expression of RFC (164). That RFC enhances folate uptake in mice was demonstrated in Folr-/1 KO (162).
The relative protein expressions of the organic anion transporters 1 and 3 (OAT1 and OAT3), which play a major role on organic anion uptake from blood circulation to renal proximal tubular cells. The expression levels of these anionic transporters are reduced following treatment with calcitriol in rats, leading to reduced clearance of cefdinir and cefadroxil, organic anion transporter substrates (62, 142, 149, 165). Although rat Oat1 protein expression was decreased (62, 149), murine Oat1 protein in kidney is unchanged with calcitriol treatment (61). Murine Oat3 protein is decreased in vitamin D deficiency, and cholecalciferol or calcitriol treatment restored murine Oat3 expression (61). Moreover, Alam et al. reported an elevated mRNA expression of murine Oat3 after calcitriol treatment (162). The VDR regulatory changes appear to be opposite in the rat and mouse, and there has been some debate that it is attributed to the co-presence of the inhibitor, protein kinase C (PKC) (61).
The efflux of bile acids is handled specifically by the protein complex formed by the organic solute transporters α and β (OSTα and OSTβ) that are expressed mostly in the colon, liver, small intestine, and kidney (169), whose major role is towards the net efflux or uptake of bile acids at the basolateral membrane of enterocytes (56), thus contributing an important role in the enterohepatic circulation of bile acids. The expression of the half-transporters, OSTα and OSTβ that must coexist for the transport of bile acids, has been shown to be induced by chenodeoxycholic acid (CDCA), a FXR ligand (70). Khan et al. found that this heteromeric transporter pair consisting of the gene products of OSTα and OSTβ is under the negative regulation of the VDR in vitro (166). Both calcitriol and LCA treatment decreased the mRNA expression of Ostα and Ostβ in rat ileum slices, while calcitriol had no effect on human ileum, and rat and human liver slices. But LCA, which is a ligand for both FXR and VDR (152), increased OSTα and OSTβ mRNA expression in rat liver slices, and human ileum and liver slices, likely because of FXR regulation (166). Treatment of mice with 1α(OH)D3 reduced renal Ostα mRNA levels by 71% and Ostβ levels by 17%, and this effect was shown to be independent of FXR, as a reduction in mRNA of both genes still occurred in fxr(− / −) mice (68).
The VDR inductive pattern for the oligopeptide transporter 1 (PEPT1) is, however, not clear. Rat intestinal Pept1 function is induced following calcitriol treatment in vivo (142), as evidenced by an increase in apical-to-basolateral (A → B) transport of glycylsarcosine, a PepT1 substrate, in the everted rat intestinal sac (167). However, no induction was found in Caco-2 cells (40), and Pept1 protein expression in rat kidney (142, 149) and mouse Pept1 (61) were decreased and unchanged, respectively.
Receptors
The VDR is known to be involved in the important function of calcium homeostasis that is vital to bone health (34). The promotors of the genes encoding the two calcium channels, the transient receptor potential vanilloid subfamily member 6 (TRPV6) of the intestine and members 5 and 6 (TRPV5 and TRPV6) of the kidney, contain VDREs that regulate their expressions (170). Among these channels, the intestinal TRPV6 is the one that is most induced by the VDR and is found responsible for the toxic side effect of hypercalcemia (125). Unfortunately, excess calcitriol will elicit the untoward effect of hypercalcemia that deters the use of vitamin D analogs as therapeutic agents.
The possible role of the VDR that targets the clearing receptors of the β-amyloid peptides or Aβ has sparked interests as a mechanism to attenuate the accumulation of Aβ monomers in Alzheimer’s disease (171). The receptors involved are RAGE, a receptor for advanced glycation end products, that allows Aβ entry into the brain, and LRP1, the low-density receptor-related protein 1, that effluxes β-amyloid peptides out of the blood–brain barrier (BBB) (172) (Fig. 5). Guo et al. (173) reported the regulatory effect of calcitriol on the expression of LRP1 and RAGE, favoring the efflux and inhibiting the influx of Aβ following calcitriol treatment in mice. Studies in rats and mice in our laboratory failed to produce any substantial changes in Lrp1 or Rage (141, 174, 175), likely due to the lower pharmacologic doses used. Our recent studies in guinea pigs that are maintained on the same calcitriol dosing regimen led to increased LRP1 and reduced RAGE (unpublished data of H.B. Peng and K.S. Pang).

VDR regulation of receptors governing the influx (RAGE) and efflux (LRP1 and P-gp) of Aβ1–40 and Aβ1–42 peptides in brain; other benefits of the VDR include anti-inflammatory activities and lowering of reactive oxygen species
Regulation of ABC Transporters: P-gp, MRPs, and BCRP
One of the first discoveries of the VDR on regulation of drug transporters is its ability to increase the multidrug resistance protein (MDR1) and its gene product, P-glycoprotein, which transports many lipophilic substrates (176). The observation has sparked much interests since this ATP-binding cassette (ABC) transporter, P-gp, poses a major obstacle in cancer chemotherapy and other pharmacotherapy (177) by promoting the cellular efflux of anticancer drugs—anthracyclines, steroids, vinca alkyloids, peptides, epipodophyllotoxins, HIV protease inhibitors, taxanes, and others via efflux transporters. The mRNA expression of MDR1, present ubiquitously in major eliminating organs, the liver, kidney (178), and BBB (179), was induced by the VDR in intestinal Caco-2 cells (150) and LS180 cells (38). Tachibana et al. (180) reported that the mRNA expression of MDR1 was induced in intestinal LS174T cells by calcitriol as well as LCA treatment, and the increased mRNA level of MDR1 by calcitriol was higher than that following treatment with LCA (22- vs. 2.5-fold). The mechanism of induction involves direct gene regulation through a response element for VDR (VDRE) on the human MDR1 gene (39). It was further shown that an increase in P-gp function was accompanied by increased P-gp protein in the Caco-2 system; basolateral to apical (B → A) transport of the P-gp substrate digoxin increased in cells treated with 1,25(OH)2D3 (40). Both MDR1 mRNA and P-gp protein expressions were increased following treatment with other vitamin D analogs, 1α(OH)D3 and 1α(OH)D2 (40).
The above in vitro results have been verified in vivo in both rats and mice, although in vivo results are highly tissue-specific. Chow et al. demonstrated that treatment with calcitriol in mice resulted in induction of Mdr1a mRNA and P-gp protein in the rat liver (51) and kidney but not in the intestine, despite the relatively high VDR protein expression levels there (142). The lack of intestinal induction was further verified by a lack of increase in P-gp function following the same treatment (167), although P-gp protein was induced following treatment with 1α(OH)D2 (149). Treatment with calcitriol increased Mdr1 mRNA and P-gp protein in the mouse in vivo as well, in both the kidney and brain, resulting in increased clearance of digoxin from these two tissues compared to vehicle-treated mice (181). Similar to 1,25(OH)2D3, 1α(OH)D3 induced Mdr1a/P-gp in the kidney even to a greater extent (eightfold vs. threefold), due to higher circulating 1,25(OH)2D3, levels elicited by 1α(OH)D3 treatment, compared to direct treatment with 1,25(OH)2D3 (68).
At the BBB, calcitriol induction of murine brain P-gp by calcitriol is found to enhance the clearance of Aβ1-40, the amyloid peptide of 40-amino acid length (182). The increased P-gp expression as well as function in the brain was also observed in vivo; brain efflux of quinidine, a P-gp substrate, was enhanced following 1,25(OH)2D3 treatment in rats due to P-gp induction in the brain (175). Because of the special interests towards brain accumulation and brain transport, induction of MDR1/P-gp in the brain was studied in the rat (RBE4) and human (hCMEC/D3) brain microvessel endothelial cell lines treated with 1,25(OH)2D3 (174). In the RBE4 cells, Mdr1b mRNA and P-gp protein were increased following 1,25(OH)2D3 treatment, accompanied by a decrease in the cellular accumulation of the P-gp substrates, rhodamine 6G (R6G) and HiLyte Fluor-labeled human Aβ1-42 (hAβ1-42). Similar results were seen in the hCMEC/D3 cell line, an increase in MDR1 mRNA and P-gp protein, followed by a decrease in R6G and Hi-Lyte Fluor hAβ1-42 accumulation (174). The same study further demonstrated that in isolated rat brain capillaries, treated with 1,25(OH)2D3, P-gp protein was induced and intraluminal efflux of the fluorescent P-gp substrate [N-ε(4-nitrobenzofurazan-7-yl)-d-Lys(8)]-cyclosporin A (NBD-CSA) was increased compared to those treated with vehicle (174).
The VDR is involved in the regulation of all of multidrug resistance-associated protein (MRP) transporters. Like P-gp, the MRPs are also ATP cassette efflux transporters that mediate the transport of a wide range of substrates. The isoforms are expressed in numerous organs. MRP1 transports organic anions and is ubiquitously expressed in the body (183), while MPR2, which transports bilirubin, conjugates, glucuronidated bile acids, and glutathione adducts (184, 185), is abundantly expressed in the liver, kidney, and intestine. MRP3 is found in the colon, kidney, and liver (186, 187). MRP4 is present in the intestine, liver, kidney (188, 189), and BBB (190), and transports nucleoside analogs (191) as well as glucuronide (192) and glutathione conjugates (189, 193). Fan et al. (40) found that calcitriol treatment of Caco-2 cells induces human MRP2 and MRP4, though post-transcriptionally for the latter. Higher expression of MRP2, which excretes drug/conjugates and endobiotics at the apical membrane, is found in numerous systems following calcitriol treatment. In Caco-2 cells, increased MRP2 expression and B → A transport activity of the MRP2 substrate 5-(and-6)-carboxy-2′,7′-dichlorofluorescein ensues following 1,25(OH)2D3 treatment (40). This effect was mirrored following treatment with other vitamin D analogs, 1α(OH)D3 and 1α(OH)D2 but not LCAa (40). In the rat intestine (142), but not the kidney and liver (51, 142), calcitriol treatment induced protein expression of Mrp2 and was accompanied by a change in function, evidenced by increased B → A efflux of 5-(and-6)-carboxy-2′,7′-dichlorofluorescein using the everted sac model (167). It was further shown that in rat and human intestine slices, LCA-induced MRP2 mRNA levels (158), and that 1α(OH)D3 induced Mrp2 in the mouse kidney (156, 157).
The inductive changes for MRP3 with VDR are not that consistent. In Caco-2 cells, no significant change in MRP3 mRNA expression was observed following treatment with 1,25(OH)2D3, 1α(OH)D2, and LCAa. In contrast, MRP3 mRNA level was increased in human intestine but not in liver slices following treatment with LCA (158). Rat Mrp3 mRNA expression was increased in liver (51) and intestine (142) following calcitriol treatment, as was mouse Mrp3 expression in kidney with 1α(OH)D3 (156); the change was once again greater for 1,25(OH)2D3 compared to 1α(OH)D3 (68). Mrp3 expression was shown to be increased in the mouse colon following treatment with LCA, the VDR, and FXR ligands (69) in mouse in vivo. It is surmised that the discrepancy in the extent of VDR induction could be due to the relative abundances of the VDR present in tissues.
MRP4 protein induction was observed in the Caco-2 cell line in response to treatment with calcitriol without alteration in MRP4 mRNA expression (40), a change attributed to post-translational regulation. Treatment of cells with cycloheximide, an inhibitor of protein synthesis, revealed that the VDR increases the stability of MRP4 protein rather than increasing it at the transcriptional level (40). The MRP4 substrate, adefovir dipivoxil, is transported into Caco-2 cells (194). An increase in MRP4 function was observed in the Caco-2 system; B → A transport of the MRP4 substrate, adefovir, was greater across the cell monolayer when cells were treated with calcitriol (167). Similar results were seen for Mrp4 in rat intestine following calcitriol treatment; Mrp4 protein increased (142) with concomitant increase in Mrp4 function—enhanced B → A transport of adefovir in the everted gut sac model (167). Vitamin D analogs elicited similar effects as calcitriol; 1α(OH)D3 induced Mrp4 in the mouse intestine (156), kidney (68, 156, 157), and liver (68); the renal induction was verified to be greater for 1α(OH)D3 compared to 1,25(OH)2D3 (68). No significant changes in MRP4 mRNA levels occurred following treatment with 1α(OH)D2 and LCAa in the Caco-2 system. Induction of mouse Mrp2, Mrp3, and Mrp4 in vivo in rat and human intestinal slices was further demonstrated to occur with vitamin D analogs, LCA, and 1α(OH)D3 (156,157,158).
The breast cancer resistance protein (BCRP) is also one of important ABC efflux transporters, and numerous compounds are substates of BCRP. Although PPARγ-mediated regulatory mechanism has been reported (195), the role of VDR in BCRP regulation has not been reviewed thoroughly. Durk et al. (175) and Fan et al. (40) reported that brain Bcrp/BCRP mRNA and protein expression was not altered in rats following repeated injections of 1,25(OH)2D3 (q2d × 4). Moreover, vitamin D-deficiency in mice did not result in significant changes in Bcrp mRNA in liver, kidney, and intestine tissue, with or without 20 µg/kg cholecalciferol treatment (q2d × 4) (61). A review by Gorczyca and Aleksunes (196) also revealed that VDRE is not found in the promoter region of ABCG2. These studies clearly suggest that the VDR is not involved in BCRP regulation.
Concluding Remarks on Significance of the Regulatory Function of the VDR
From this short communication, we learn to appreciate that the role of the VDR is not limited to its own synthesis and degradation, but is associated with other therapeutic and regulatory functions. These noteworthy functions of the VDR on NR crosstalk and drug dispostion should be recognized, in particular on the maintenace of bone health, proper kidney and bone functions, and brain fitness. The VDR strongly exerts tight control on circulating plasma concentrations of the active ligand, calcitriol (Fig. 2), and is paramount in maintaining calcium, phosphate, and bone homeostasis (Fig. 3). VDR-mediated alterations in enzymes, transporters, and receptors (Tables I, II, III, and IV) showing the changes in CYPs, SULT2A1, SLCs, and ABCs such as P-gp and MRPs will affect the disposition of xenobiotics. VDR suppresses SHP in upregulating CYP7A1 expression in the liver (17) (Fig. 4). Increased P-gp activity in brain (Fig. 5) suggests a therapeutic potential in Alzheimer’s disease (AD) due to the anti-inflammatory properties of the VDR (197, 198). The reduction in Aβ plaque (142) and suppression of proinflammatory cytokines in the brain with the VDR are important sequelae towards the preservation of brain health. Of note is that VDR enhances Aβ efflux via LRP1 and P-gp and suppresses influx via RAGE to reduce Aβ accumulation (Fig. 5). Accumulation of Aβ in the brain and siRNA to VDR significantly downregulated the nerve growth factor, showing the importance of the VDR (199). Among rats replenished with low to high doses of vitamin D, dietary vitamin D deficiency increased iNOS expression and oxidative stress in the brain and contributed to significant nitrosative stress in brain and may promote cognitive decline in middle-aged and elderly adults (200). It is surmised that vitamin D deficiency or lowered serum 25(OH)D3 levels increase the prevalence of dementia including AD (201). Vitamin D supplementation with memantine showed a better effect on cognition in AD patients (202). The use of calcitriol treatment has been implicated via clinical studies, indicating a significant correlation between VDR gene polymorphism and AD susceptibility (203, 204). Lastly, the vitamin D receptor is of vital importance in the maintenance of kidney health and offers benefits towards preservation of calcium and phosphate homeostasis and tissue repair, reducing renal fibrosis and systemic and renal inflammation (205). Moreover, antiproliferative properties of calcitriol are appreciated by many cell types, particularly epithelial cells, providing the rationale for adjunct therapy to treat cancer (206, 207).
Given these beneficial effects of the VDR, it would be unreasonable for us to ignore calcitriol as a therapeutic modality. Unfortunately, it is well known that pharmacologic doses of calcitriol cause hypercalcemia (15, 17, 45, 51, 61, 68, 142, 149) due to overstimulation of intestinal TRPV6. Because of its lipophilic nature, calcitriol is able to induce TRPV6 rapidly after intravenous, intraportal, or oral administration, and all routes are equally toxic. Hypercalcemia leads to dehydration, diarrhea, and death, and curtails the use of 1,25(OH)2D3. The need for vitamin D receptor analogs for the treatment of CKD is undisputable (208,209,210). Indeed, the less hypercalcemic paricalcitol (211, 212) and doxercalciferol (213,214,215,216) are used therapeutically in CKD. It is expected that the development of safer, non-calcemic vitamin D analog drugs will broaden the therapeutic landscape for medications that act on the VDR.
Abbreviations
- 1α(OH)D2 :
-
1α-Hydroxyvitamin D2
- 1α(OH)D3 :
-
1α-Hydroxyvitamin D3 or Alfacalcidol®
- 1,24(OH)2D3 :
-
1α,24-Dihydroxyvitamin D3 or Tacalcitol®
- 1,24,25(OH)3D3 :
-
1α,24,25-Trihydroxyvitamin D3
- 1,25(OH)2D2 :
-
1α,25-Dihydroxyvitamin D2
- 1,25(OH)2D3 :
-
1α,25-Dihydroxyvitamin D3 or calcitriol (active VDR ligand)
- 24,25(OH)2D3 :
-
24,25-Dihydroxyvitamin D3
- 25(OH)D3 :
-
25-Hydroxyvitamin D3
- Aβ:
-
β-Amyloid peptide
- ABC:
-
ATP-binding cassette
- ADL:
-
Alzheimer’s disease
- ASBT:
-
Apical sodium-dependent bile acid transporter
- BBB:
-
Blood–brain barrier
- BCRP:
-
Breast cancer resistance protein
- CAR:
-
Constitutive androstane receptor
- CDCA:
-
Chenodeoxycholic acid
- CKD:
-
Chronic kidney disease
- CYP2R1:
-
Cytochrome P450 isoform 2R1, microsomal vitamin D 25-hydroxylase
- CYP3A4:
-
Cytochrome P450 isoform 3A4
- CYP7A1:
-
Cytochrome P450 isoform 7A1, cholesterol 7α-hydroxylase
- CYP24A1:
-
Cytochrome P450 isoform 24A1, 24-hydroxylase
- CYP27A1:
-
Cytochrome P450 isoform 27A1, mitochondrial vitamin D 25-hydroxylase
- CYP27B11:
-
Cytochrome P450 isoform 27B1, mitochondrial 1α-hydroxylase
- DBD:
-
DNA-binding domain
- DBP:
-
Vitamin D-binding protein
- Fgf15:
-
Fibroblast growth factor 15
- FGF19:
-
Fibroblast growth factor 19
- FGF23:
-
Fibroblast growth factor 23
- FGFR4:
-
Fibroblast growth factor receptor 4
- FRα:
-
Folate receptor-α
- FXR:
-
Farnesoid X receptor
- LBD:
-
Ligand-binding domain
- LBP:
-
Ligand-binding pocket
- LCA:
-
Lithocholic acid
- LRH1:
-
Liver receptor homolog 1
- LRP1:
-
LDL receptor-related protein 1
- LXRα:
-
Liver X receptor-α
- MDR1:
-
Multidrug resistance protein 1
- MRP2:
-
Multidrug resistance-associated protein 2
- MRP3:
-
Multidrug resistance-associated protein 3
- MRP4:
-
Multidrug resistance-associated protein 4
- NaPi :
-
Sodium-dependent phosphate cotransporter
- NR:
-
Nuclear receptor
- OAT1:
-
Organic anion transporter 1
- OAT3:
-
Organic anion transporter 3
- OSTα-OSTβ:
-
Organic solute transporter-α-β
- PCFT:
-
Proton-coupled folate transporter
- PEPT1:
-
Oligopeptide transporter 1
- P-gp:
-
P-Glycoprotein
- PPARα:
-
Peroxisome proliferator-activated receptor-α
- PPARγ:
-
Peroxisome proliferator-activated receptor-γ
- PTH:
-
Parathyroid hormone
- PXR:
-
Pregnane X-receptor
- RAGE:
-
Receptor for advanced glycation end products
- RFC:
-
Reduced folate carrier
- RXR:
-
Retinoid X receptor
- SHP:
-
Small heterodimer partner
- SHPT:
-
Secondary hyperparathyroidism
- SLC:
-
Solute carrier
- SULT2A1:
-
Sulfotransferase 2A1
- TRPV5:
-
Transient receptor potential cation channel, subfamily V, member 5
- TRPV6:
-
Transient receptor potential cation channel, subfamily V, member 6
- UGT:
-
UDP glucuronosyltransferase
- VDR:
-
Vitamin D receptor
- VDRE:
-
Vitamin D response element
References
Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23(5):687–702. https://doi.org/10.1210/er.2001-0038.
Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol. 2004;66(3):413–9. https://doi.org/10.1124/mol.66.3.
Prakash C, Zuniga B, Song CS, Jiang S, Cropper J, Park S, et al. Nuclear receptors in drug metabolism, drug response and drug interactions. Nucl Receptor Res. 2015;2. doi: https://doi.org/10.11131/2015/101178.
Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–6. https://doi.org/10.1126/science.1070477.
Thompson PD, Jurutka PW, Whitfield GK, Myskowski SM, Eichhorst KR, Dominguez CE, et al. Liganded VDR induces CYP3A4 in small intestinal and colon cancer cells via DR3 and ER6 vitamin D responsive elements. Biochem Biophys Res Commun. 2002;299(5):730–8. https://doi.org/10.1016/s0006-291x(02)02742-0.
Xu Y, Iwanaga K, Zhou C, Cheesman MJ, Farin F, Thummel KE. Selective induction of intestinal CYP3A23 by 1α,25-dihydroxyvitamin D3 in rats. Biochem Pharmacol. 2006;72(3):385–92. https://doi.org/10.1016/j.bcp.2006.04.033.
Echchgadda I, Song CS, Roy AK, Chatterjee B. Dehydroepiandrosterone sulfotransferase is a target for transcriptional induction by the vitamin D receptor. Mol Pharmacol. 2004;65(3):720–9. https://doi.org/10.1124/mol.65.3.720.
Chatterjee B, Echchgadda I, Song CS. Vitamin D receptor regulation of the steroid/bile acid sulfotransferase SULT2A1. Methods Enzymol. 2005;400:165–91. https://doi.org/10.1016/S0076-6879(05)00010-8.
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835–9. https://doi.org/10.1016/0092-8674(95)90199-x.
Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53(3):1023–34. https://doi.org/10.1002/hep.24148.
Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276(40):36863–4. https://doi.org/10.1074/jbc.R100047200.
Sandgren ME, Bronnegard M, DeLuca HF. Tissue distribution of the 1,25-dihydroxyvitamin D3 receptor in the male rat. Biochem Biophys Res Commun. 1991;181(2):611–6. https://doi.org/10.1016/0006-291x(91)91234-4.
Liesegang A, Singer K, Boos A. Vitamin D receptor amounts across different segments of the gastrointestinal tract in Brown Swiss and Holstein Frisean cows of different age. J Anim Physiol Anim Nutr (Berl). 2008;92(3):316–23. https://doi.org/10.1111/j.1439-0396.2007.00782.x.
Colston K, Hirt M, Feldman D. Organ distribution of the cytoplasmic 1,25-dihydroxycholecalciferol receptor in various mouse tissues. Endocrinology. 1980;107(6):1916–22. https://doi.org/10.1210/endo-107-6-1916.
Chow EC, Quach HP, Vieth R, Pang KS. Temporal changes in tissue 1α,25-dihydroxyvitamin D3, vitamin D receptor target genes, and calcium and PTH levels after 1,25(OH)2D3 treatment in mice. Am J Physiol Endocrinol Metab. 2013;304(9):E977–89. https://doi.org/10.1152/ajpendo.00489.2012.
Gascon-Barre M, Demers C, Mirshahi A, Neron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003;37(5):1034–42. https://doi.org/10.1053/jhep.2003.50176.
Chow EC, Magomedova L, Quach HP, Patel R, Durk MR, Fan J, et al. Vitamin D receptor activation down-regulates the small heterodimer partner and increases CYP7A1 to lower cholesterol. Gastroenterology. 2014;146(4):1048–59. https://doi.org/10.1053/j.gastro.2013.12.027.
Prufer K, Veenstra TD, Jirikowski GF, Kumar R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. 1999;16(2):135–45. https://doi.org/10.1016/s0891-0618(99)00002-2.
Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1α-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21–30. https://doi.org/10.1016/j.jchemneu.2004.08.006.
Kamei Y, Kawada T, Fukuwatari T, Ono T, Kato S, Sugimoto E. Cloning and sequencing of the gene encoding the mouse vitamin D receptor. Gene. 1995;152(2):281–2. https://doi.org/10.1016/0378-1119(94)00735-b.
Shaffer PL, McDonnell DP, Gewirth DT. Characterization of transcriptional activation and DNA-binding functions in the hinge region of the vitamin D receptor. Biochemistry. 2005;44(7):2678–85. https://doi.org/10.1021/bi0477182.
Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289(1):F8-28. https://doi.org/10.1152/ajprenal.00336.2004.
Hourai S, Fujishima T, Kittaka A, Suhara Y, Takayama H, Rochel N, et al. Probing a water channel near the A-ring of receptor-bound 1α,25-dihydroxyvitamin D3 with selected 2α-substituted analogues. J Med Chem. 2006;49(17):5199–205. https://doi.org/10.1021/jm0604070.
Vanhooke JL, Benning MM, Bauer CB, Pike JW, DeLuca HF. Molecular structure of the rat vitamin D receptor ligand binding domain complexed with 2-carbon-substituted vitamin D3 hormone analogues and a LXXLL-containing coactivator peptide. Biochemistry. 2004;43(14):4101–10. https://doi.org/10.1021/bi036056y.
Feldman D, P JM. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bonekey Rep. 2014;3:510. doi: https://doi.org/10.1038/bonekey.2014.5.
Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173–9. https://doi.org/10.1016/s1097-2765(00)80413-x.
Maestro MA, Molnar F, Carlberg C. Vitamin D and its synthetic analogs. J Med Chem. 2019;62(15):6854–75. https://doi.org/10.1021/acs.jmedchem.9b00208.
Wan LY, Zhang YQ, Chen MD, Du YQ, Liu CB, Wu JF. Relationship between structure and conformational change of the vitamin D receptor ligand binding domain in 1alpha,25-dihydroxyvitamin D3 signaling. Molecules. 2015;20(11):20473–86. https://doi.org/10.3390/molecules201119713.
Yamada S, Yamamoto K. Ligand recognition by vitamin D receptor: total alanine scanning mutational analysis of the residues lining the ligand binding pocket of vitamin D receptor. Curr Top Med Chem. 2006;6(12):1255–65. https://doi.org/10.2174/156802606777864881.
Brumbaugh PF, Haussler MR. Specific binding of 1α,25-dihydroxycholecalciferol to nuclear components of chick intestine. J Biol Chem. 1975;250(4):1588–94.
Walters MR, Hunziker W, Norman AW. Unoccupied 1,25-dihydroxyvitamin D3 receptors. Nuclear/cytosol ratio depends on ionic strength. J Biol Chem. 1980;255(14):6799–805.
Taketani Y, Segawa H, Chikamori M, Morita K, Tanaka K, Kido S, et al. Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3. Identification of a vitamin D-responsive element in the human NAPi-3 gene. J Biol Chem. 1998;273(23):14575–81. doi: https://doi.org/10.1074/jbc.273.23.14575.
Dawson PA, Markovich D. Transcriptional regulation of the sodium-sulfate cotransporter NaSi-1 gene. Cell Biochem Biophys. 2002;36(2–3):175–82. https://doi.org/10.1385/CBB:36:2-3:175.
den Dekker E, Hoenderop JG, Nilius B, Bindels RJ. The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium. 2003;33(5–6):497–507. https://doi.org/10.1016/s0143-4160(03)00065-4.
Haussler MR, Jurutka PW, Mizwicki M, Norman AW. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)2vitamin D3: genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab. 2011;25(4):543–59. https://doi.org/10.1016/j.beem.2011.05.010.
Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Watkins PB. Induction of CYP3A4 by 1α,25-dihydroxyvitamin D3 is human cell line-specific and is unlikely to involve pregnane X receptor. Drug Metab Dispos. 2001;29(11):1446–53.
Pfrunder A, Gutmann H, Beglinger C, Drewe J. Gene expression of CYP3A4, ABC-transporters (MDR1 and MRP1-MRP5) and hPXR in three different human colon carcinoma cell lines. J Pharm Pharmacol. 2003;55(1):59–66. https://doi.org/10.1111/j.2042-7158.2003.tb02434.x.
Aiba T, Susa M, Fukumori S, Hashimoto Y. The effects of culture conditions on CYP3A4 and MDR1 mRNA induction by 1α,25-dihydroxyvitamin D3 in human intestinal cell lines, Caco-2 and LS180. Drug Metab Pharmacokinet. 2005;20(4):268–74. https://doi.org/10.2133/dmpk.20.268.
Saeki M, Kurose K, Tohkin M, Hasegawa R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem Pharmacol. 2008;76(4):531–42. https://doi.org/10.1016/j.bcp.2008.05.030.
Fan J, Liu S, Du Y, Morrison J, Shipman R, Pang KS. Up-regulation of transporters and enzymes by the vitamin D receptor ligands, 1α,25-dihydroxyvitamin D3 and vitamin D analogs, in the Caco-2 cell monolayer. J Pharmacol Exp Ther. 2009;330(2):389–402. https://doi.org/10.1124/jpet.108.149815.
Jin CH, Kerner SA, Hong MH, Pike JW. Transcriptional activation and dimerization functions in the human vitamin D receptor. Mol Endocrinol. 1996;10(8):945–57. https://doi.org/10.1210/mend.10.8.8843411.
Colnot S, Lambert M, Blin C, Thomasset M, Perret C. Identification of DNA sequences that bind retinoid X receptor-1,25(OH)2D3-receptor heterodimers with high affinity. Mol Cell Endocrinol. 1995;113(1):89–98. https://doi.org/10.1016/0303-7207(95)03618-h.
Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol. 2002;16(11):2538–46. https://doi.org/10.1210/me.2002-0152.
Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Wang Y, Busby SA, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat Struct Mol Biol. 2011;18(5):556–63. https://doi.org/10.1038/nsmb.2046.
Chen X, Chen F, Liu S, Glaeser H, Dawson PA, Hofmann AF, et al. Transactivation of rat apical sodium-dependent bile acid transporter and increased bile acid transport by 1α,25-dihydroxyvitamin D3 via the vitamin D receptor. Mol Pharmacol. 2006;69(6):1913–23. https://doi.org/10.1124/mol.105.020792.
Reschly EJ, Krasowski MD. Evolution and function of the NR1I nuclear hormone receptor subfamily (VDR, PXR, and CAR) with respect to metabolism of xenobiotics and endogenous compounds. Curr Drug Metab. 2006;7(4):349–65. https://doi.org/10.2174/138920006776873526.
Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005;115(1):177–86. https://doi.org/10.1172/JCI21867.
Moreau A, Maurel P, Vilarem MJ, Pascussi JM. Constitutive androstane receptor-vitamin D receptor crosstalk: consequence on CYP24 gene expression. Biochem Biophys Res Commun. 2007;360(1):76–82. https://doi.org/10.1016/j.bbrc.2007.06.003.
Honjo Y, Sasaki S, Kobayashi Y, Misawa H, Nakamura H. 1,25-dihydroxyvitamin D3 and its receptor inhibit the chenodeoxycholic acid-dependent transactivation by farnesoid X receptor. J Endocrinol. 2006;188(3):635–43. https://doi.org/10.1677/joe.1.06105.
Jiang W, Miyamoto T, Kakizawa T, Nishio SI, Oiwa A, Takeda T, et al. Inhibition of LXRα signaling by vitamin D receptor: possible role of VDR in bile acid synthesis. Biochem Biophys Res Commun. 2006;351(1):176–84. https://doi.org/10.1016/j.bbrc.2006.10.027.
Chow EC, Maeng HJ, Liu S, Khan AA, Groothuis GM, Pang KS. 1α,25-Dihydroxyvitamin D3 triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm Drug Dispos. 2009;30(8):457–75. https://doi.org/10.1002/bdd.682.
Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem. 2010;285(19):14486–94. https://doi.org/10.1074/jbc.M110.116004.
Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–26. https://doi.org/10.1016/s1097-2765(00)00051-4.
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217–25. https://doi.org/10.1016/j.cmet.2005.09.001.
Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, et al. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem. 2003;278(22):19909–16. https://doi.org/10.1074/jbc.M207903200.
Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter α-β, Ostα-Ostβ, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280(8):6960–8. https://doi.org/10.1074/jbc.M412752200.
Li H, Chen F, Shang Q, Pan L, Shneider BL, Chiang JY, et al. FXR-activating ligands inhibit rabbit ASBT expression via FXR-SHP-FTF cascade. Am J Physiol Gastrointest Liver Physiol. 2005;288(1):G60–6. https://doi.org/10.1152/ajpgi.00170.2004.
Sertznig P, Dunlop T, Seifert M, Tilgen W, Reichrath J. Cross-talk between vitamin D receptor (VDR)- and peroxisome proliferator-activated receptor (PPAR)-signaling in melanoma cells. Anticancer Res. 2009;29(9):3647–58.
Sertznig P, Seifert M, Tilgen W, Reichrath J. Activation of vitamin D receptor (VDR)- and peroxisome proliferator-activated receptor (PPAR)-signaling pathways through 1,25(OH)2D3 in melanoma cell lines and other skin-derived cell lines. Dermatoendocrinol. 2009;1(4):232–8. https://doi.org/10.4161/derm.1.4.9629.
Alimirah F, Peng X, Yuan L, Mehta RR, von Knethen A, Choubey D, et al. Crosstalk between the peroxisome proliferator-activated receptor γ (PPARγ) and the vitamin D receptor (VDR) in human breast cancer cells: PPARγ binds to VDR and inhibits 1α,25-dihydroxyvitamin D3 mediated transactivation. Exp Cell Res. 2012;318(19):2490–7. https://doi.org/10.1016/j.yexcr.2012.07.020.
Quach HP, Noh K, Hoi SY, Bruinsma A, Groothuis GMM, Li AP, et al. Alterations in gene expression in vitamin D-deficiency: down-regulation of liver Cyp7a1 and renal Oat3 in mice. Biopharm Drug Dispos. 2018;39(2):99–115. https://doi.org/10.1002/bdd.2118.
Kim YC, Kim IB, Noh CK, Quach HP, Yoon IS, Chow ECY, et al. Effects of 1α,25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J Pharm Sci. 2014;103(11):3793–805. https://doi.org/10.1002/jps.24195.
Simboli-Campbell M, Franks DJ, Welsh J. 1,25(OH)2D3 increases membrane associated protein kinase C in MDBK cells. Cell Signal. 1992;4(1):99–109. https://doi.org/10.1016/0898-6568(92)90011-v.
Simboli-Campbell M, Gagnon A, Franks DJ, Welsh J. 1,25-Dihydroxyvitamin D3 translocates protein kinase C beta to nucleus and enhances plasma membrane association of protein kinase C alpha in renal epithelial cells. J Biol Chem. 1994;269(5):3257–64.
Takeda M, Sekine T, Endou H. Regulation by protein kinase C of organic anion transport driven by rat organic anion transporter 3 (rOAT3). Life Sci. 2000;67(9):1087–93. https://doi.org/10.1016/s0024-3205(00)00694-9.
Redling S, Pfaff IL, Leitges M, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta I, beta II, delta, and epsilon in mouse kidney. Am J Physiol Renal Physiol. 2004;287(2):F289–98. https://doi.org/10.1152/ajprenal.00273.2003.
Pfaff IL, Wagner HJ, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta1 and betaII in rat kidney. J Am Soc Nephrol. 1999;10(9):1861–73. https://doi.org/10.1681/ASN.V1091861.
Chow EC, Durk MR, Maeng HJ, Pang KS. Comparative effects of 1α-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 on transporters and enzymes in fxr(+/+) and fxr(-/-) mice. Biopharm Drug Dispos. 2013;34(7):402–16. https://doi.org/10.1002/bdd.1856.
McCarthy TC, Li X, Sinal CJ. Vitamin D receptor-dependent regulation of colon multidrug resistance-associated protein 3 gene expression by bile acids. J Biol Chem. 2005;280(24):23232–42. https://doi.org/10.1074/jbc.M411520200.
Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-α and -β genes. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G476–85. https://doi.org/10.1152/ajpgi.00430.2005.
Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40(1):149–56. https://doi.org/10.1002/hep.20295.
Xu S, Sun AQ, Suchy FJ. A novel RARalpha/CAR-mediated mechanism for regulation of human organic solute transporter-beta gene expression. Am J Physiol Gastrointest Liver Physiol. 2014;306(2):G154–62. https://doi.org/10.1152/ajpgi.00138.2013.
Park S, Cheng SL, Cui JY. Characterizing drug-metabolizing enzymes and transporters that are bona fide CAR-target genes in mouse intestine. Acta Pharm Sin B. 2016;6(5):475–91. https://doi.org/10.1016/j.apsb.2016.07.004.
Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121(1):140–7. https://doi.org/10.1053/gast.2001.25503.
Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276(31):28857–65. https://doi.org/10.1074/jbc.M011610200.
Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai SY, Moustafa T, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1124–30. https://doi.org/10.1152/ajpgi.00539.2005.
Zollner G, Fickert P, Fuchsbichler A, Silbert D, Wagner M, Arbeiter S, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. 2003;39(4):480–8. https://doi.org/10.1016/s0168-8278(03)00228-9.
Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42(2):420–30. https://doi.org/10.1002/hep.20784.
Brommage R, DeLuca HF. Evidence that 1,25-dihydroxyvitamin D3 is the physiologically active metabolite of vitamin D3. Endocr Rev. 1985;6(4):491–511. https://doi.org/10.1210/edrv-6-4-491.
Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78(4):1193–231. https://doi.org/10.1152/physrev.1998.78.4.1193.
Bouillon R, Schuit F, Antonio L, Rastinejad F. Vitamin D binding protein: a historic overview. Front Endocrinol (Lausanne). 2019;10:910. https://doi.org/10.3389/fendo.2019.00910.
Cooke NE, Haddad JG. Vitamin D binding protein (Gc-globulin). Endocr Rev. 1989;10(3):294–307. https://doi.org/10.1210/edrv-10-3-294.
Heaney RP, Horst RL, Cullen DM, Armas LA. Vitamin D3 distribution and status in the body. J Am Coll Nutr. 2009;28(3):252–6. https://doi.org/10.1080/07315724.2009.10719779.
Sakaki T, Kagawa N, Yamamoto K, Inouye K. Metabolism of vitamin D3 by cytochromes P450. Front Biosci. 2005;10:119–34. https://doi.org/10.2741/1514.
Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxylase. J Biol Chem. 2003;278(39):38084–93. https://doi.org/10.1074/jbc.M307028200.
DeLuca HF. The vitamin D story: a collaborative effort of basic science and clinical medicine. FASEB J. 1988;2(3):224–36.
Reddy GS, Tserng KY. Calcitroic acid, end product of renal metabolism of 1,25-dihydroxyvitamin D3 through C-24 oxidation pathway. Biochemistry. 1989;28(4):1763–9. https://doi.org/10.1021/bi00430a051.
Noh K, Yang QJ, Sekhon L, Quach HP, Chow ECY, Pang KS. Noteworthy idiosyncrasies of 1α,25-dihydroxyvitamin D3 kinetics for extrapolation from mouse to man: commentary. Biopharm Drug Dispos. 2020;41(3):126–48. https://doi.org/10.1002/bdd.2223.
Meyer MB, Pike JW. Mechanistic homeostasis of vitamin D metabolism in the kidney through reciprocal modulation of Cyp27b1 and Cyp24a1 expression. J Steroid Biochem Mol Biol. 2020;196: 105500. https://doi.org/10.1016/j.jsbmb.2019.105500.
Gao C, Bergagnini-Kolev MC, Liao MZ, Wang Z, Wong T, Calamia JC, et al. Simultaneous quantification of 25-hydroxyvitamin D3–3-sulfate and 25-hydroxyvitamin D3–3-glucuronide in human serum and plasma using liquid chromatography-tandem mass spectrometry coupled with DAPTAD-derivatization. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1060:158–65. https://doi.org/10.1016/j.jchromb.2017.06.017.
Wang Z, Schuetz EG, Xu Y, Thummel KE. Interplay between vitamin D and the drug metabolizing enzyme CYP3A4. J Steroid Biochem Mol Biol. 2013;136:54–8. https://doi.org/10.1016/j.jsbmb.2012.09.012.
Wang Z, Wong T, Hashizume T, Dickmann LZ, Scian M, Koszewski NJ, et al. Human UGT1A4 and UGT1A3 conjugate 25-hydroxyvitamin D3: metabolite structure, kinetics, inducibility, and interindividual variability. Endocrinology. 2014;155(6):2052–63. https://doi.org/10.1210/en.2013-2013.
Cooke NE, Walgate J, Haddad JG, Jr. Human serum binding protein for vitamin D and its metabolites. II. Specific, high affinity association with a protein in nucleated tissue. J Biol Chem. 1979;254(13):5965–71.
Bikle DD, Siiteri PK, Ryzen E, Haddad JG. Serum protein binding of 1,25-dihydroxyvitamin D: a reevaluation by direct measurement of free metabolite levels. J Clin Endocrinol Metab. 1985;61(5):969–75. https://doi.org/10.1210/jcem-61-5-969.
Bikle DD, Halloran BP, Gee E, Ryzen E, Haddad JG. Free 25-hydroxyvitamin D levels are normal in subjects with liver disease and reduced total 25-hydroxyvitamin D levels. J Clin Invest. 1986;78(3):748–52. https://doi.org/10.1172/JCI112636.
Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. 1999;103(2):239–51. https://doi.org/10.1172/JCI5244.
Yamane T, Takeuchi K, Yamamoto Y, Li YH, Fujiwara M, Nishi K, et al. Legumain from bovine kidney: its purification, molecular cloning, immunohistochemical localization and degradation of annexin II and vitamin D-binding protein. Biochim Biophys Acta. 2002;1596(1):108–20. https://doi.org/10.1016/s0167-4838(02)00209-1.
Fraser DR, Kodicek E. Unique biosynthesis by kidney of a biological active vitamin D metabolite. Nature. 1970;228(5273):764–6. https://doi.org/10.1038/228764a0.
Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, et al. Cloning of human 25-hydroxyvitamin D-1α-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961–70. https://doi.org/10.1210/mend.11.13.0035.
Anderson PH, Hendrix I, Sawyer RK, Zarrinkalam R, Manavis J, Sarvestani GT, et al. Co-expression of CYP27B1 enzyme with the 1.5kb CYP27B1 promoter-luciferase transgene in the mouse. Mol Cell Endocrinol. 2008;285(1–2):1–9. doi: https://doi.org/10.1016/j.mce.2007.12.018.
Hollis BW. Assessment of vitamin D nutritional and hormonal status: what to measure and how to do it. Calcif Tissue Int. 1996;58(1):4–5. https://doi.org/10.1007/BF02509538.
Vieth R. What is the optimal vitamin D status for health? Prog Biophys Mol Biol. 2006;92(1):26–32. https://doi.org/10.1016/j.pbiomolbio.2006.02.003.
Ganji V, Zhang X, Tangpricha V. Serum 25-hydroxyvitamin D concentrations and prevalence estimates of hypovitaminosis D in the U.S. population based on assay-adjusted data. J Nutr. 2012;142(3):498–507. doi: https://doi.org/10.3945/jn.111.151977.
van Schoor NM, Lips P. Worldwide vitamin D status. Best Pract Res Clin Endocrinol Metab. 2011;25(4):671–80. https://doi.org/10.1016/j.beem.2011.06.007.
Cianferotti L, Marcocci C. Subclinical vitamin D deficiency. Best Pract Res Clin Endocrinol Metab. 2012;26(4):523–37. https://doi.org/10.1016/j.beem.2011.12.007.
Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int. 2005;16(7):713–6. https://doi.org/10.1007/s00198-005-1867-7.
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911–30. https://doi.org/10.1210/jc.2011-0385.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266–81. https://doi.org/10.1056/NEJMra070553.
Brown AJ. Therapeutic uses of vitamin D analogues. Am J Kidney Dis. 2001;38(5 Suppl 5):S3–S19. https://doi.org/10.1053/ajkd.2001.28111.
Brown AJ, Slatopolsky E. Vitamin D analogs: therapeutic applications and mechanisms for selectivity. Mol Aspects Med. 2008;29(6):433–52. https://doi.org/10.1016/j.mam.2008.04.001.
Sjoden G, Smith C, Lindgren U, DeLuca HF. 1α-Hydroxyvitamin D2 is less toxic than 1α-hydroxyvitamin D3 in the rat. Proc Soc Exp Biol Med. 1985;178(3):432–6. https://doi.org/10.3181/00379727-178-42028.
Nehring JA, Zierold C, DeLuca HF. Lithocholic acid can carry out in vivo functions of vitamin D. Proc Natl Acad Sci U S A. 2007;104(24):10006–9. https://doi.org/10.1073/pnas.0703512104.
Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116(8):2062–72. https://doi.org/10.1172/JCI29449.
Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117(4):503–11. https://doi.org/10.1161/CIRCULATIONAHA.107.706127.
Andress DL. Vitamin D in chronic kidney disease: a systemic role for selective vitamin D receptor activation. Kidney Int. 2006;69(1):33–43. https://doi.org/10.1038/sj.ki.5000045.
Bhattarai HK, Shrestha S, Rokka K, Shakya R. Vitamin D, calcium, parathyroid hormone, and sex steroids in bone health and effects of aging. J Osteoporos. 2020;2020:9324505. https://doi.org/10.1155/2020/9324505.
Silver J, Levi R. Regulation of PTH synthesis and secretion relevant to the management of secondary hyperparathyroidism in chronic kidney disease. Kidney Int Suppl. 2005;95:S8-12. https://doi.org/10.1111/j.1523-1755.2005.09501.x.
Slatopolsky E, Weerts C, Thielan J, Horst R, Harter H, Martin KJ. Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxy-cholecalciferol in uremic patients. J Clin Invest. 1984;74(6):2136–43. https://doi.org/10.1172/JCI111639.
Takeda E, Taketani Y, Sawada N, Sato T, Yamamoto H. The regulation and function of phosphate in the human body. BioFactors. 2004;21(1–4):345–55. https://doi.org/10.1002/biof.552210167.
Goyal R, Jialal I. Hyperphosphatemia. StatPearls. Treasure Island (FL)2021.
Goretti Penido M, Alon US. Phosphate homeostasis and its role in bone health. Pediatr Nephrol. 2012;27(11):2039–48. https://doi.org/10.1007/s00467-012-2175-z.
Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. https://doi.org/10.1146/annurev.med.051308.111339.
Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297(2):F282–91. https://doi.org/10.1152/ajprenal.90742.2008.
Silver J, Naveh-Many T. FGF23 and the parathyroid. Adv Exp Med Biol. 2012;728:92–9. https://doi.org/10.1007/978-1-4614-0887-1_6.
Yang QJ, Bukuroshi P, Quach HP, Chow ECY, Pang KS. Highlighting vitamin D receptor-targeted activities of 1α,25-dihydroxyvitamin D3 in mice via physiologically based pharmacokinetic-pharmacodynamic modeling. Drug Metab Dispos. 2018;46(1):75–87. https://doi.org/10.1124/dmd.117.077271.
Enomoto H, Hendy GN, Andrews GK, Clemens TL. Regulation of avian calbindin-D28K gene expression in primary chick kidney cells: importance of posttranscriptional mechanisms and calcium ion concentration. Endocrinology. 1992;130(6):3467–74. https://doi.org/10.1210/endo.130.6.1375904.
Murer H, Hernando N, Forster I, Biber J. Molecular aspects in the regulation of renal inorganic phosphate reabsorption: the type IIa sodium/inorganic phosphate co-transporter as the key player. Curr Opin Nephrol Hypertens. 2001;10(5):555–61. https://doi.org/10.1097/00041552-200109000-00002.
Hildmann B, Storelli C, Danisi G, Murer H. Regulation of Na+-Pi cotransport by 1,25-dihydroxyvitamin D3 in rabbit duodenal brush-border membrane. Am J Physiol. 1982;242(5):G533–9. https://doi.org/10.1152/ajpgi.1982.242.5.G533.
Hamano T, Nakano C, Obi Y, Fujii N, Matsui I, Tomida K, et al. Fibroblast growth factor 23 and 25-hydroxyvitamin D levels are associated with estimated glomerular filtration rate decline. Kidney Int Suppl (2011). 2013;3(5):469–75. doi: https://doi.org/10.1038/kisup.2013.97.
Koizumi M, Komaba H, Fukagawa M. Parathyroid function in chronic kidney disease: role of FGF23-Klotho axis. Contrib Nephrol. 2013;180:110–23. https://doi.org/10.1159/000346791.
Tani T, Orimo H, Shimizu A, Tsuruoka S. Development of a novel chronic kidney disease mouse model to evaluate the progression of hyperphosphatemia and associated mineral bone disease. Sci Rep. 2017;7(1):2233. https://doi.org/10.1038/s41598-017-02351-6.
Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contrib Nephrol. 2013;180:47–63. https://doi.org/10.1159/000346778.
Thorne J, Campbell MJ. The vitamin D receptor in cancer. Proc Nutr Soc. 2008;67(2):115–27. https://doi.org/10.1017/S0029665108006964.
Voutsadakis IA. Vitamin D baseline levels at diagnosis of breast cancer: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2021;14(1):16–26. https://doi.org/10.1016/j.hemonc.2020.08.005.
Negri M, Gentile A, de Angelis C, Monto T, Patalano R, Colao A, et al. Vitamin D-induced molecular mechanisms to potentiate cancer therapy and to reverse drug-resistance in cancer cells. Nutrients. 2020;12(6). doi: https://doi.org/10.3390/nu12061798.
Henner WD, Beer TM. A new formulation of calcitriol (DN-101) for high-dose pulse administration in prostate cancer therapy. Rev Urol. 2003;5(Suppl 3):S38-44.
Beer TM, Eilers KM, Garzotto M, Egorin MJ, Lowe BA, Henner WD. Weekly high-dose calcitriol and docetaxel in metastatic androgen-independent prostate cancer. J Clin Oncol. 2003;21(1):123–8. https://doi.org/10.1200/jco.2003.05.117.
Muindi JR, Potter DM, Peng Y, Johnson CS, Trump DL. Pharmacokinetics of liquid calcitriol formulation in advanced solid tumor patients: comparison with caplet formulation. Cancer Chemother Pharmacol. 2005;56(5):492–6. https://doi.org/10.1007/s00280-005-1015-2.
Fakih MG, Trump DL, Muindi JR, Black JD, Bernardi RJ, Creaven PJ, et al. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(4):1216–23. https://doi.org/10.1158/1078-0432.CCR-06-1165.
Barrea L, Savanelli MC, Di Somma C, Napolitano M, Megna M, Colao A, et al. Vitamin D and its role in psoriasis: an overview of the dermatologist and nutritionist. Rev Endocr Metab Disord. 2017;18(2):195–205. https://doi.org/10.1007/s11154-017-9411-6.
Durk MR, Han K, Chow EC, Ahrens R, Henderson JT, Fraser PE, et al. 1α,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J Neurosci. 2014;34(21):7091–101. https://doi.org/10.1523/JNEUROSCI.2711-13.2014.
Chow EC, Sun H, Khan AA, Groothuis GM, Pang KS. Effects of 1α,25-dihydroxyvitamin D3 on transporters and enzymes of the rat intestine and kidney in vivo. Biopharm Drug Dispos. 2010;31(1):91–108. https://doi.org/10.1002/bdd.694.
Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 “pie.” Drug Metab Dispos. 2006;34(5):880–6. https://doi.org/10.1124/dmd.105.008672.
Galetin A, Gertz M, Houston JB. Potential role of intestinal first-pass metabolism in the prediction of drug-drug interactions. Expert Opin Drug Metab Toxicol. 2008;4(7):909–22. https://doi.org/10.1517/17425255.4.7.909.
Khan AA, Chow EC, van Loenen-Weemaes AM, Porte RJ, Pang KS, Groothuis GM. Comparison of effects of VDR versus PXR, FXR and GR ligands on the regulation of CYP3A isozymes in rat and human intestine and liver. Eur J Pharm Sci. 2009;37(2):115–25. https://doi.org/10.1016/j.ejps.2009.01.006.
Zierold C, Mings JA, Deluca HF. 19nor-1,25-Dihydroxyvitamin D2 specifically induces CYP3A9 in rat intestine more strongly than 1,25-dihydroxyvitamin D3 in vivo and in vitro. Mol Pharmacol. 2006;69(5):1740–7. https://doi.org/10.1124/mol.105.019851.
Kutuzova GD, DeLuca HF. 1,25-Dihydroxyvitamin D3 regulates genes responsible for detoxification in intestine. Toxicol Appl Pharmacol. 2007;218(1):37–44. https://doi.org/10.1016/j.taap.2006.10.005.
Wang K, Chen S, Xie W, Wan YJ. Retinoids induce cytochrome P450 3A4 through RXR/VDR-mediated pathway. Biochem Pharmacol. 2008;75(11):2204–13. https://doi.org/10.1016/j.bcp.2008.02.030.
Chow EC, Sondervan M, Jin C, Groothuis GM, Pang KS. Comparative effects of doxercalciferol (1α-hydroxyvitamin D2) versus calcitriol (1α,25-dihydroxyvitamin D3) on the expression of transporters and enzymes in the rat in vivo. J Pharm Sci. 2011;100(4):1594–604. https://doi.org/10.1002/jps.22366.
Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Lown KS, Watkins PB. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1α,25-dihydroxyvitamin D3. Mol Pharmacol. 1997;51(5):741–54. https://doi.org/10.1124/mol.51.5.741.
Thummel KE, Brimer C, Yasuda K, Thottassery J, Senn T, Lin Y, et al. Transcriptional control of intestinal cytochrome P-4503A by 1α,25-dihydroxy vitamin D3. Mol Pharmacol. 2001;60(6):1399–406. https://doi.org/10.1124/mol.60.6.1399.
Adachi R, Honma Y, Masuno H, Kawana K, Shimomura I, Yamada S, et al. Selective activation of vitamin D receptor by lithocholic acid acetate, a bile acid derivative. J Lipid Res. 2005;46(1):46–57. https://doi.org/10.1194/jlr.M400294-JLR200.
Jurutka PW, Thompson PD, Whitfield GK, Eichhorst KR, Hall N, Dominguez CE, et al. Molecular and functional comparison of 1,25-dihydroxyvitamin D3 and the novel vitamin D receptor ligand, lithocholic acid, in activating transcription of cytochrome P450 3A4. J Cell Biochem. 2005;94(5):917–43. https://doi.org/10.1002/jcb.20359.
Matsubara T, Yoshinari K, Aoyama K, Sugawara M, Sekiya Y, Nagata K, et al. Role of vitamin D receptor in the lithocholic acid-mediated CYP3A induction in vitro and in vivo. Drug Metab Dispos. 2008;36(10):2058–63. https://doi.org/10.1124/dmd.108.021501.
Drocourt L, Ourlin JC, Pascussi JM, Maurel P, Vilarem MJ. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem. 2002;277(28):25125–32. https://doi.org/10.1074/jbc.M201323200.
Nishida S, Ozeki J, Makishima M. Modulation of bile acid metabolism by 1α-hydroxyvitamin D3 administration in mice. Drug Metab Dispos. 2009;37(10):2037–44. https://doi.org/10.1124/dmd.109.027334.
Ogura M, Nishida S, Ishizawa M, Sakurai K, Shimizu M, Matsuo S, et al. Vitamin D3 modulates the expression of bile acid regulatory genes and represses inflammation in bile duct-ligated mice. J Pharmacol Exp Ther. 2009;328(2):564–70. https://doi.org/10.1124/jpet.108.145987.
Khan AA, Chow EC, Porte RJ, Pang KS, Groothuis GM. The role of lithocholic acid in the regulation of bile acid detoxication, synthesis, and transport proteins in rat and human intestine and liver slices. Toxicol In Vitro. 2011;25(1):80–90. https://doi.org/10.1016/j.tiv.2010.09.011.
Song CS, Echchgadda I, Seo YK, Oh T, Kim S, Kim SA, et al. An essential role of the CAAT/enhancer binding protein-alpha in the vitamin D-induced expression of the human steroid/bile acid-sulfotransferase (SULT2A1). Mol Endocrinol. 2006;20(4):795–808. https://doi.org/10.1210/me.2005-0428.
Seo YK, Chung YT, Kim S, Echchgadda I, Song CS, Chatterjee B. Xenobiotic- and vitamin D-responsive induction of the steroid/bile acid-sulfotransferase Sult2A1 in young and old mice: the role of a gene enhancer in the liver chromatin. Gene. 2007;386(1–2):218–23. https://doi.org/10.1016/j.gene.2006.10.006.
Eloranta JJ, Zair ZM, Hiller C, Hausler S, Stieger B, Kullak-Ublick GA. Vitamin D3 and its nuclear receptor increase the expression and activity of the human proton-coupled folate transporter. Mol Pharmacol. 2009;76(5):1062–71. https://doi.org/10.1124/mol.109.055392.
Alam C, Aufreiter S, Georgiou CJ, Hoque MT, Finnell RH, O’Connor DL, et al. Upregulation of reduced folate carrier by vitamin D enhances brain folate uptake in mice lacking folate receptor alpha. Proc Natl Acad Sci U S A. 2019;116(35):17531–40. https://doi.org/10.1073/pnas.1907077116.
Brandsch C, Zibolka J, Frommhagen M, Lehmann U, Dierkes J, Kuhne H, et al. Vitamin D is not linked to folate status and mRNA expression of intestinal proton-coupled folate transporter. Eur J Nutr. 2014;53(4):1115–22. https://doi.org/10.1007/s00394-013-0614-7.
Alam C, Hoque MT, Finnell RH, Goldman ID, Bendayan R. Regulation of reduced folate carrier (RFC) by vitamin D receptor at the blood-brain barrier. Mol Pharm. 2017;14(11):3848–58. https://doi.org/10.1021/acs.molpharmaceut.7b00572.
Miao Q, Liu Q, Wang C, Meng Q, Guo X, Sun H, et al. Inhibitory effect of 1α,25-dihydroxyvitamin D3 on excretion of JBP485 via organic anion transporters in rats. Eur J Pharm Sci. 2013;48(1–2):351–9. https://doi.org/10.1016/j.ejps.2012.11.008.
Khan AA, Chow EC, Porte RJ, Pang KS, Groothuis GM. Expression and regulation of the bile acid transporter, OSTα-OSTβ in rat and human intestine and liver. Biopharm Drug Dispos. 2009;30(5):241–58. https://doi.org/10.1002/bdd.663.
Maeng HJ, Durk MR, Chow EC, Ghoneim R, Pang KS. 1α,25-Dihydroxyvitamin D3 on intestinal transporter function: studies with the rat everted intestinal sac. Biopharm Drug Dispos. 2011;32(2):112–25. https://doi.org/10.1002/bdd.742.
Zhao R, Min SH, Wang Y, Campanella E, Low PS, Goldman ID. A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J Biol Chem. 2009;284(7):4267–74. https://doi.org/10.1074/jbc.M807665200.
Seward DJ, Koh AS, Boyer JL, Ballatori N. Functional complementation between a novel mammalian polygenic transport complex and an evolutionarily ancient organic solute transporter. OSTα-OSTβ. J Biol Chem. 2003;278(30):27473–82. https://doi.org/10.1074/jbc.M301106200.
Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol. 2006;20(6):1447–61. https://doi.org/10.1210/me.2006-0031.
Patel P, Shah J. Role of vitamin D in amyloid clearance via LRP-1 upregulation in Alzheimer’s disease: a potential therapeutic target? J Chem Neuroanat. 2017;85:36–42. https://doi.org/10.1016/j.jchemneu.2017.06.007.
Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35(11 Suppl 1):2628–31. https://doi.org/10.1161/01.STR.0000143452.85382.d1.
Guo YX, He LY, Zhang M, Wang F, Liu F, Peng WX. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Aβ1-40 brain-to-blood efflux and peripheral uptake transport. Neuroscience. 2016;322:28–38. https://doi.org/10.1016/j.neuroscience.2016.01.041.
Durk MR, Chan GN, Campos CR, Peart JC, Chow EC, Lee E, et al. 1α,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J Neurochem. 2012;123(6):944–53. https://doi.org/10.1111/jnc.12041.
Durk MR, Fan J, Sun H, Yang Y, Pang H, Pang KS, et al. Vitamin D receptor activation induces P-glycoprotein and increases brain efflux of quinidine: an intracerebral microdialysis study in conscious rats. Pharm Res. 2015;32(3):1128–40. https://doi.org/10.1007/s11095-014-1524-y.
Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455(1):152–62. https://doi.org/10.1016/0005-2736(76)90160-7.
Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34(1–2):47–54. https://doi.org/10.1081/dmr-120001389.
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84(21):7735–8. https://doi.org/10.1073/pnas.84.21.7735.
Miller DS, Bauer B, Hartz AM. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60(2):196–209. https://doi.org/10.1124/pr.107.07109.
Tachibana S, Yoshinari K, Chikada T, Toriyabe T, Nagata K, Yamazoe Y. Involvement of vitamin D receptor in the intestinal induction of human ABCB1. Drug Metab Dispos. 2009;37(8):1604–10. https://doi.org/10.1124/dmd.109.027219.
Chow EC, Durk MR, Cummins CL, Pang KS. 1α,25-Dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(-/-) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J Pharmacol Exp Ther. 2011;337(3):846–59. https://doi.org/10.1124/jpet.111.179101.
Ito S, Ohtsuki S, Nezu Y, Koitabashi Y, Murata S, Terasaki T. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1–40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS. 2011;8:20. https://doi.org/10.1186/2045-8118-8-20.
Schinkel AH. The roles of P-glycoprotein and MRP1 in the blood-brain and blood-cerebrospinal fluid barriers. Adv Exp Med Biol. 2001;500:365–72. https://doi.org/10.1007/978-1-4615-0667-6_60.
Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461(2):377–94. https://doi.org/10.1016/s0005-2736(99)00169-8.
Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011;201:299–323. https://doi.org/10.1007/978-3-642-14541-4_8.
Englund G, Rorsman F, Ronnblom A, Karlbom U, Lazorova L, Grasjo J, et al. Regional levels of drug transporters along the human intestinal tract: co-expression of ABC and SLC transporters and comparison with Caco-2 cells. Eur J Pharm Sci. 2006;29(3–4):269–77. https://doi.org/10.1016/j.ejps.2006.04.010.
Rost D, Mahner S, Sugiyama Y, Stremmel W. Expression and localization of the multidrug resistance-associated protein 3 in rat small and large intestine. Am J Physiol Gastrointest Liver Physiol. 2002;282(4):G720–6. https://doi.org/10.1152/ajpgi.00318.2001.
Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell AL, Karlsson J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos. 2007;35(8):1333–40. https://doi.org/10.1124/dmd.107.014902.
van Aubel R, Smeets PHE, Peters JGP, Bindels RJM, Russel FGM. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol. 2002;13(3):595–603. https://doi.org/10.1681/ASN.V133595.
Nies AT, Jedlitschky G, Konig J, Herold-Mende C, Steiner HH, Schmitt HP, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129(2):349–60. https://doi.org/10.1016/j.neuroscience.2004.07.051.
Schuetz JD, Connelly MC, Sun D, Paibir SG, Flynn PM, Srinivas RV, et al. MRP4: a previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat Med. 1999;5(9):1048–51. https://doi.org/10.1038/12487.
Mizuno N, Takahashi T, Kusuhara H, Schuetz JD, Niwa T, Sugiyama Y. Evaluation of the role of breast cancer resistance protein (BCRP/ABCG2) and multidrug resistance-associated protein 4 (MRP4/ABCC4) in the urinary excretion of sulfate and glucuronide metabolites of edaravone (MCI-186; 3-methyl-1-phenyl-2-pyrazolin-5-one). Drug Metab Dispos. 2007;35(11):2045–52. https://doi.org/10.1124/dmd.107.016352.
Lai L, Tan TM. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem J. 2002;361(Pt 3):497–503. https://doi.org/10.1042/0264-6021:3610497.
Maeng HJ, Chapy H, Zaman S, Pang KS. Effects of 1α,25-dihydroxyvitamin D3 on transport and metabolism of adefovir dipivoxil and its metabolites in Caco-2 cells. Eur J Pharm Sci. 2012;46(3):149–66. https://doi.org/10.1016/j.ejps.2012.02.018.
Szatmari I, Vamosi G, Brazda P, Balint BL, Benko S, Szeles L, et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281(33):23812–23. https://doi.org/10.1074/jbc.M604890200.
Gorczyca L, Aleksunes LM. Transcription factor-mediated regulation of the BCRP/ABCG2 efflux transporter: a review across tissues and species. Expert Opin Drug Metab Toxicol. 2020;16(3):239–53. https://doi.org/10.1080/17425255.2020.1732348.
Masuda S, Jones G. Promise of vitamin D analogues in the treatment of hyperproliferative conditions. Mol Cancer Ther. 2006;5(4):797–808. https://doi.org/10.1158/1535-7163.MCT-05-0539.
Ma D, Zhang RN, Wen Y, Yin WN, Bai D, Zheng GY, et al. 1,25(OH)2D3-induced interaction of vitamin D receptor with p50 subunit of NF-kappaB suppresses the interaction between KLF5 and p50, contributing to inhibition of LPS-induced macrophage proliferation. Biochem Biophys Res Commun. 2017;482(2):366–74. https://doi.org/10.1016/j.bbrc.2016.11.069.
Gezen-Ak D, Dursun E, Yilmazer S. The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS ONE. 2011;6(3): e17553. https://doi.org/10.1371/journal.pone.0017553.
Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PH, et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology. 2014;83(10):920–8. https://doi.org/10.1212/WNL.0000000000000755.
Keeney JTR, Forster S, Sultana R, Brewer LD, Latimer CS, Cai J, et al. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med. 2013;65:324–34. https://doi.org/10.1016/j.freeradbiomed.2013.07.019.
Annweiler C, Rolland Y, Schott AM, Blain H, Vellas B, Herrmann FR, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205–11. https://doi.org/10.1093/gerona/gls107.
Laczmanski L, Jakubik M, Bednarek-Tupikowska G, Rymaszewska J, Sloka N, Lwow F. Vitamin D receptor gene polymorphisms in Alzheimer’s disease patients. Exp Gerontol. 2015;69:142–7. https://doi.org/10.1016/j.exger.2015.06.012.
Lee YH, Kim JH, Song GG. Vitamin D receptor polymorphisms and susceptibility to Parkinson’s disease and Alzheimer’s disease: a meta-analysis. Neurol Sci. 2014;35(12):1947–53. https://doi.org/10.1007/s10072-014-1868-4.
Eleftheriadis T, Antoniadi G, Liakopoulos V, Antoniadis N, Stefanidis I, Galaktidou G. Vitamin D receptor activators and response to injury in kidney diseases. J Nephrol. 2010;23(5):514–24.
Zhuang SH, Burnstein KL. Antiproliferative effect of 1α,25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin-dependent kinase 2 activity and persistent G1 accumulation. Endocrinology. 1998;139(3):1197–207. https://doi.org/10.1210/endo.139.3.5770.
Muindi JR, Peng Y, Potter DM, Hershberger PA, Tauch JS, Capozzoli MJ, et al. Pharmacokinetics of high-dose oral calcitriol: results from a phase 1 trial of calcitriol and paclitaxel. Clin Pharmacol Ther. 2002;72(6):648–59. https://doi.org/10.1067/mcp.2002.129305.
Kovesdy CP, Kalantar-Zadeh K. Vitamin D receptor activation and survival in chronic kidney disease. Kidney Int. 2008;73(12):1355–63. https://doi.org/10.1038/ki.2008.35.
Moorthi RN, Kandula P, Moe SM. Optimal vitamin D, calcitriol, and vitamin D analog replacement in chronic kidney disease: to D or not to D: that is the question. Curr Opin Nephrol Hypertens. 2011;20(4):354–9. https://doi.org/10.1097/MNH.0b013e3283470450.
Salusky IB, Kuizon BD, Belin TR, Ramirez JA, Gales B, Segre GV, et al. Intermittent calcitriol therapy in secondary hyperparathyroidism: a comparison between oral and intraperitoneal administration. Kidney Int. 1998;54(3):907–14. https://doi.org/10.1046/j.1523-1755.1998.00045.x.
Coyne D, Acharya M, Qiu P, Abboud H, Batlle D, Rosansky S, et al. Paricalcitol capsule for the treatment of secondary hyperparathyroidism in stages 3 and 4 CKD. Am J Kidney Dis. 2006;47(2):263–76. https://doi.org/10.1053/j.ajkd.2005.10.007.
Greenbaum LA, Benador N, Goldstein SL, Paredes A, Melnick JZ, Mattingly S, et al. Intravenous paricalcitol for treatment of secondary hyperparathyroidism in children on hemodialysis. Am J Kidney Dis. 2007;49(6):814–23. https://doi.org/10.1053/j.ajkd.2007.03.008.
Moe SM, Saifullah A, LaClair RE, Usman SA, Yu Z. A randomized trial of cholecalciferol versus doxercalciferol for lowering parathyroid hormone in chronic kidney disease. Clin J Am Soc Nephrol. 2010;5(2):299–306. https://doi.org/10.2215/CJN.07131009.
Dennis VC, Albertson GL. Doxercalciferol treatment of secondary hyperparathyroidism. Ann Pharmacother. 2006;40(11):1955–65. https://doi.org/10.1345/aph.1G523.
Noonan W, Koch K, Nakane M, Ma J, Dixon D, Bolin A, et al. Differential effects of vitamin D receptor activators on aortic calcification and pulse wave velocity in uraemic rats. Nephrol Dial Transplant. 2008;23(12):3824–30. https://doi.org/10.1093/ndt/gfn375.
Mizobuchi M, Finch JL, Martin DR, Slatopolsky E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int. 2007;72(6):709–15. https://doi.org/10.1038/sj.ki.5002406.
Acknowledgements
KSP acknowledges the support of NSERC and CIHR.
Author information
Authors and Affiliations
Contributions
Wrote or contributed to the writing of the manuscript: KN, HPQ, ECYC, GMMG, RGT, and KSP.
Contributed data in manuscripts: HPQ, ECYC, KN, GMMG, RGT, and KSP
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Noh, K., Chow, E.C.Y., Quach, H.P. et al. Significance of the Vitamin D Receptor on Crosstalk with Nuclear Receptors and Regulation of Enzymes and Transporters. AAPS J 24, 71 (2022). https://doi.org/10.1208/s12248-022-00719-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-022-00719-9