
Abstract
Background
The evolutionally conserved homeobox transcription factor NKX2-5 has been at the forefront in the field of cardiac biology, providing molecular insights into the mechanisms of cardiac development and disease. This homodomain transcription factor is a central regulator of cardiac development and is expressed in both the first and second heart fields (FHF and SHF). Mutations in the NKX2-5 gene have been linked to sporadic cases of congenital heart disease (CHD), making it a significant target for research and study. While several studies have been conducted on Caucasian populations, there is a dearth of knowledge on the effects of NKX2-5 gene mutations in other settings, underscoring the need for further investigation. Due to differences in geographical and ancestral origin, we hypothesize that mutations may vary across different populations. Understanding the genetic factors that cause CHD is essential for providing effective genetic counseling and developing strategies for risk reduction. Additionally, identification of NKX2-5 mutations in individuals with CHDs is crucial because patients with CHDs are at a higher risk of progressive conduction disease and sudden cardiac death, and genetic information is taken into consideration while making decisions regarding pacemakers and implantable cardiac defibrillators. To determine the risk of congenital heart disease among infants, we conducted a study where we sequenced the exon 1 and exon 2 of NKX 2.5 in patients with sporadic CHDs, with the aim of identifying mutations in the NKX2.5 gene.
Results
In this study, a novel frame-shift disease-causing mutation was discovered in patients with atrial-ventricular septal defect. The mutation, identified as c95_95 del A; cDNA.369–369 delA; g 369–369 delA, resulted in the substitution of phenylalanine to leucine (F295L), which in turn caused a truncated NKX2.5 protein. In addition, a non-synonymous mutation, g 316C > T; cDNA 316C > T leucine to arginine (L37R) substitution, was found in a patient with the tetralogy of Fallot, affecting protein function. Furthermore, a novel non-synonymous mutation identified as g 2295–2298; cDNA 755–758 delins AGGG, was predicted by mutation taster to be disease-causing in a ventricular septal defect. It is worth noting that none of these mutations were found among the control subjects, highlighting their potential significance in the pathogenesis of these cardiac defects.
Conclusion
Mutations in the NKX2.5 gene are associated with congenital heart diseases and provide molecular insight into the pathogenesis of congenital heart diseases. We recommend that patients with NKX2.5 mutations have periodic screening for cardiac conduction abnormalities and be evaluated for potential implanted cardiac defibrillators and pacemakers.
Similar content being viewed by others

Background
Congenital heart disease (CHD) is a type of structural abnormality present in the heart and great vessels since birth. It is a common congenital anomaly that affects approximately 8 out of 1000 live births globally[1]. CHD is a significant cause of birth defect-related mortality [2, 3]. The prevalence of CHD is increasing globally, with unmet diagnostic needs observed in Africa [4]. CHD has a multifactorial basis that involves genetic and environmental components [5, 6].
The NKX 2.5 transcription factor is an important protein that plays a vital role in the development of the heart. It is a highly conserved homeobox transcription factor and consists of two exons that encode 324 amino acids. It is situated on chromosome 5q34 [7, 8] and is expressed in the early cardiac mesoderm [9]. It is a homo-domain transcription factor that acts as a central regulator of cardiac development and is expressed in both the first and second heart fields, known as FHF and SHF, respectively [10, 11]. Various studies conducted on animal models have shown that NKX2.5 knockout mice are embryonically lethal, highlighting the critical role of this factor in cardiac development [12, 13].
Over the past decade, it has been the focus of extensive research, providing valuable molecular insights into the mechanisms underlying heart development and related disorders. The proper functioning of NKX2-5 is essential for normal cardiac development, and any abnormalities in this gene have been linked to congenital heart disorders in humans [14]. In fact, NKX2.5 mutations have been identified as key factors responsible for various forms of CHD, making this transcription factor a critical target for further study and potential therapeutic interventions [15].
People with CHD have been found to have over 40 heterozygous NKX2-5 germline mutations; these mutations are dispersed throughout the coding area and affect protein function [16]. Heterozygous mutation of human NKX2.5 has been associated with various congenital heart diseases such as atrial septal defect (ASD), ventricular septal defect, tetralogy of Fallot, and tricuspid valve abnormalities, including Ebstein's anomaly [17]. Furthermore, NKX2.5 mutations have been associated with atrial septal defects and hypoplastic left heart syndrome (ASD and HLHS) and have also been found in patients with tetralogy of Fallot, secundum ASD, truncus arteriosus, double-outlet right ventricle, TOF, and familial ASD [18,19,20,21,22,23]. Mutation of human NKX2.5 has been associated with various congenital heart diseases and can also cause atrioventricular (AV) conduction block in a patient with CHDs [17].
Patients with atrial septal defect (ASD) who have mutations in the NKX2-5 gene are more likely to develop conduction disturbances, which can eventually lead to sudden cardiac death. For such patients, it is highly recommended to consider implantation of an implantable cardioverter defibrillator that can effectively prevent sudden cardiac death [24]. Studies have reported that point mutations in the NKX2-5 gene can lead to the development of familial ASD (autosomal dominant form)[25]. Additionally, heterozygous mutations in the same gene have been linked to the occurrence of atrial septal defect (ASD) and/or atrioventricular (AV) conduction disturbance in certain families [26, 27]. The NKX2.5 gene is also associated with congenital heart diseases, with two specific single nucleotide polymorphisms (SNPs), rs2277923 and rs703752, having been identified in previous research [28,29,30,31,32]. However, the prevalence of these SNPs and their correlation with congenital heart diseases remain unknown in our current setting. The NKX 2.5 mutations and their relationship to CHD characteristics are summarized in Table 1.
In our country, advanced diagnostic facilities have become more widely available and as a result, the prevalence of congenital heart diseases (CHDs) has increased. Although both genetic and environmental factors have been suggested as possible causes of this condition, the exact etiology is still unknown. This lack of understanding about the factors contributing to CHDs can impede risk control measures, genetic counseling, and the ability to predict disease outcomes. Therefore, it is crucial to gain a better understanding of the etiological factors underlying CHDs as this knowledge can enhance the clinical management of the condition by enabling accurate prognosis predictions.
In individuals with CHDs, identifying mutations in the NKX2-5 gene is of utmost clinical significance as it helps in predicting the risk of progressive conduction disease and sudden cardiac death. Such genetic information is crucial for making informed decisions regarding the use of pacemakers and implantable cardiac defibrillators. Additionally, understanding the genetic basis of CHDs is essential for providing appropriate genetic counseling and developing risk-reduction strategies. In this study, we sequenced exon 1 and exon 2 of NKX 2.5 in patients with sporadic congenital heart diseases to determine NKX2.5 transcription factor gene mutations and the risk of congenital heart diseases among infants.
Methods
A retrospective case–control study was conducted among infants at the Jakaya Kikwete Cardiac Institute (JKCI), Tanzania . We used a convenience sampling technique to obtain 70 cases with isolated congenital heart diseases and 70 health control subjects. Echocardiography was performed by a pediatric cardiologist.
Blood sample collection and DNA extraction
Blood samples were collected for the purpose of DNA extraction using DBS paper. The blood was collected by filling each well on the DBS paper with a single drop of blood, which was then left to dry in the open air for one hour. The dried DBS paper was then packed in specialized air-tight parcels for transfer and subsequent DNA extraction.
For genomic DNA extraction, the Quick-DNA Miniprep Kit protocol was followed. The extracted DNA was then quantified and its quality was assayed by the use of a computer-based Thermo Scientific NanoDrop™ 1000 Spectrophotometer. This apparatus is capable of measuring microvolumes of dilute and concentrated DNA samples of varying concentrations with high accuracy and reproducibility. The DNA samples were then diluted accordingly to equal concentrations of 10 ng/µl for the next step of analysis.
Primer design and PCR amplification of NKX2.5 gene (Exon 1 and Exon 2) for mutation screening
To identify the reference gene sequence of NKX2.5, the National Center for Biotechnology Information (NCBI) was used. Based on published NKX2-5 coding regions (exon 1 and exon 2) sequences, allele-specific oligonucleotide primers kit was designed using bioinformatics pipelines Primer 3 software. The primers were validated using the Program Basic Local Alignment Search Tool (BLAST) and were also checked for hairpin, homodimer, and heterodimer using Integrated DNA Technologies (IDT) oligo Analyzer software.
Due to alternative splicing NKX2.5 has five transcript variants. The variant considered for this study was Homo sapiens NK2 homeobox 5 (NKX2-5), transcript variant 1, mRNA (NM_004387.4). Exon 1 and Exon 2 of the NKX2.5 gene were amplified using the designed primers. The primers for exon 1 were Forward Primer 5’- CGCCCTTCTCAGTCAAAGAC-3’ (20 bp), GC content 55%, and Reverse primer 3’- AAAGGCAGACGCACACTTG-5’ (19 bp), GC content 52.6%. The primers for exon 2 were Forward Primer 5’- CCTCAACAGCTCCCTGACTC-3’ (20 bp), GC content 60% and Reverse primer 3’- CTCATTGCACGCTGCATAAT-5’ (20 bp), GC content 45%. The designed primers (forward and reverse) were manufactured by Microgen Europe B.V. Company in Amsterdam, Netherlands.
For PCR amplification, AccuPower® PCR PreMix from Bioneer (Bioneer Corporation, 8–11 Munpyeongseo-ro, Daedeok-gu, Daejeon 306–220, Republic of Korea) was used on GeneAmp® PCR System 9700 (Applied Biosystems). The PCR mixture consisted of 2 μl of extracted DNA, 1 μl of forward primer, 1 μl of reverse primer, and 16 μl of nuclease-free water in a micro-tube containing AccuPower® PCR PreMix concentrate making a total reaction volume of 20 μl. Cycling conditions consisted of initial denaturation at 95◦C for 5 min followed by 35 cycles of 95◦C for 30 s, 57 ◦C for 30 s, and 72 ◦C for 45 min. A final extension at 72 ◦C for 5 min was performed to complete the extension.
Gel electrophoresis of PCR products
For the analysis of PCR products, a gel electrophoresis technique was used. A 1.5% agarose gel was prepared by dissolving 1.5 g agarose in 100 ml of 1X Sodium borate buffer and heating it on a hot plate until it was completely dissolved. The gel was then stained with 4 μl of GelRed® Nucleic Acid Stain. In each well of the gel, 4 μl of every sample and 4 μl of the 100-bp DNA ladder were loaded. The first well was loaded with the DNA ladder to indicate the size of any fragments. The voltage was set to 100 V, and electrophoresis was allowed to run for 40 min. The DNA fragments were observed as gray bands against a black background on Bio-Rad’s Gel Doc™ EZ Imaging System. Figures 1 and 2 show the gel electrophoresis results for exon 1 and exon 2, respectively.

Gel electrophoresis exon 1

Gel electrophoresis exon 2
DNA sequencing
The amplicons, which are amplified segments of DNA, were sent to Macrogen Europe (Meibergdreef 57, 1105 BA, Amsterdam, the Netherlands) for sequencing. The sequencing was performed on samples from cases and controls, with a sample size of 70 for each (Case: Control; n = 70 + 70). To conduct the sequencing, gene-specific primers were used to amplify the DNA segments. The PCR products were then purified and sequenced directly using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). The sequencing was conducted using a genetic analyzer (ABI 3730xl System from Applied Biosystems). After the sequencing was completed, the raw sequence data were cleaned, edited, and assembled using the Geneious Prime software, which is version 2021.2.2. The software was used to obtain consensus sequences, which are sequences that represent the most common base at each position of the DNA segment. Finally, the sequences were aligned to a reference genome known as hg37, using ClustalW, which is a software used to align multiple DNA or protein sequences. The alignment allowed for the identification of any mutations present in the DNA segments (Figs. 3 and 4).

Multiple sequence Alignment exon 1

Multiple sequence Alignment exon 2
Single nucleotide polymorphisms and the subsequent changes in the amino acids were detected. The obtained nucleotide sequences were also subjected to pair-wise alignment using the heuristic sequence alignment approach, Basic Alignment Search Tool to determine the identity and similarity of the study nucleotide sequences and protein sequences by comparing them with other published sequences available in the GenBank database.
The possible effects of genetic variants (mutation pathogenicity analysis) were predicted whether they are pathogenic or tolerated using computational analysis methods such as SIFT (Sorting Intolerant from Tolerant) [43, 44]; PolyPhen-2 (Polymorphism Phenotyping, version 2)[45, 46] MutationTaster [47]. The protein secondary structure was predicted using a Swiss model [48, 49].
For the SIFT score, amino acids with probabilities of less than 0.05 are predicted to be deleterious, and a SIFT score of greater than 0.05 is considered to signify a tolerated variant. PolyPhen-2 results are presented with qualitative levels of benign, potentially detrimental, and probably damaging. PolyPhen-2 prediction results have a numerical score range of 0 to 1. PolyPhen-2 variations with scores equal to or greater than 0.5 are anticipated to be deleterious. For the Mutation Taster, we classified an alteration as either a disease-causing polymorphism, a polymorphism probably harmless, or a polymorphism known to be harmless.
Results: for exon 1:
A non-synonymous mutation g 316C > T; cDNA 316C > T Leucine 37 Arginine in a patient with the tetralogy of Fallot was predicted by mutation taster to be disease-causing and by using PolyPhen; the mutation was predicted to be possibly damaging with a score of 0.655, sensitivity of 0.79, and specificity of 0.84. Another novel synonymous mutation g 2295–2298; cDNA 755–758 delins AGGG: in a ventral septal defect was predicted by mutation taster to be disease-causing. Not found in ExAC (exome aggregation consortium) or 1000G (Fig. 5).

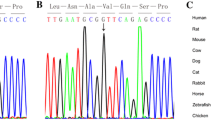
Polyphen multiple sequence alignment prediction: amino acid leucine at mutation position 37 is highly conserved in the NKX2.5 protein in different species
Another non-synonymous g 225G > T; g 245C > A; g 257C > A; g 410G > T; 429 T > C, resulted in leucine48 Methionine substitution in a patient with patent ductous arteriosus. Using SIFT, the substitution leucine48 Methionine is predicted to affect protein function with a score of 0.04 and median sequence conservation of 3.37, and using polyphen, the mutation was predicted to be probably damaging with a score of 0.963, sensitivity of 0.62, and specificity of 0.92(Fig. 6).

Polyphen multiple sequence alignment prediction: the amino acid leucine at the mutation site is highly conserved in the NKX2.5 protein in different species
We found synonymous mutations g 457 T > G; g 63 T > G in a patient with atrial septal defects resulting in K110Q, F92V, and L41R. Using the SIFT algorithm, substitution at position 110 from K to Q is predicted to be TOLERATED with a score of 0.42. Median sequence conservation: 3.18 Sequences represented at this position. Substitution at position 92 from F to V is predicted to be TOLERATED with a score of 0.38. Median sequence conservation: 3.18. Sequences represented at this position:9. Substitution at position 41 from L to R is predicted to be TOLERATED with a score of 0.15. Median sequence conservation: 3.41 Sequences represented at this position also found synonymous mutations, g 11G > C; g 19G > A; g 71 insertion T was found in a patient with ventral septal.
For exon 2: We found an alteration in 3UTR;cDNA 1282 T > C;g2822T > C which was predicted to be polymorphism in a patient with ventral septal defect. In another sample, we found a novel frameshift, disease-causing mutation; c95_95 del A; cDNA.369–369 delA;g 369-369delA which resulted in phenylalanine 295 leucine substitution in patients with atrial-ventricular septal defect which resulted in a truncated NKX2.5 protein (Figs. 7,8 and 9).

Polyphen multiple sequence alignment prediction: amino acid phenylalanine at mutation position 295 is highly conserved in the NKX2.5 protein in different species

PSIPRED.A wild-type NKX2.5 PROTEIN

PSIPED.A mutant NKX2.5 PROTEIN (c95_95 del A in exon 2)
Discussion
The early development of the vertebrate heart is a complex and coordinated process that involves the specification and differentiation of myocardial and endocardial cells in the anterior lateral mesoderm shortly after gastrulation. This culminates in the formation and rightward looping of the early heart tube, which is the precursor to the mature heart[50].This intricate process involves multiple cell types and a highly regulated machinery of genetic events involving transcriptional [51, 52]. One of the key transcriptional factors involved in cardiac development is NKX2-5, which is a homeodomain-containing transcription factor that plays a critical role in the development of heart [37]. It is indispensable for normal cardiac development, and mutations of the gene are associated with human congenital heart diseases (CHD)[14]. NKX2-5 mutations alter a section of the protein called the homeodomain, which interferes with the ability of NKX2-5 to bind to DNA or associate with other important cardiac proteins called cofactors [53]. Humans who are born with congenital heart disease carry mutations in the gene that encodes this protein [54].
In a recent study, researchers have identified a single nucleotide polymorphism (SNP) of the NKX2.5 gene, which is associated with congenital heart defects (CHD) [55]. The study detected five different variants, including the previously documented p.R25C variant, in eleven families. Interestingly, the p.R25C variant was found in seven patients from different families and one healthy individual [56]. Other studies have also identified various genetic variations in different CHD phenotypes. For instance, a novel heterozygous DNA sequence variant 1433A > G was discovered in a patient with tetralogy of Fallot (TOF), while another was found in a patient with a persistent left superior vena cava [57]. Another study identified several single nucleotide polymorphisms (SNPs) (rs2277923, rs3729753, rs703752, and rs202071628), and a novel heterozygous DNA sequence variant (1500G > C) in the 3'UTR region of the NKX2-5 gene, in two [2] VSD patients [32]. In our study, we identified a novel single nucleotide polymorphism (SNP) in 3UTR of NKX2.5; cDNA 1282 T > C; g2822T > C in a patient with ventral septal defect. These findings provide valuable insights into the genetic architecture of CHD and may have important implications for the diagnosis and treatment of these conditions.
Furthermore, various mutation analyses carried out in different research studies have discovered a new kind of heterozygous NKX2.5 mutation, known as p.Q181X, in a patient who suffers from ASD [38]. Additionally, another study has identified two different types of NKX2-5 missense mutations—T178M—in a family with ASD but without AV (atrioventricular) conduction block. Moreover, the same study also found a missense change, E21Q, in a patient with ASD who did not have any AV block [23]. In our study, however, we found non-synonymous mutations c 457 T > G and c 63 T > G in a patient with atrial septal defects without AV block, which resulted in K110Q,F92V and L41R amino acid changes that were tolerated and not disease-causing.
In a previous study, researchers identified several genetic mutations associated with different cardiac conditions. Among the mutations identified were missense nucleotide substitutions, deletions, and a premature termination of translation. One of the mutations identified was a missense mutation (R190L) observed in patients with atrial septal defect type II (ASD-II), and a frame-shift mutation (A255fsX38) observed in a patient with hypoplastic left heart syndrome (HLHS)[20]. In our study, we discovered a novel frame-shift mutation in exon 2; c95_95 del A; cDNA.369-369 delA; g 369-369delA which resulted in a substitution of phenylalanine to leucine (F295L) in a patient with atrial-ventricular septal defect (AVSD). This genetic mutation caused a truncated NKX2.5 protein, which is typically associated with AVSD.
A common genetic variation, known as single nucleotide polymorphism (SNP), c.63 A > G (E21E), and c.606 G > C (L202L), has been identified among individuals with congenital heart diseases in the Chinese population [58]. Additionally, a study conducted on CHD patients found four NKX2.5 mutations. Among those, two were novel genetic variations in the coding region of exon 1 (c. 95 A > T and c. 93 A > T), while the other two were previously reported as single nucleotide polymorphisms (SNPs)—rs72554028 (c. 2357 G > A) and rs3729753 (c. 606 G > C) in exon [59].
In our study, however, we found a novel non-synonymous mutation in exon 2, g 316C > T; cDNA 316C > T Leucine 37 Arginine in a patient with the tetralogy of Fallot, which was predicted by mutation taster to be disease-causing. We also found a novel non-synonymous mutation g 2295–2298; cDNA 755–758 delins AGGG: in a ventral septal defect was predicted by mutation taster to be disease-causing. Another sequence variation c 225G > T; c 245C > A; c 257C > A; c 410G > T; c429T > C, which resulted in leucine48 methionine substitution in a patient with patent ductous arteriosus, was detected in our study.
These variations have never been reported from other studies. These findings could play a crucial role in understanding the underlying genetic mechanisms of these conditions and may help in the development of new treatment options.
Conclusions
Our research has led us to discover specific genetic variants that are linked to various phenotypes of congenital heart diseases. Through our analysis, we have identified that mutations in the NKX2.5 gene could play a crucial role in the development of structural cardiac defects in humans. Our findings have significant implications in the clinical management of patients with these mutations. We advise that individuals with NKX2.5 mutations should undergo regular screening to detect any potential cardiac conduction abnormalities. Additionally, they should be evaluated for the possibility of implanted cardiac defibrillators and pacemakers to manage any cardiac complications that may arise.
Availability of data and materials
Patient data were obtained from patient files from inpatient and outpatient departments and reproductive and child health cards. Blood samples were collected using DBS paper. All the references in this manuscript were cited from PubMed and Google Scholar. For data management and referencing, we use Endnote software version 8.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- PCR:
-
Polymerase chain reaction
- SNP:
-
Single nucleotide polymorphism
- DBS:
-
Dry blood sample
- ASD:
-
Atrial septal defect
- VSD:
-
Ventral septal defects
- AVSD:
-
Atrial ventral septal defects
- TOF:
-
Tetralogy of Fallot
- PDA:
-
Persistent ductus arteriosus
- JKCI:
-
Jakaya Kikwete Cardiac Institute
- CHD:
-
Congenital heart disease
- MUHAS:
-
Muhimbili University of Health and Allied Sciences
References
van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ et al (2011) Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247
Nees SN, Chung WK (2020) Genetic Basis of Human Congenital Heart Disease. Cold Spring Harb Perspect Biol 12(9):036749
Cowan JR, Ware SM (2015) Genetics and genetic testing in congenital heart disease. Clin Perinatol 42(2):373–393
Liu Y, Chen S, Zühlke L, Black GC, Choy MK, Li N et al (2019) Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol 48(2):455–463
Shabana NA, Shahid SU, Irfan U (2020) Genetic Contribution to Congenital Heart Disease (CHD). Pediatr Cardiol 41(1):12–23
Zaidi S, Brueckner M (2017) Genetics and Genomics of Congenital Heart Disease. Circ Res 120(6):923–940
Banerjee-Basu S, Baxevanis AD (2001) Molecular evolution of the homeodomain family of transcription factors. Nucleic Acids Res 29(15):3258–3269
Turbay D, Wechsler SB, Blanchard KM, Izumo S (1996) Molecular cloning, chromosomal mapping, and characterization of the human cardiac-specific homeobox gene hCsx. Molecular medicine (Cambridge, Mass) 2(1):86–96
Harvey RP (1996) NK-2 homeobox genes and heart development. Dev Biol 178(2):203–216
Colombo S, de Sena-Tomás C, George V, Werdich AA, Kapur S, MacRae CA et al (2018) Nkx genes establish second heart field cardiomyocyte progenitors at the arterial pole and pattern the venous pole through Isl1 repression. Development. https://doi.org/10.1242/dev.161497
Guner-Ataman B, Paffett-Lugassy N, Adams MS, Nevis KR, Jahangiri L, Obregon P et al (2013) Zebrafish second heart field development relies on progenitor specification in anterior lateral plate mesoderm and nkx2.5 function. Development 140(6):1353–1363
Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L et al (1995) Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 9(13):1654–1666
Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D (2001) The combinatorial activities of Nkx25 and dHAND are essential for cardiac ventricle formation. Dev Biol 239(2):190–203
Akazawa H, Komuro I (2005) Cardiac transcription factor Csx/Nkx2-5: Its role in cardiac development and diseases. Pharmacol Ther 107(2):252–268
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2009) Screening NKX2.5 mutation in a sample of 230 Han Chinese children with congenital heart diseases. Genet Test Mol Biomark 13(2):159–162
Reamon-Buettner SM, Borlak J (2010) NKX2-5: an update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD). Hum Mutat 31(11):1185–1194
Ikeda Y, Hiroi Y, Hosoda T, Utsunomiya T, Matsuo S, Ito T et al (2002) Novel point mutation in the cardiac transcription factor CSX/NKX25 associated with congenital heart disease. Circul J Off J Jpn Circul Soc 66(6):561–563
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H et al (2005) Phenotypes with GATA4 or NKX25 mutations in familial atrial septal defect. Am J Med Genet Part A 135(1):47–52
Goldmuntz E, Geiger E, Benson DW (2001) NKX2.5 mutations in patients with tetralogy of fallot. Circulation 104(21):2565–2568
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E (2003) NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 42(9):1650–1655
Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S et al (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104(11):1567–1573
Stallmeyer B, Fenge H, Nowak-Göttl U, Schulze-Bahr E (2010) Mutational spectrum in the cardiac transcription factor gene NKX25 (CSX) associated with congenital heart disease. Clinical Genet 78(6):533–540
Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P et al (2003) Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol 41(11):2072–2076
Ellesøe SG, Johansen MM, Bjerre JV, Hjortdal VE, Brunak S, Larsen LA (2016) Familial Atrial septal defect and sudden cardiac death: identification of a novel NKX2-5 mutation and a review of the literature. Congenit Heart Dis 11(3):283–290
Hosoda T, Komuro I, Shiojima I, Hiroi Y, Harada M, Murakawa Y et al (1999) Familial atrial septal defect and atrioventricular conduction disturbance associated with a point mutation in the cardiac homeobox gene CSX/NKX2-5 in a Japanese patient. Jpn Circ J 63(5):425–426
Gutierrez-Roelens I, De Roy L, Ovaert C, Sluysmans T, Devriendt K, Brunner HG et al (2006) A novel CSX/NKX2-5 mutation causes autosomal-dominant AV block: are atrial fibrillation and syncopes part of the phenotype? Eur J Human Genet EJHG 14(12):1313–1316
Gutierrez-Roelens I, Sluysmans T, Gewillig M, Devriendt K, Vikkula M (2002) Progressive AV-block and anomalous venous return among cardiac anomalies associated with two novel missense mutations in the CSX/NKX2-5 gene. Hum Mutat 20(1):75–76
Ashiq S, Ashiq K, Sabar MF (2021) The role of NKX2–5 gene polymorphisms in congenital heart disease (CHD): a systematic review and meta-analysis. Egypt Heart J Egypt Soc Cardiol 73(1):72
Zhao M, Diao J, Huang P, Li J, Li Y, Yang Y et al (2020) Association of maternal diabetes mellitus and polymorphisms of the NKX25 gene in children with congenital heart disease: a single centre-based case-control study. J Diabet Res 2020:3854630
González-Castro TB, Tovilla-Zárate CA, López-Narvaez ML, Juárez-Rojop IE, Calderón-Colmenero J, Sandoval JP et al (2020) Association between congenital heart disease and NKX2.5 gene polymorphisms: systematic review and meta-analysis. Biomark Med 14(18):1747–1757
Yin J, Qian J, Dai G, Wang C, Qin Y, Xu T et al (2019) Search of Somatic Mutations of NKX2-5 and GATA4 genes in Chinese patients with sporadic congenital heart disease. Pediatr Cardiol 40(1):17–22
Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H et al (2016) Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene 575(1):29–33
Peng T, Wang L, Zhou SF, Li X (2010) Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica 138(11–12):1231–1240
Salazar M, Consoli F, Villegas V, Caicedo V, Maddaloni V, Daniele P et al (2011) Search of somatic GATA4 and NKX2.5 gene mutations in sporadic septal heart defects. Eur J Med Genet 54(3):306–309
Wang J, Lu Y, Chen H, Yin M, Yu T, Fu Q (2011) Investigation of somatic NKX2-5, GATA4 and HAND1 mutations in patients with tetralogy of Fallot. Pathology 43(4):322–326
Gioli-Pereira L, Pereira AC, Mesquita SM, Xavier-Neto J, Lopes AA, Krieger JE (2010) NKX2.5 mutations in patients with non-syndromic congenital heart disease. Int J Cardiol 138(3):261–265
Reamon-Buettner SM, Hecker H, Spanel-Borowski K, Craatz S, Kuenzel E, Borlak J (2004) Novel NKX2-5 mutations in diseased heart tissues of patients with cardiac malformations. Am J Pathol 164(6):2117–2125
Xu YJ, Qiu XB, Yuan F, Shi HY, Xu L, Hou XM et al (2017) Prevalence and spectrum of NKX2.5 mutations in patients with congenital atrial septal defect and atrioventricular block. Mol Med Rep 15(4):2247–2254
Reamon-Buettner SM, Borlak J (2004) Somatic NKX2-5 mutations as a novel mechanism of disease in complex congenital heart disease. J Med Genet 41(9):684–690
Liu XY, Wang J, Yang YQ, Zhang YY, Chen XZ, Zhang W et al (2011) Novel NKX2-5 mutations in patients with familial atrial septal defects. Pediatr Cardiol 32(2):193–201
Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G et al (2005) Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 42(2):e16
Watanabe Y, Benson DW, Yano S, Akagi T, Yoshino M, Murray JC (2002) Two novel frameshift mutations in NKX25 result in novel features including visceral inversus and sinus venosus type ASD. J Med Genet 39(11):807–811
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40:W452–W457
Hu J, Ng PC (2013) SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS ONE 8(10):e77940
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protocols Hum Genet. https://doi.org/10.1002/0471142905.hg0720s76
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep 7(1):10480
Stephenson A, Adams JW, Vaccarezza M (2017) The vertebrate heart: an evolutionary perspective. J Anat 231(6):787–797
Miquerol L, Kelly RG (2013) Organogenesis of the vertebrate heart. Wiley Interdiscip Rev Dev Biol 2(1):17–29
Floriani MA, Glaeser AB, Dorfman LE, Agnes G, Rosa RFM, Zen PRG (2021) GATA 4 deletions associated with congenital heart diseases in South Brazil. J. Pediatr. Genet. 10(2):92–97
Bouveret R, Waardenberg AJ, Schonrock N, Ramialison M, Doan T, de Jong D et al (2015) NKX2-5 mutations causative for congenital heart disease retain functionality and are directed to hundreds of targets. Elife 4:06942
Chung IM, Rajakumar G (2016) Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 7(2):6
Draus JM Jr, Hauck MA, Goetsch M, Austin EH 3rd, Tomita-Mitchell A, Mitchell ME (2009) Investigation of somatic NKX2-5 mutations in congenital heart disease. J Med Genet 46(2):115–122
Abou Hassan OK, Fahed AC, Batrawi M, Arabi M, Refaat MM, DePalma SR et al (2015) NKX2-5 mutations in an inbred consanguineous population: genetic and phenotypic diversity. Sci Rep 5:8848
Cao Y, Lan W, Li Y, Wei C, Zou H, Jiang L (2015) Single nucleotide polymorphism of NKX2-5 gene with sporadic congenital heart disease in Chinese Bai population. Int J Clin Exp Pathol 8(11):14917–14924
Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q et al (2015) Investigation of somatic NKX2-5 mutations in Chinese children with congenital heart disease. Int J Med Sci 12(7):538–543
Khatami M, Mazidi M, Taher S, Heidari MM, Hadadzadeh M (2018) Novel point mutations in the NKX2.5 gene in pediatric patients with non-familial congenital heart disease. Medicina (Kaunas) 54(3):46
Acknowledgements
We would like to thank Shandong University, the University of Muhimbili, and the Jakaya Kikwete Cardiac Center for providing the necessary facilities and guidance for effective composition and submission of the manuscript for publication.
Funding
The corresponding author received funds for writing this manuscript from the Chinese Government Scholarship (CSC).
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the manuscript. The manuscript has been read and approved by all of the authors. ES helped in concept drafting of the manuscript, literature review, data analysis, and manuscript writing; JM was involved in research methods, literature review; EM contributed to manuscript corrections, molecular methods; MM helped in molecular methods, data analysis; JM was involved in clinical method review; HM helped in literature review; LS contributed to technical support, guidelines, drafting, manuscript corrections, and supervision.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Muhimbili University Institutional Review Board provided ethical approval for this work. Permission to gather data was acquired from the Director of the Jakaya Kikwete Cardiac Institute Research and Publication Committee (Ref.No.DA.282/298/01.C/MUHAS-REC-06–2021-666) and (Ref: AB.123/307/01E/16), respectively. We asked participants for permission to participate in this study with their children. Participants were informed of the study's purpose and their rights, including the opportunity to withdraw from the study at any time without losing any benefits.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest or competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suluba, E., Masaganya, J., Mbugi, E. et al. NKX2.5 coding exons sequencing reveals novel non-synonymous mutations in patients with sporadic congenital heart diseases among the Tanzanian population. Egypt J Med Hum Genet 25, 86 (2024). https://doi.org/10.1186/s43042-024-00557-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00557-8