
Abstract
Atrial septal defect (ASD) is a common cardiovascular malformation and an important contributor to substantial morbidity and mortality. Increasing evidence demonstrates that mutated NKX2-5, a gene encoding a homeobox transcription factor crucial to cardiogenesis, is a significant genetic determinant for congenital ASD. Nevertheless, the genetic basis for ASD in a majority of ASD patients remains largely unknown. In the current study, the entire coding region of NKX2-5 was sequenced initially for 58 unrelated probands with familial ASD. The relatives of the probands harboring identified mutations and 200 unrelated control individuals were subsequently genotyped. Three novel heterozygous NKX2-5 mutations (p.P43GfsX59, p.C46 W, and p.S179F) were identified respectively in three families with autosomal dominantly inherited ASD. These mutations, absent in 200 control individuals, cosegregated with ASD in the families that had complete penetrance. The findings expand the spectrum of mutations in NKX2-5 linked to ASD and contribute to genetic counseling, clinical interventions, and prenatal prevention of ASD for individuals with genetic susceptibility.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Congenital heart disease, the most prevalent form of birth defect in the structure of a neonate’s heart and great vessels, affecting nearly 1% of newborns, is the most common cause of infant death resulting from birth abnormality. More than 29% of the infants who die of a birth defect have a heart defect [24].
Atrial septal defect (ASD), accounting for approximately 33% of all congenital cardiovascular malformations, affects more than 3 in 1,000 live births [25]. Due to an anatomically deficient interatrial septum, ASD allows blood to flow directly between the left and right atria. Categorized by whether they involve other structures of the heart and how they are formed during the cardiac developmental process, ASDs are classified clinically into five types: ostium secundum ASD, patent foramen ovale, ostium primum ASD, sinus venosus ASD, and common or single atrium. Of these five types, ostium secundum ASD is the most frequent defect, representing 85% of all ASDs [18].
Congenital ASD may be isolated or associated with other cardiac anomalies such as ventricular septal defect, pulmonary valve stenosis, and conduction defects. Irrespective of other malformations that may accompany ASD, interatrial communication bidirections and thus persistent blood shunt between two atria may result in cardiac enlargement, congestive heart failure, pulmonary hypertension, Eisenmenger’s syndrome, arrhythmias, and even sudden cardiac death in the absence of surgical or catheter-based repair [37].
Obviously, ASD is an important contributor to morbidity and mortality in infancy. Nevertheless, the etiology responsible for ASD in most patients remains largely unknown [19, 30]. An abnormally developed atrial septum is implicated in a heterogeneous, complex biologic process associated with environmental and genetic risk factors [19, 30].
Increasing evidence has highlighted the crucial role of several transcription factors, including NKX2-5, in the septogenesis [30]. The human NKX2-5 gene maps to chromosome 5q34 and consists of two exons encoding a protein of 324 amino acids. This homeobox transcription factor NKX2-5, expressed during early cardiac morphogenesis, is indispensible for normal cardiac development. Therefore, NKX2-5 has been a prime candidate gene in studies to identify the genetic determinants of structural congenital heart defects [1, 3].
To date, more than 40 mutations within the NKX2-5 gene have been identified in patients with a variety of congenital heart malformations including ASDs with normal or abnormal atrioventricular conduction, ventricular septal defects, conotruncal abnormalities such as tetralogy of Fallot, double-outlet right ventricle, L-transposition of the great arteries, and hypoplastic left heart syndrome [34, 38]. These observations strongly suggest that NKX2-5 is important in the later stages of heart development and maturation in addition to its role in cardiac progenitor commitment and patterning in the developing heart [32].
In this study, the coding exons of NKX2-5 were sequenced in 58 unrelated probands with familial ASD, and three mutations (p.P43GfsX59, p.C46 W, and p.S179F) were identified in three ASD probands, respectively. Genetic analysis of the families carrying mutations demonstrated that in each family, the mutation cosegregated with ASD.
Materials and Methods
Study Subjects
A total of 58 unrelated kindreds with familial ASD were identified among the Han Chinese population. The subjects were evaluated by individual and familial history, review of medical records, complete physical examination, 12-lead electrocardiogram (ECG), and two-dimensional transthoracic echocardiography with color flow Doppler. All the patients had a classic form of ASD with a defect diameter larger than 5 mm. Nearly all the patients underwent cardiac catheterization and, if required, cardiac surgery.
Family history was obtained by multiple interviews with varied family members, and the phenotypes of family members were determined by medical records, physical examination, echocardiogram, and ECG. All the family members recruited underwent an ECG. A total of 200 ethnically matched, unrelated healthy individuals derived from the general Han Chinese population were used as control subjects to screen for identified mutations in NKX2-5.
Peripheral venous blood specimens from the subjects and the control individuals were prepared. The study protocol was reviewed and approved by the local institutional ethics committee, and written informed consent was obtained from all the participants or their guardians before the investigation began.
Genetic Studies
Genomic DNA for all the participants was extracted from blood lymphocytes with the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). Initially, the coding exons and exon/intron boundaries of the candidate gene NKX2-5 for 58 unrelated probands with familial ASD were screened in their entirety. Subsequently, genotyping NKX2-5 for the available relatives of probands carrying mutations and the 200 ethnically matched unrelated healthy control individuals was conducted for the presence of mutations identified in the probands.
The referential genomic DNA sequence of NKX2-5 was derived from GenBank (accession no. NT_023133). By the aid of online Primer 3 software (http://frodo.wi.mit.edu), the primer pairs used to amplify the complete coding region of NKX2-5 by polymerase chain reaction (PCR) were designed as follows: primer 1 forward 5′-CAC, GAT, GCA, GGG, AAG, CTG-3′ and reverse 5′-AGT, TTC, TTG, GGG, ACG, AAA, GC-3′ (the PCR product was 477 base pairs [bp] in size); primer 2 forward 5′-CCT, CCA, CGA, GGA, TCC, CTT, AC-3′ and reverse 5′-CGA, GTC, CCC, TAG, GCA, TGG-3′ (the product was 463 bp); and primer 3 forward 5′-AGA, ACC, GGC, GCT, ACA, AGT, G-3′ and reverse 5′-GAG, TCA, GGG, AGC, TGT, TGA, GG-3′ (the product was 473 bp).
The PCR was carried out using HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a PE 9700 Thermal Cycler (Applied Biosystems, Foster, CA, USA) with standard conditions and concentrations of reagents. Amplified products were analyzed on 1% agarose gels stained with ethidium bromide and purified with the QIAquick Gel Extraction Kit (Qiagen). Both strands of each PCR product were sequenced with a BigDye Terminator v 3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems).
The sequencing primers used had been designed previously for specific region sequencing. The DNA sequences were viewed and analyzed with the DNA Sequencing Analysis Software v 5.1 (Applied Biosystems). The variant was validated by resequencing of an independent PCR-generated amplicon from the subject and met our quality control threshold with a call rate exceeding 99%.
For family 1, the deletion mutation was originally identified as two apparently different sequences overlapping at the deletion point in direct sequencing analysis. To ascertain the actual sequence variation on the mutant allele, the PCR products amplified with primer 1F and 1R were cloned by a TA cloning kit (TaKaRa, Dalian, Liaoning, China) and sequenced. Six individual clones were sequenced for the wild-type allele, and the mutant allele was identified.
Multiple Sequence Alignments
The multiple NKX2-5 protein sequences across various species were aligned using the MUSCLE program, version 3.6 (an online program at http://www.ncbi.nlm.nih.gov/).
Prediction of the Disease-Causing Potential of a Sequence Alteration
The disease-causing potential of a sequence alteration detected in NKX2-5 was predicted automatically by MutationTaster (an online program at http://www.mutationtaster.org/), which gave a probability for the alteration to be either a disease mutation or a harmless polymorphism. Notably, the p value used in this study was the probability of the prediction rather than the probability of error as used in t test statistics (i.e., a value close to 1 indicates a high “security” of the prediction).
Results
Characteristics of the Study Subjects
A total of 58 unrelated kindreds with familial ASD were recruited and clinically evaluated in contrast to a cohort of 200 ethnically matched unrelated healthy individuals used as control subjects. None of them had traditional risk factors for ASD. Among the 58 probands (26 females and 32 males; age range, 0.25–16 years; mean, 3.52 years), 51 (87.93%) had ostium secundum ASD, 5 (8.62%) had ostium primum ASD, and 2 (3.45%) had sinus venosus ASD. Within the proband cohort, 7 (12.07%) also had a single first-degree atrioventricular conduction block, 1 (1.72%) had first-degree atrioventricular conduction block and paroxysmal atrial fibrillation, and 9 (15.52%) had some other form of congenital heart disease. The clinical characteristics of the 58 unrelated probands with familial ASD are summarized in Table 1.
NKX2-5 Mutations
The probands in 58 kindreds with familial ASD were genetically evaluated. Direct sequencing of the coding regions of the NKX2-5 gene was performed after PCR amplification of genomic DNA from the 58 index patients. Three heterozygous mutations in NKX2-5 were identified in 3 of 58 unrelated probands. The total population prevalence of NKX2-5 mutations based on the probands was approximately 5.17%.
A 17-bp (GCCCTCCTCCTGCATGC) deletion in exon 1 from coding nucleotides 126 to 142 (alternatively, c.126_142del) of the NKX2-5 gene, resulting in a frameshift from amino acid 43 of downstream codons, was identified in the proband from family 1. The deletion frameshift arises in amino acid 43, with proline 43 as the first affected amino acid, changing into a glycine, and the new reading frame ending in a stop codon at position 59. The position of the stop in the new reading frame is calculated starting at the first amino acid changed by the frameshift and ending at the first stop codon. This also may be described as a p.P43GfsX59 mutation, predicting a truncated protein (100 amino acids) without a homeodomain.
A substitution of guanine (G) for cytosine (C) in the third nucleotide of codon 46 of the NKX2-5 gene (c.138C > G) corresponding to the transversion of cysteine to tryptophane at amino acid position 46 (p.C46 W) was identified in the proband from family 2.
A change of C into thymine (T) at nucleotide position 536 from the translation starting point (c.536C > T) of the NKX2-5 gene, equivalent to the transition of serine into phenylalanine at amino acid 179 (p.S179F), was identified in the proband from family 3.
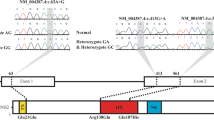
The sequence chromatograms showing the detected heterozygous NKX2-5 variations of c.126_142del, c.175C > G, and c.536C > T compared with control sequences are shown in Fig. 1. A diagram of NKX2-5 showing the locations of novel mutations relative to the amino-terminus, homeodomain, and carboxyl-terminus is presented in Fig. 2.

Sequence chromatograms of NKX2-5 in index patients and control subjects. The heterozygous NKX2-5 mutations of c.126_142del (alternatively p.P43GfsX59), c.175C > G (p.C46 W), and c.536C > T (p.S179F) compared with control sequences are shown in a, b, and c, respectively. The underlined nucleotides of GCCCTCCTCCTGCATGC in an allele of the control subject are those deleted in a counterpart of the patient (a). The arrow indicates the heterozygous nucleotides of C/G (b) and C/T (c) in the probands from families 2 and 3 (patients), respectively, or the homozygous nucleotides of C/C (b) and C/C (c) in the corresponding control subjects (controls). The square denotes the nucleotides comprising a codon of NKX2-5

Diagram of NKX2-5 depicting the locations of novel mutations. The mutations identified in this study are noted above the diagram of the NKX2-5 protein. NH 2 amino-terminus, TN tinman domain, HD homeodomain, NK nucleotide kinase domain, COOH carboxyl-terminus
All three probands carrying identified mutations presented with isolated ostium secundum ASD and without evidence of ECG-documented atrioventicular conduction block or atrial fibrillation. The three variants were neither found in the control population nor reported in the SNP database at the Web site (http://www.ncbi.nlm.nih.gov/SNP). The genetic scan of the three unrelated families showed that the gene variant was present in all the affected family members alive but absent in the unaffected family members tested in each family. Analysis of the pedigrees demonstrated that the mutation cosegregated with ASD transmitted as an autosomal dominant trait in the three families with complete penetrance. The pedigree structures of the three families are shown in Fig. 3.

Pedigree structures of families with atrial septal defects, designated as families 1, 2, and 3, respectively. Family members are identified by generations and numbers. Squares = male family members; circles = female members; symbols with a slash = the deceased members; closed symbols = affected members; open symbols = unaffected members; arrow = proband; + = carriers of the heterozygous mutations; − = noncarriers
Clinical evaluation of the family members from the three studied families showed that 10 (38.46%) of 26 individuals had ostium secundum ASD and that half of the affected individuals also had ECG-documented first-degree atrioventricular conduction block. Other structural cardiovascular malformations identified in affected family members included ventricular septal defect (I-1 in family 2), pulmonary artery stenosis (I-2 in family 3), and left ventricular hypertrophy and mitral valve fenestration (I-1 in family 1).
At the time of the study, no surviving family members available had atrial fibrillation during monitoring of 12-lead ECG. All the affected family members underwent surgical repair except for individual I-1 in family 1, who died of ASD complications before the study and had medical records of ASD and atrioventricular block. The phenotypic characteristics and the results of the genetic screening for the affected pedigree members are listed in Table 2.
Multiple Alignments of the NKX2-5 Protein Sequences Across Species
A cross-species alignment of NKX2-5 protein sequences showed that the altered amino acids were highly conserved evolutionarily, as presented in Fig. 4, suggesting that these amino acids are functionally important.

Alignment of multiple NKX2-5 protein sequences across species. The altered amino acids of P43 (the first amino acid altered by a deletion frameshift mutation), C46, and S179 are completely conserved evolutionarily
Disease-Causing Potential of a Sequence Alteration
The sequence alterations of c.175C > G and c.536C > T detected in NKX2-5 were predicted to be disease-causing, with p values of 0.99953 for c.175C > G and 0.99999 for c.536C > T, providing evidence that the two alterations were disease-causing mutations rather than benign polymorphisms. The sequence alteration of c.126_142de could not be handled because of an excessively long deletion (currently, MutationTaster handles only Ins/Del up to 12 bases).
Discussion
This report describes three previously unrecognized mutations of NKX2-5 identified in three families with familial ASD. Half of the affected family members had atrioventricular block. These novel heterozygous mutations were present in all the affected family members alive but absent in unaffected relatives tested and 400 normal chromosomes from a matched control population.
A cross-species alignment of NKX2-5 protein sequences showed that the altered amino acids were highly conserved evolutionarily. Prediction of the causative potential of a sequence alteration demonstrated that two missense mutations of p.C46 W and p.S179F were disease-causing with probability values as high as 1. The pathogenic likelihood of the deletion mutation could not be predicted by the MutationTaster due to the excessively long deletion. However, this sequence variation resulted in a frameshift starting from amino acid 43 and a premature termination of the translation (p.P43GfsX59). This would predict a truncated protein for only 100 amino acids lacking all known domains important for its functionality and thus probably a disease-causing mutation with haploinsufficiency, with loss of function being the pathophysiologic mechanism. Therefore, it is very likely that the three mutations were responsible for the ASD in these families.
Although the mechanisms by which the three novel NKX2-5 mutations cause ASD with or without atrioventricular block have not been defined, previous functional studies suggest that these mutations may exert loss of function or a dominant negative effect [8, 17, 20, 21, 28, 41]. Notably, the NKX2-5 mutations affecting the homeodomain resulted in reduced or loss of DNA binding, transcriptional activation activity, and protein-protein interactions, particularly with GATA4 and TBX5, in all the biochemical assays. The mutations also gave rise to an anomalous nuclear location in some functional analyses [17, 20, 21, 28, 41].
In contrast, the mutations with an intact homeodomain exhibited normal DNA binding to the monomeric binding site but displayed remarkable reduction in DNA binding to the dimeric sites, thus reducing the transcriptional activation activity [8, 21]. However, the mutations located at the carboxyl-terminus to the homeodomain of the NKX2-5 protein may have enhanced transcriptional activation activity, inducing apoptosis of cardiomyocytes by upregulating NKX2-5-target genes such as ANP and MEF2 [28, 41]. Therefore, the mutations p.P43GfsX59 and p.S179F, which affect the homeodomain of the NKX2-5, probably have little or no DNA binding activity or transcriptional activation function, whereas the mutation p.C46 W located at the amino-terminus to the homeodomain of the NKX2-5 likely has normal DNA-binding activity but reduced transcriptional activation activity.
In addition to production of truncated proteins, mRNAs harboring premature termination codons may be selectively degraded by a surveillance mechanism called nonsense-mediated mRNA decay, resulting in decreased abundance of mutant mRNA transcripts [12]. Hence, the p.P43GfsX59 mutation of NKX2-5 also may be implicated with haploinsufficiency by failure to yield protein.
Since the first report of the NKX2-5 mutations leading to congenital heart disease [36], at least 41 different heterozygous germline NKX2-5 mutations have been identified in patients with congenital cardiovascular disease [34, 38]. Of these 41 mutations, 33 are single-nucleotide substitutions, 6 are deletions, and 2 are insertions.
The NKX2-5 mutations are spread along the gene, and except for one at a splice site, all are located in the coding region, 18 of which affect conserved regions, including 2 in the TN domain, 14 in the homeodomain, and 2 in the NK2 domain [34, 38]. Moreover, NKX2-5 mutations have been identified in patients with either familial or sporadic cardiac defects. Of the 41 mutations, 25 are familial, 14 are sporadic, and 2 are sporadic/familial cases.
Although NKX2-5 mutations are involved in a long list of cardiac malformations, the most frequent phenotype in patients with NKX2-5 mutations is ASD with or without atrioventricular block. Specifically, 29 of the 41 mutations were detected in patients with ASD: 26 in the atrioventricular block and 25 in the ASD combined with atrioventricular block [34, 38]. However, the prevalence of NKX2-5 mutations varies significantly in different cohorts of individuals with congenital cardiovascular diseases [2, 4, 7, 8, 10, 11, 13, 14, 16, 27, 31, 38–40]. According to the 14 reports on the prevalence of NKX2-5 mutations in different cohorts of patients with cardiac defects, the detection frequencies of NKX2-5 mutations are 20% (7/35) [4], 18.75% (3/16) [13], 4.80% (6/126) [8], 2.96% (18/608) [27], 2.48% (3/121) [38], 1.39% (1/72) [2], 1.37% (2/146) [7], 1.26% (2/159) [10], 0.92% (1/109) [16], 0% (0/230) [39], 0% (0/227) [14], 0% (0/205) [31], 0% (0/104) [11], and 0% (0/62) [39], respectively, and the prevalence in the compound population is 1.94% (43/2219).
Similar to these findings, the mutational prevalence of 5.17% (3/58) in our cohort suggests that NKX2-5 mutations are an infrequent cause of ASD. Furthermore, remarkable genetic heterogeneity of ASD was proved by an inability to detect mutations in nearly 95% of our index patients despite somatic NKX2-5 mutations as a likely mechanism of ASD in some patients [33]. Hence, the contribution of genes other than NKX2-5 to ASD pathogenesis appears likely.
Mutations in other transcription factors associated with cardiogenesis, such as TBX5 [22], GATA4 [5, 9], and GATA6 [23], also have been detected in patients with ASD. Also, mutations in cardiac structural proteins such as alpha myosin heavy chain (MYH6) and alpha cardiac actin (ACTC1) were identified in familial ASD [6, 26]. However, to date, only NKX2-5 mutations are reported to cause an ASD phenotype and development of atrioventricular block [34, 38]. The most common two phenotypes caused by mutated NKX2-5 are ASD and atrioventricular conduction disturbance [1, 34, 38], indicating the pivotal role of NKX2-5 not only in the morphogenesis of the heart but also in the construction of the cardiac conduction system.
In the current study, a compound phenotype of ASD and atrioventricular block was observed in half of family members who carried the identified mutations of NKX2-5. Moreover, the atrioventricular block seems to be a progredient with increasing age in each individual, as described previously [13, 15, 29, 35, 36], whereas it is not observed soon after birth.
Atrioventricular block is a possible cause of sudden death, and molecular genetic screening appears to be the most helpful in identifying individuals at risk for the life-threatening arrhythmia. When a person harboring a NKX2-5 mutation is identified, it is necessary for medical staff to monitor this patient carefully, who may currently present with no symptoms or may have spontaneously closed or surgically corrected ASD. Monitoring enables offering accurate and early therapy.
In conclusion, the current study links novel mutations in the cardiac transcription factor NKX2-5 to familial ASD as well as atrioventricular block, which has potential implications in gene-specific prophylaxis and therapy of this common congenital heart disease.
References
Akazawa H, Komuro I (2005) Cardiac transcription factor Csx/NKX2-5: its role in cardiac development and diseases. Pharmacol Ther 107:252–268
Akcaboy MI, Cengiz FB, Inceoglu B, Ucar T, Atalay S, Tutar E, Tekin M (2008) The effect of p.Arg25Cys alteration in NKX2-5 on conotruncal heart anomalies: mutation or polymorphism? Pediatr Cardiol 29:126–129
Bartlett H, Veenstra GJ, Weeks DL (2010) Examining the cardiac NK-2 genes in early heart development. Pediatr Cardiol 31:335–341
Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573
Chen MW, Pang YS, Guo Y, Pan JH, Liu BL, Shen J, Liu TW (2010) GATA4 mutations in Chinese patients with congenital cardiac septal defects. Pediatr Cardiol 31:85–89
Ching YH, Ghosh TK, Cross SJ, Packham EA, Honeyman L, Loughna S, Robinson TE, Dearlove AM, Ribas G, Bonser AJ, Thomas NR, Scotter AJ, Caves LS, Tyrrell GP, Newbury-Ecob RA, Munnich A, Bonnet D, Brook JD (2005) Mutation in myosin heavy chain 6 causes atrial septal defect. Nat Genet 37:423–428
Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, Feneley M, Harvey RP (2003) Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol 41:2072–2076
Esposito G, Grutter G, Drago F, Costa MW, De Santis A, Bosco G, Marino B, Bellacchio E, Lepri F, Harvey RP, Sarkozy A, Dallapiccola B (2009) Molecular analysis of PRKAG2, LAMP2, and NKX2-5 genes in a cohort of 125 patients with accessory atrioventricular connection. Am J Med Genet A 149A:1574–1577
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447
Gioli-Pereira L, Pereira AC, Mesquita SM, Xavier-Neto J, Lopes AA, Krieger JE (2010) NKX2.5 mutations in patients with nonsyndromic congenital heart disease. Int J Cardiol 138:261–265
Hamanoue H, Rahayuningsih SE, Hirahara Y, Itoh J, Yokoyama U, Mizuguchi T, Saitsu H, Miyake N, Hirahara F, Matsumoto N (2009) Genetic screening of 104 patients with congenitally malformed hearts revealed a fresh mutation of GATA4 in those with atrial septal defects. Cardiol Young 19:482–485
Hentze MW, Kulozik AE (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96:307–310
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, Matsuoka R (2005) Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 135:47–52
Hobbs CA, Cleves MA, Keith C, Ghaffar S, James SJ (2005) NKX2.5 and congenital heart defects: a population-based study. Am J Med Genet A 134:223–225
Hosoda T, Komuro I, Shiojima I, Hiroi Y, Harada M, Murakawa Y, Hirata Y, Yazaki Y (1999) Familial atrial septal defect and atrioventricular conduction disturbance associated with a point mutation in the cardiac homeobox gene CSX/NKX2-5 in a Japanese patient. Jpn Circ J 63:425–426
Ikeda Y, Hiroi Y, Hosoda T, Utsunomiya T, Matsuo S, Ito T, Inoue J, Sumiyoshi T, Takano H, Nagai R, Komuro I (2002) Novel point mutation in the cardiac transcription factor CSX/NKX2.5 associated with congenital heart disease. Circ J 66:561–563
Inga A, Reamon-Buettner SM, Borlak J, Resnick MA (2005) Functional dissection of sequence-specific NKX2-5 DNA binding domain mutations associated with human heart septation defects using a yeast-based system. Hum Mol Genet 14:1965–1975
Jacobs JP, Quintessenza JA, Burke RP, Mavroudis C (2000) Congenital Heart Surgery Nomenclature and Database Project: atrial septal defect. Ann Thorac Surg 69:S18–S24
Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL, American Heart Association Council on Cardiovascular Disease in the Young (2007) Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:2995–3014
Kasahara H, Benson DW (2004) Biochemical analyses of eight NKX2.5 homeodomain missense mutations causing atrioventricular block and cardiac anomalies. Cardiovasc Res 64:40–51
Kasahara H, Lee B, Schott JJ, Benson DW, Seidman JG, Seidman CE, Izumo S (2000) Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest 106:299–308
Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, Bonnet D, Lyonnet S, Young ID, Raeburn JA, Buckler AJ, Law DJ, Brook JD (1997) Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15:21–29
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao H, Yan B, Liu Y, Yang Y, Chen YH (2010) A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J Hum Genet 55:662–667
Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y, American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2009) Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 119:e21–e181
Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L (2007) Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 115:163–172
Matsson H, Eason J, Bookwalter CS, Klar J, Gustavsson P, Sunnegårdh J, Enell H, Jonzon A, Vikkula M, Gutierrez I, Granados-Riveron J, Pope M, Bu’Lock F, Cox J, Robinson TE, Song F, Brook DJ, Marston S, Trybus KM, Dahl N (2008) Alpha-cardiac actin mutations produce atrial septal defects. Hum Mol Genet 17:256–265
McElhinney DB, Geiger E, Blinder J, Woodrow BD, Goldmuntz E (2003) NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 42:1650–1655
Monzen K, Zhu W, Kasai H, Hiroi Y, Hosoda T, Akazawa H, Zou Y, Hayashi D, Yamazaki T, Nagai R, Komuro I (2002) Dual effects of the homeobox transcription factor Csx/NKX2-5 on cardiomyocytes. Biochem Biophys Res Commun 298:493–500
Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR (2004) NKX2-5 pathways and congenital heart disease: loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117:373–386
Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL, American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young (2007) Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:3015–3038
Posch MG, Perrot A, Schmitt K, Mittelhaus S, Esenwein EM, Stiller B, Geier C, Dietz R, Gessner R, Ozcelik C, Berger F (2008) Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A 146:251–253
Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham ME, Harvey RP (2007) An NKX2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 128:947–959
Reamon-Buettner SM, Borlak J (2004) Somatic NKX2-5 mutations as a novel mechanism of disease in complex congenital heart disease. J Med Genet 41:684–690
Reamon-Buettner SM, Borlak J (2010) NKX2-5: an update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD). Hum Mutat 31:1185–1194
Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B (2005) Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 42:e16
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281:108–111
Sommer RJ, Hijazi ZM, Rhodes JF Jr (2008) Pathophysiology of congenital heart disease in the adult: part I: shunt lesions. Circulation 117:1090–1099
Stallmeyer B, Fenge H, Nowak-Göttl U, Schulze-Bahr E (2010) Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet 78:533–540
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2009) Screening NXK2.5 mutation in a sample of 230 Han Chinese children with congenital heart diseases. Genet Test Mol Biomarkers 13:159–162
Zhang WM, Li XF, Ma ZY, Zhang J, Zhou SH, Li T, Shi L, Li ZZ (2009) GATA4 and NKX2.5 gene analysis in Chinese Uygur patients with congenital heart disease. Chin Med J Engl 122:416–419
Zhu W, Shiojima I, Hiroi Y, Zou Y, Akazawa H, Mizukami M, Toko H, Yazaki Y, Nagai R, Komuro I (2000) Functional analyses of three Csx/Nkx-2.5 mutations that cause human congenital heart disease. J Biol Chem 275:35291–35296
Acknowledgments
We are indebted to the participants for their dedication to the study. This project was made possible through grants from the Natural Science Fund of Shanghai, China (10ZR1433100), the National Natural Science Fund of China (30570768 and 30700776), and the National Basic Research Program of China (2010CB912604).
Author information
Authors and Affiliations
Corresponding author
Additional information
X.-Y. Liu and J. Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Liu, XY., Wang, J., Yang, YQ. et al. Novel NKX2-5 Mutations in Patients With Familial Atrial Septal Defects. Pediatr Cardiol 32, 193–201 (2011). https://doi.org/10.1007/s00246-010-9859-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-010-9859-6