
Abstract
The review describes the CRISPR/CAS system and its adaptation for the genome editing in filamentous fungi commonly used for production of enzyme complexes, enzymes, secondary metabolites, and other compounds used in industrial biotechnology and agriculture. In the second part of this review, examples of the CRISPR/CAS technology application for improving properties of the industrial strains of fungi from the Trichoderma, Aspergillus, Penicillium, and other genera are presented. Particular attention is given to the efficiency of genome editing, as well as system optimization for specific industrial producers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
For centuries, filamentous fungi have been used by humans for production of useful metabolites and enzymes. The advantage of fungi over other industrial microorganisms is their ability to secrete large amounts of proteins (up to 120-150 g/liter) using relatively simple, cheap, and easily scaled fermentation schemes. Because of their genetic diversity, filamentous fungi can serve as sources of new genes coding for enzymes with unique properties, as well as in the development of recombinant strains for heterologous expression of industrially important proteins [1, 2].
Filamentous fungi commonly used in biotechnology belong to the genera Aspergillus [3-5], Trichoderma [6, 7], Penicillium [8-10], Acremonium [11], etc. Enzymatic preparations produced by the industrial fungal strains are used in the production of organic acids, biofuel, antibiotics, biologically active supplements, food products, and fodder. Fungal enzyme preparations are commonly added to detergents and used in the textile, paper, and waste management industries [12-15].
The main objectives in biotechnology involve identification of new enzymes, improvement of their operational stability, development of highly productive strains, and their metabolic engineering. Recombinant DNA technology is used to solve these problems worldwide [4]. Development of optimal expression systems for each strain of filamentous fungi with adapted transformation method are at the core of the recombinant DNA technology. In the case of fungi, the development of expression system involves creation of the recipient strain with dominant or auxotrophic trait for selection and construction of integration plasmid for the transfer of exogenous DNA to the fungal protoplasts [16]. As a rule, heterologous genes are placed under control of strong inducible promoters, i.e., those providing maximum levels of the synthesized protein in the culture liquid of the recipient strain [17]. The cbhI promoter in Trichoderma reesei and Penicillium verruculosum [18-22], glaA promoter in Aspergillus niger and P. verruculosum [19, 20], xylA promoter in Penicillium canescens [8], and alcA promoter in Aspergillus nidulans [21] are examples of the strong promoters worth mentioning. When these promoters are used in the optimized fermentation schemes, content of the target heterologous protein can reach up to 50-80% of the total secreted protein [22].
However, even the optimized procedures of heterologous expression in the industrial superproducer strains do not always solve the problems that emerge during expression optimization, such as inhibition of microbial proteolytic activity in order to prevent proteolysis of the heterologous proteins [23], reduction of the mycotoxin content and generation of safer strains [24], development of new auxotrophic strains for subsequent transformation and selection, etc. [25]. Other issues in the transcriptional regulation of heterologous expression essential for industrial application are reduction of the catabolic repression in industrial strain [26, 27], identification of transcriptional activators [28, 29], and optimization of promoter regions involved in heterologous expression [30-32]. Increase of the efficiency of homologous recombination is another important task [33-36]. Finding solutions for these problems will allow to increase productivity of the recombinant strains and efficiency of the technological processes in general due to reduction of expenses associated with the synthesis and purification of enzyme preparations [37].
Until recently, approaches for the strain improvement had been limited to the classic methods of random mutagenesis [38, 39], directed evolution [40, 42], and plasmid development for deletion/substitution of the target genes [43], followed by transformation and selection of the recombinant clones. Random mutagenesis increases the chance of additional unwanted mutations in the fungal genome (especially when several rounds of mutagenesis are performed) that often slow down fungal growth, impair sporulation, and induce genomic instability [44]. Directed evolution also involves the stage of random mutagenesis to generate libraries of target mutant clones by polymerase chain reaction (PCR) [45]. Main limitation of this method is the necessity for screening of mutant clones and, therefore, is used for the improvement of properties of the particular enzyme rather than the producer strain itself [46-48]. The third method involves inactivation of the undesired gene by deletion/mutagenesis or its substitution with another gene (e.g., antibiotic resistance gene) for providing an additional dominant trait for further selection [43]. However, construction of plasmids for the gene substitution requires cloning of the flanking regions (1500 to 2000 bp long) at the 5′- and 3′-ends of the target gene for efficient homologous recombination, which might be time-consuming if the fungal genome sequence is unavailable.
The recently developed genome editing technology, which allows rapid and precise correction of both prokaryotic and eukaryotic genomes is a promising alternative to the existing methods.
Genome editing is a molecular genetic tool that allows to add, remove, or move DNA fragments in an organism’s genome. These manipulations allow to turn off genes determined by the experimenter, introduce new genes into pre-selected loci of the genome, or adjust the gene sequences in the body. [49]. Genome editing in combination with genome sequencing mediated rise of a new direction in genetics – reversed genetics, which studies functions of genes with known nucleotide sequences by analyzing consequences of their knock-out or overexpression in an organism [50]. Genome editing has significantly facilitated genetic studies, as it is much less labor-consuming than the traditional methods used for the search of mutants and their genetic analysis. Besides, genome editing promotes the development of biotechnology methods, because it allows to inactivate selected genes (e.g., those involved in the biosynthesis of toxins [51]) and to alter metabolic pathways for creating superproducer strains or producers of new compounds, etc. [52].
The CRISPR/Cas technology (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) is a new and promising genome editing tool [53]. The first articles on application of the CRISPR system were published in 2013 [54-56]. At present, this technology is widely used in the genetic modification of both prokaryotic and eukaryotic organisms, from bacteria to humans [52].
The year of CRISPR discovery is 1987, when Yoshizumi Ishino and colleagues found unusual sequence repeats in the Escherichia coli genome [57]. From 1993 to 2000, Francisco Mojica had identified similar motifs in the genomes of 20 microorganisms [58], although functional significance of these sequences had remained unknown. In 2002, Jansen and colleagues revealed that CRISPR cassettes are flanked by the loci coding for proteins homologous with the nuclease and helicase activities [59]. It was shown in 2007 that CRISPR/Cas systems protect bacteria from bacteriophages [60].
The first studies on the genome editing with the CRISPR/Cas system in filamentous fungi were published in 2015 [61], following experiments on the yeast genome editing [62-64]. At present, the studies on the CRISPR/Cas system optimization for its application in filamentous fungi continue in parallel with the attempts to improve the editing system itself by searching for new and optimizing its already known components. Below, we describe the action mechanism of the CRISPR/Cas system and provide examples of its successful application for the genome editing in filamentous fungi.
CRISPR/Cas SYSTEM: COMPONENTS AND MECHANISM OF ACTION
The CRISPR/Cas system includes genomic CRISPR loci and Cas proteins encoded by the genes flanking the CRISPR loci. A CRISPR locus contains direct DNA repeats connected by short (on average, 20 bp) varying nucleotide sequences (spacers). The spacers are inserted into the genome DNA during an adaptive immune response. Cas proteins are divided into two functional modules: the more conserved adaptation module and the variable interference module (effector complex). Proteins of the adaptation module provide insertion of new spacers into the CRISPR cassette, while effector complex protein are involved in degradation of the DNA or RNA molecules containing fragments complementary to the spacer sequence (Fig. 1a) [65].

a) Principle design of the CRISPR/Cas system. DNA fragment is incorporated as a new spacer in the CRISPR cassette after the first attack. When the same DNA enters the cell again, the spacer is expressed in the crRNA-tracrRNA complex and directs Cas9 to the corresponding DNA for its cleavage. b) CRISPR/Cas system type II. sgRNA binds to Cas9 and catalyzes DSB formation in the DNA targeted by the 20-nt protospacer.
There are two classes of the CRISPR/Cas systems: class 1 systems involve multisubunit effector complexes, while the effector complexes of the class 2 systems use a single type of Cas protein. Each class is subdivided into several types and subtypes based on the presence of a “signature” protein found almost exclusively in this category [66]. Effector complexes currently used for genome editing are mostly of type II because they are easier to use (Fig. 1b) [67].
Despite the significant differences in the composition of effector complexes in microbial species, the mechanism of action of the CRISPR/Cas system is the same. First, phage genomes sequences, mobile elements, or fragments of the cell DNA formed during DNA repair or transcription termination are inserted into the CRISPR cassette. The process of interference starts with transcription of the CRISPR cassette with formation of immature pre-CRISPR RNA (pre-crRNA) composed of multiple repeats and spacers. The primary transcript is processed with generation of the short CRISPR RNAs (crRNAs), each containing a spacer flanked by the fragments of repeats. crRNAs form the effector complex with Cas proteins, which recognizes its target via interaction of the crRNA spacer with the complementary sequence in the foreign DNA (protospacer). This recognition induces hydrolysis of the phosphodiester bond between the bases in the target sequence and degradation of the latter. If the target sequence is present in the cell genome, this might result in the cell death (Fig. 1a) [68, 69].
Type II CRISPR/Cas systems. Based on the differences between Cas proteins, the class 2 CRISPR/Cas systems are classified into 3 types (II, V, VI) [70, 71]. Unlike type I, III, and IV CRISPR/Cas systems (which utilize complex mechanisms of Cas protein interactions), type II CRISPR/Cas system containing Cas9 protein from Streptococcus pyogenes is simple, which makes it easy to use for editing microbial genomes [66, 72]. The effector complex of type II systems contains only two components – Cas9 nuclease and single guide RNA (sgRNA) consisting of the fragment that recognizes DNA target (crRNA) and auxiliary trans-activating RNA (tracrRNA) that forms hydrogen bonds with crRNA and interacts with Cas9 [73, 74]. The 5′-end of crRNA contains a spacer that determines its specificity toward a particular genomic DNA locus (Fig. 1b) [75, 76].
Structure and function of Cas9 from Streptococcus pyogenes. Cas9 from S. pyogenes (SpCas9) is a type II protein from the class 2 effector complexes. It is a multidomain, multifunctional protein composed of 1368 amino acid residues. The 3D structure of SpCas9 in complex with DNA and sgRNA was solved by X-ray diffraction analysis and cryo electron microscopy (Fig. 2) [77]. SpCas9 consists of the nuclease (NUC) and recognition (REC) segments. The NUC segment is composed of the HNH-like and RuvC-like nuclease domains and the C-terminal domain (CTD) that interacts with the protospacer adjacent motif (PAM). The REC segment includes three α-helical domains (REC1-3) (Fig. 2).

3D structure of SpCas9 in the complex with sgRNA (based on the data of XRD analysis; PDB:4ZT9). Lower panel, SpCas9 domain structure.
After the SpCas9/sgRNA complex binds to DNA, the HNH domain cleaves the DNA strand complementary to the sgRNA protospacer, while the RuvC-like domain cleaves the other strand. The Cas9 protein globule completely surrounds the DNA molecule, which enters the tunnel between the CTD and REC1 domain, where it is separated into two strands. One strand binds in the narrow cleft between the RuvC-like domain, HNH domain, and CTD; the other strand forms heteroduplex with sgRNA in the space between the REC and NUC segments. After circumventing the RuvC-like and HNH domains, the DNA duplex reforms behind the protospacer end opposite to the PAM [77, 78].
To facilitate genetic engineering procedures, it was suggested to use common sgRNA (Fig. 3). The 5′-end of this sgRNA is a variable sequence (spacer) of 20 nucleotides (nt); the 3′-end is a constitutive fragment (76 nt) required for Cas9 binding. sgRNA nucleotides 21-41 are part of the native crRNA, while the rest of the molecule is part of the tracrRNA. All fragments of sgRNA bind to the corresponding domains in Cas9, except the spacer and nucleotides 29-59 that form a loop due to complementary interactions between the repeat and antirepeat. The loop protrudes from the Cas9 protein globule [73].

Secondary structure of sgRNA interacting with the target DNA sequence.
According to the biochemical and biophysical data, DNA recognition and cleavage by Cas9/sgRNA occurs in several steps. First, sgRNA forms a complex with the Cas9 apoenzyme. The active complex randomly (via 3D diffusion) binds to the DNA. If DNA lacks PAM at the binding site, the complex rapidly dissociates from the DNA and continues “search” for the target. Inability of Cas9 to function via processive mechanism (sliding along the DNA molecule) typical for many other DNA proteins exhibiting affinity to certain sequences, increases significantly (by several orders of magnitude) the time required for the target location and recognition in large eukaryotic genomes [79].
Recognition of PAM in the DNA strand by the Cas9/sgRNA complex is followed by separation of the DNA strands and inspection of the seed sequence [80]. At the same time, the C-terminus of Cas9 stabilizes the phosphate group between the last deoxyribonucleotide of the sequence complementary to the sgRNA protospacer and the first PAM deoxyribonucleotide. As a result, the DNA strand complementary to the protospacer rotates toward the sgRNA for the formation of complementary interaction, while Cas9 stabilizes DNA in the molten state by interacting with the sugar-phosphate backbone in the seed part of the non-target strand [81]. This leads to formation of the RNA protospacer/DNA heteroduplex, whose stability is critical for the target sequence identification. If the DNA sequence is not complementary to the sgRNA in the first eight nucleotides following PAM, the protospacer does not bind to DNA, which prevents interaction between the DNA and the Cas9/sgRNA complex. However, if formation of the heteroduplex occurs in the case of nonspecific binding to the protospacer, the off-target effects in genome editing are possible. Heteroduplex formation induces conformational rearrangements in the Cas9 molecule, resulting in its activation. The catalytic mechanism of the phosphodiester bond hydrolysis is poorly understood. It is only known that the HNH domain acquires its stable conformation after formation of the complete heteroduplex that results in significant conformational changes in the linkers targeting the non-target strand for cleavage in the catalytic site of RuvC [82]. The non-target stand is cleaved by exonucleases in the 3′→5′ direction after hydrolysis, while the target strand is degraded [73]. Cas9 remains bound to the cleaved DNA until it is replaced by other enzymes, such as DNA or RNA polymerases. Next, the double-strand breaks (DSBs) can be repaired via two mechanisms: (i) non-homologous end joining (NHEJ) with possible emergence of deletions or insertion at the site of DNA break and (ii) homologous recombination (HR) via insertion of the donor DNA flanked by the homologous sequences, which might be used for insertion of the target gene.
Expression of the Cas9 nuclease for genome editing in filamentous fungi. Expression of the Cas9 nuclease is an important element in optimization the CRISPR/Cas system for the genome editing in filamentous fungi. The first step in the SpCas9 adaptation to the expression in fungi is optimization of the spCas9 codons with respect to the codon frequency in fungi [61, 83]. Another important issue is selection of the nuclear localization sequence (NLS). The most commonly used NLS is the SV40 sequence [61]; however, in some fungi, e.g., Fusarium oxysporum, this typical NLS cannot be used, so homologous NLS sequences are employed [84].
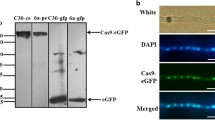
To detect expression and to assess intracellular localization of the optimized Cas9, most researchers use fusion constructs composed of Cas9 and eGFP (enhanced green fluorescent protein), so that nuclear or cytosolic location of Cas9 can be recorded by fluorescence microscopy [84-87].
Cas9 expression also strongly depends on the promoter used. Constitutive promoters are most commonly employed, since they provide stable and induction-independent expression of heterologous genes. The three promoters used for the Cas9 expression in filamentous fungi are the trpC promoter of the tryptophan synthesis gene from A. niduans, gpdA promoter of the glyceraldehyde 3-phosphate dehydrogenase gene from A. nidulans and P. verruculosum, and TEF1 promoter of the translation elongation factor 1 α-subunit from A. nidulans [61, 88-89]. The less common promoters, which are still used for the Cas9 expression in fungi are xlnA, Ham34, amyB, niiA, Otef, and hsp70 [90-93]. It should be noted that heterologous expression of Cas9 may be toxic for the recipient strain; hence, it is important to study its effect on standard morphological and physiological parameters (strain growth and development, sporogenesis, stress resistance, virulence, etc.) during each adaptation of the CRISPR/Cas system in a filamentous fungus strain [87, 94-96]. The data on the developed systems for the Cas9 expression in fungi are summarized in table.
Use of SpCas9 orthologs and other Cas proteins in genome editing. The widespread use of SpCas9 can be likely explained by the fact that this protein was the first discovered second class nuclease. Although SpCas9 has been studied in detail and is widely applied in genome editing, the search for new effector proteins continues. Many Cas9 orthologs exceed SpCas9 in their properties. For example, the proteins from Streptococcus thermophiles (StCas9) and Neisseria meningitides (NmeCas9) are more specific. The Cas9 gene from Staphylococcus aureus is shorter, which facilitates its delivery to the edited organism. In addition, the Cas9 orthologs have different PAM sequences that allow editing different DNA sites and, in some cases, reducing the number of off-target deletions in the edited genome [97].
In addition to the Cas9 proteins, the Cas12a protein (Cpf1) from Franciella novicida, which belongs to the class 2 CRISPR/Cas type V system, has been widely used for genome editing. Its advantages include smaller size (1300 amino acids vs. 1629 in SpCas9) and smaller sgRNA (40 nt vs. 100 nt in the SpCas9 sgRNA). Also, the PAM for Cpf1 is located upstream of the protospacer and has the 5′-TTN-3′ sequence, which expands possibilities of the method in general. The cleavage takes place after the 18th nucleotide from PAM in the non-target strand and after the 23rd nucleotide in the target sequence resulting in the formation of 5′-protruding sticky ends [98]. The first article on the successful use of Cpf1 for genome editing in Saccharomyces cerevisiae was published in 2017 [99] confirming that Cpf1 can be a promising protein for the genome editing in yeast and other fungi.
Cas13a is an RNA-guided RNA ribonuclease of the type VI-A CRISPR/Cas system that degrades invasive/viral RNAs targeted by crRNA [100]. The crystal structure of Cas13a from Leptotrichia buccalis was resolved, and its mechanism of action was elucidated. It was found that Cas13a stays inactive in the absence of viral RNAs complementary to the targeting sequence. However, following phage attack the host and the targeting sequence of crRNA forms a duplex with the RNA-guided target, Cas13a switches to the catalytically active state and randomly cleaves all collateral and single-stranded RNA targets exposed by the ssRNA. These studies demonstrated how the type VI CRISPR/Cas systems containing Cas13a protect cells from the RNA-containing phages and can serve as a basis for developing tools for RNA manipulation.
sgRNA expression. RNA polymerase III promoters. sgRNA in the CRISPR/Cas9 system lacks the cap and the polyA tail and requires clear transcription initiation start. In most cases, stable transcription of sgRNA is guided by the U6 promoter of RNA polymerase III [101]. To investigate if the U6 promoter can be used in filamentous fungi, snRNA (small nuclear RNA) sequences from various organisms (from yeast to humans) were compared. Bioinformatic analysis revealed high degree of homology (65%) between the snRNA U6 sequences of all eukaryotes [91]. Identified U6 promoters successfully controlled sgRNA transcription and directed Cas9 to the DSB site [92, 94].
The authors of [93] tested efficiency of the CRISPR/Cas9 systems controlled by the tRNA promoters (ptRNALeu-TAA, ptRNAGlyGCC, ptRNATyrGTA, and ptRNAGlyTCC). All four tRNA promoters were more efficient than the U6 promoter, indicating the possibility of their application in an alternative expression systems [102].
The search for more efficient promoters resulted in the discovery of a highly conserved region of the 5S rRNA gene. The use of this fragment and 328-nt region directed toward the 5′-end as an sgRNA promoter increased the efficiency of DNA cleavage up to 96% (vs. 25% in the case of the U6 promoter) [103].
RNA polymerase II promoters. The use of U6 and U3 promoters has some limitations. Firstly, snRNA U6 and snRNA U3 are constitutive genes expressed almost in all cell types, therefore, they cannot be used for a tissue-specific expression of sgRNAs. Secondly, the U6 and U3 promoters cannot be used for in vitro sgRNA transcription, because RNA polymerase III is commercially unavailable. Besides, variety of the spacers compatible with the U6 and U3 promoters is limited to the G(N)20GG and A(N)20GG sequences, respectively [101, 104].
The abovementioned limitations can be avoided by using ribozymes, which are RNA molecules with catalytic activity. The authors of [102] used the 5′-HH (hammerhead) and 3′-HDV (hepatitis D virus) ribozymes with nuclease activity for the cassette design. The resulting construct was introduced into yeast cells under the control of the ADH1 promoter transcribed by RNA polymerase II. In this case, the CRISR/Cas9 system was not limited to the use of the G(N)20GG and A(N)20GG sequences. Later, an analogous construct using the strong constitutive gpdA promoter and the trpC terminator was obtained and successfully tested in Aspergillus strains [83].
The above-described methods are relatively complicated and technically difficult; they can also introduce toxic compounds into the cells. For this reason, researchers have constructed the microRNA-shRNA-sgRNA (miRsh-sgRNA) cassette that was also controlled by the RNA polymerase II promoter [105]. Efficiency of this cassette in sgRNA transcription was proven experimentally using the cleavage of the p53 gene as a readout [106].
Hence, the RNA II polymerase promoter may be used in future for controlled and safe CRISR/Cas9-mediated editing of genes, including genes in filamentous fungi.
STRATEGIES FOR APPLICATION OF THE CRISPR/Cas SYSTEM IN GENOME EDITING OF FILAMENTOUS FUNGI
The process of genome editing with the CRISPR/Cas system includes: (i) selection of the target gene or locus; (ii) development of the appropriate selection method; (iii) selection of the spacer sequence; (iv) cloning of the sgRNA with the selected spacer; (v) delivery of the CRSPR/Cas system to the edited organism (cell); (vi) selection of the recombinant clones.
The first step is unique for each organism and depends on the purpose of genome editing. The only prerequisite is knowledge of the nucleotide sequence of the edited DNA fragment. So far, very few studies have been devoted to the use of genome editing for metabolic engineering, as the goal of most investigations has been adaptation of the CRISPR/Cas9 system for filamentous fungi [107].
Approaches to selection. Efficiency of the CRISPR/Cas9 system can be easily evaluated by introducing changes in the functional genes in order to alter phenotypic traits of the edited strains (table). Thus, Katayama et al. [92] used the wA and yA genes as targets to study expression from the snRNA U6 promoters in the CRISPR/Cas9 system. Deletion of these genes results in the formation of colored conidia. Similar approach was used in the Aspergillus [61, 83, 108], Trichoderma [109], Talaromyces atroroseus [110], Penicillium subrubescens [111], Magnaporte oryzae [112], and other fungi (table).
Special attention in the CRISPR/Cas system adaptation is given to the search for efficient selection markers, e.g., antibiotic resistance genes, such as hph (resistance to hygromycin B), ble (resistance to bleomycin), bar (resistance to bialaphos), and neo (resistance to geneticin), that have been successfully used for the selection of positive clones. For example, bialaphos-containing medium was used for the selection of Aspergillus [61], N. crassa [88], and Myceliophthora [113] transformants. In [114] and [61], positive clones were selected on the bleomycin-containing medium. Geneticin (G418) was used for the selection of transformed Phytophthora sojae clones [115]. Hygromycin B, which is a commonly used selective marker, was applied for the selection of various filamentous fungi, including A. fumigatus [114], A. niger [116], A. carbonarius [117], Alternaria alternata [118], and Shiraia bambusicola [95].
Orotidine 5-phosphate decarboxylase (PyrG), an enzyme of uridine biosynthesis, is inhibited by 5-fluoroorotic acid (5-FOA). Generation of auxotrophic 5-FAO-resistant strains mutant by the PyrG gene was used for the selection of A. oryzae, T. reesei, A. fumigatus [91, 109, 119, 120]. The amdS, gene (coding for acetamidase) ensured the growth of positive P. chrysogenum transformants on the medium containing acetamide as a nitrogen and carbon source [90].
However, variety of the selective markers is not limitless because of the individual metabolic features of filamentous fungi. For this reason, the phage Cre recombinase is often used to restore selection markers, so the same marker can be employed for the selection of positive clones. Zhang et al. [121] developed a method for one-step marker-free genetic modification of industrial eukaryotic microorganisms using the CRISPR/Cas9 system based on Cre/loxP. The target gene is knocked-out or deleted by CRISPR/Cas9, while the light-induced Cre recombinase removes selection markers (such as genes providing resistance to hygromycin B, phosphinothricin, and chlorimuron ethyl) to allow the use of the same marker in the following genetic manipulations. The CRISPR/Cas9 system based on Cre/loxP. was successfully used in Hypocrea jecorina (T. reesei) Qm6a, N. crassa, A. niger, and Metarhizium anisopliae [120]. This system can be also applied for simultaneous removal of several gene clusters via reversible conversion of autotrophs into auxotrophs.
Spacer selection and sgRNA cloning. Selection of the spacer sequence is the most important step, which determines not only efficiency of the genome editing, but also probability of the off-target effects (alterations in the non-target DNA regions). As a rule, the spacer has to satisfy the following requirements: it should not form secondary structures (although some studies used spacers with the hairpin at the 5′-end to reduce the off-target activity [122]); the GC content should be 35 to 75%; the spacer should lack the poly(T) sequences (when polymerase III is used for sgRNA transcription as they act as termination signals in this case); the off-target sites should contain at least three nonpaired bases, preferably closer to the 3′-end. At present, spacer selection is done with special programs, many of which are open-source programs (E-CRISP: http://www.e-crisp.org/E-CRISP/; CHOPCHOP: https://chopchop.cbu.uib.no/; CRISPRdirect: https://crispr.dbcls.jp/). These programs use various algorithms to estimate the spacer specificity and to search for the possible off-target sites in the edited genome [123].
Cloning of the sgRNA with the selected promoter is a purely technical task comprising a required step in the experimental protocol of the CRISPR/Cas mediated genome editing. There are several approaches for solving this problem (e.g., cloning or PCR), which are beyond the scope of our review. As a rule, cloning involves two steps: annealing of two oligonucleotides with sticky ends corresponding to the spacer sequence with each other and their ligation into the plasmid that had been hydrolyzed by a class IIS restriction endonuclease (e.g., BbsI) between the promoter and the sgRNA sequence with formation of the sticky non-complementary ends [103]. This method is universal and allows replacing protospacers for the following genome editing in the particular mycelial fungus.
CRISPR/Cas delivery to the cells. Delivery of the CRISPR/Cas system into the cells of filamentous fungi is the most variable stage in the process of system adaptation for the use in fungi. The system can be introduced into the cells as DNA (with the following in vivo assembly), as in vitro assembled complex, or in the intermediate state (e.g., as in vitro synthesized RNA or sgRNA and the Cas9-encoding DNA). Each method has its own advantages and drawbacks, and the choice for the delivery scheme depends primarily on the edited species and available laboratory resources.
Most often, expression cassettes with the Cas9 and sgRNA genes are introduced into the edited microorganisms in the content of the same vector or in two separate vectors (table). Zhang et al. [91] investigated efficiency of two different delivery methods using the model fungus A. fumigatus and found that the single-vector-based delivery was much more efficient than the delivery with two vectors, which was likely associated with difficulties to control the exact ratio between the Cas9- and sgRNA-containing expression cassettes.
The most commonly used delivery method is the step-wise introduction of Cas9 and sgRNA. In the first step, the recipient fungal strain is stably transformed with a cassette carrying the Cas9 gene, after which the cells are screened for the nuclease expression. In the second stage, these Cas9-positive cells are transformed with the in vitro transcribed sgRNA [85, 109, 116, 124]. Transformation with polyethylene glycol (PEG) and Agrobacterium-mediated transformation (AMT) are the two most common methods for the introduction of CRISPR/Cas9 systems into fungal cells. The PEG-mediated transformation is the simplest, so this approach is generally preferred for transformation of numerous fungi [87, 125, 126]. However, the long-term Cas9 expression can be toxic for fungal cells and may promote off-target effects, which could be difficult to identify [62]. Moreover, the use of plasmids for the Cas9 and sgRNA expression could cause random integration of the plasmid DNA into the host genome.
Delivery of the in vitro assembled Cas9/sgRNA complexes allows to perform genome editing without incorporation of the Cas9 and sgRNA genes into the host genome, which reduces the possibility of nonspecific editing because of the short lifetime of the complex and keeps the genome “clean”. It was shown that the delivery of sgRNA and purified recombinant Cas9 protein to the cells results in the doubling of recombinant P. chrysogenum colonies (vs. transformation with a plasmid carrying the Cas9 and sgRNA genes), thus reducing the associated toxicity and off-target effects and providing site-specificity of the introduced mutations [90]. The same study demonstrated that the cell cycle synchronization and timely delivery of the ribonucleoprotein (RNP) complexes of Cas9 represent a simple and efficient method for enhancing the site-specific genome editing in P. chrysogenum [90]. Similar methods were used in T. reesei [127] and A. niger [128]. However, RNP delivery to the cells is challenging and so far is rarely used in filamentous fungi.
Most often, the genome editing of filamentous fungi involves delivery of the Cas9 and sgRNA as the corresponding genes that integrate into the genome or as elements of the plasmid carrying the AMA1 loci, which provides its autonomous replication during selection [61, 91, 108, 110, 111, 118, 129, 130]. The drawbacks of this approach include the problems with transcription of the sgRNA gene. This gene is commonly placed under control of the RNA polymerase III promoter; however, many species of filamentous fungi lack this promoter. For this reason, researches use the RNA polymerase II promoters and ribozymes to flank the sgRNA sequence [83, 110, 111, 118, 119, 129-132] or employ the conserved 5S rRNA promoter, which is recognized by the RNA polymerase III [103]. The problems with the sgRNA transcription can be avoided by using in vitro transcription. However, this requires generation of strains expressing the Cas9 gene [109].
As mentioned above, edited clones are initially selected based on their phenotype or ability to grow on a selective medium. However, it is often required in practice to edit genes, whose knockouts have no phenotypic manifestations. In this case, a marker gene is introduced by homologous recombination to the site of cleavage. In any case, the editing event should be confirmed by sequencing after initial analysis of phenotypic changes or selection. If several sites are edited simultaneously, sequencing is the only reliable method for evaluating efficiency of the CRISPR/Cas system.
EXAMPLES OF GENOME EDITING IN FILAMENTOUS FUNGI
Genome editing of Trichoderma fungi. T. reesei is the major source of commercial preparation of cellulolytic enzymes. This microorganism can be also used for production of heterologous enzymes. The first successful application of the CRISPR/Cas9 system was the knockout of the lae1, vib1, and clr2 genes involved in the regulation of expression of cellulase genes. In the first stages of adaptation, codon composition of the S. pyogenes Cas9 gene containing the SV40 NLS was optimized, and the codon-optimized Cas9 gene was integrated into the T. reesei genome under control of the constitutive pyruvate decarboxylase (Ppdc) promoter or inducible cellobiohydrolase (Pcbh) promoter. The resulting strains were transformed with the in vitro synthesized sgRNA with the spacer corresponding to the target gene. Simultaneously, the marker ura5 gene was knocked out to provide survival of the transformants on the 5′-FOA-containing medium. The authors demonstrated that the knockout efficiency was much higher when the donor DNA fragments with long (200 bp and over) flanking sequences were used, and that DNA repair via NHEJ was more efficient than via HR [109].
In [133], the procedure developed in [109] was used to knockout the sxlR gene coding for the transcription factor negatively regulating the activity of xylanase in T. reesei.
In [120], two variants of the promoter region corresponding to the U6 promoter were cloned for intracellular transcription of sgRNA. The sgRNA contained the protospacer against the marker ura5 gene. The T. reesei cells with basal Cas9 expression were transformed using the AMT method. Efficiency of the genome editing in this case was only 8-10%.
Rantasalo et al. [127] used the nucleoprotein complexes for simultaneous deletion of the cellobiohydrolase 2 (cbh2), endogluconase 1 (egl1), and endogluconase 2 (egl2) genes. The authors transformed T. reesei protoplasts with the in vitro assembled Cas9:crRNA complex, i.e., used the native crRNA/tracrRNA duplex, rather than in vitro transcribed sgRNA. To increase efficiency of HR, the authors used the T. reesei strain with mutation in the pyr4 gene and deleted mus53 ligase gene. The targeted genes were deleted simultaneously using equimolar amounts of crRNA with different protospacers (two crRNAs per each gene targeted to the start and the end of the gene). The deleted DNA fragments were substituted with the donor DNA added during the transformation. The donor DNA contained the pyr4 gene flanked at the 5′- and 3′-ends with homologous 1000-bp sequences. As a result, 16 colonies out of 139 transformants contained the triple deletion (12% efficiency). The same procedure was repeated using the donor DNA cassettes containing synthetic expression system (SES) with the lipase calB gene from Candida antarctica. The resulting strain produced CaLB lipase preparations with low content of non-target proteins and 1.5-fold higher specific activity compared to the standard expression system using the cbh1 promoter [127].
Genome editing of Aspergillus fungi. Many species of filamentous fungi from the Aspergillus genus are widely used in biotechnology for waste treatment, production of organic acids and alcoholic drinks, and other processes. At the same time, some Aspergillus fungi synthesize mycotoxin causing diseases in humans and animals.
In 2015, the Uffe Mortensen’s group reported the CRISPR/Cas system adapted for genome editing in six Aspergillus species: A. aculeatus, A. brasiliensis, A. carbonarius, A. lichuensis, A. nidulans, and A. niger [61]. The genes coding for the CRISPR/Cas system components were delivered to the cells in the content of the same plasmid. In order to be used in different species, four variants of the plasmid were produced that contained different marker genes commonly used in filamentous fungi: AFUM_pyrG, AN_argB, bleR, and hygR. The plasmids also carried AMA1 sequence to ensure its replication. The optimized SpCas9 gene with theSV40 NLS was expressed in A. niger using the strong constitutive promoter of the A. niger translation elongation factor 1 gene (tef1) and tef1 terminator. The sgRNA was transcribed under control of the strong A. niger glyceraldehyde 3-phosphate dehydrogenase constitutive promoter (gpdA) and the trpC terminator. Since sgRNA should lack 5′-cap and 3′-poly(A) tail, its sequence was flanked by the HH ribozyme at the 5′-end and HDV ribozyme at the 3′-end (Fig. 4; table).

sgRNA release from the transcript synthesized by RNA polymerase II. Light green and green, sgRNA; red and blue, HH ribozyme; orange, HDV ribozyme; scissors, cleavage site. HH fragment marked with blue color hybridizes with the protospacer for efficient cleavage (adapted from [61]).
The developed system was tested with the protospacer against the albA gene in five fungal species: A. aculeatus, A. niger, A. carbonarius, A. luchuensis, and A. brasiliensis. The knockout of the albA gene in filamentous fungi results in formation of white colonies. The highest editing efficiency was observed in A. brasiliensis and A. niger (approximately, 100%); the lowest efficiency was found in A. luchuensis (25%), which could be related to a lower survival rate of the transformants. Editing efficiency of the adapted CRISPR/Cas system was verified with the protospacer against the yA gene in A. nidulans and protospacer against the pyrG genes in A. aculeatus. The authors concluded that the efficiency of the adapted editing system differed in the fungal species of the same genus; they suggested that these differences could be eliminated by cloning of homologous promoters for the expression of Cas9 and sgRNA in each edited species. Another problem in the editing of filamentous Aspergillus fungi was multinuclearity of the protoplasts, which also decreased efficiency of the genome editing [61].
The study of Kuivanen et al. [116] is another example of using genome editing for metabolic engineering. The authors modified the A. niger genome to obtain a producer of galactaric acid. First, they sequenced and compared A. niger transcriptomes before and after cultivation for 5 and 18 h on a galactaric acid-containing medium in order to identify the genes involved in catabolism of this compound and found that expression of 7 genes was upregulated. Six out of seven of these genes were sequentially knocked out using the CRISPR/Cas system. For this, the corresponding strain was transformed with a plasmid carrying the Cas9 gene, the AMA1 sequence, and the hyg selective marker for the growth on hygromycin-containing medium. The synthesized sgRNAs (two per gene, targeted to its 5′- and 3′-regions) were added together with the plasmid. The knockout efficiency was 27 to 100%, depending on the deleted gene. In parallel, the same genes were knocked out using deletion cassettes with the homologous 1.5-kb shoulders and pyrG gene as a selective marker. In this case, the efficiency of genome editing was significantly less (0 to 43%) [61].
Adaptation of the CRISPR/Cas9B system for A. niger performed in [128] included minimization of the in vitro protoplast transformation method by the Cas9::sgRNA ribonucleoprotein (RNP) complex. Transformation was carried out in 96-well microplates instead of 15-ml tubes, which significantly reduced the time of procedure and the amount of reagents used. Efficiency of the target gene deletion by this optimized method was 100%. It was demonstrated that the developed method was suitable for multiplex editing of two or three targets, which is very convenient for the metabolic engineering of fungi. As a result, the authors obtained an A. niger strain with enhanced galactarate production.
In 2018, Zheng et al. published a paper on the in vivo sgRNA expression in A. niger [103]. In this work, the authors used various promoters for the sgRNA expression: human RNU6-1 promoter (PhU6), A. niger RNU6 promoter (PanU6), and A. niger 5S rRNA internal promoter with different length of the 5′-untranscribed region (-338, -160, -106, -65, -35) or lacking this sequence. When the 5S rRNA internal promoter was used, the transcript contained the 5S rRNA sequence at the 5′-end; therefore, a construct containing the HDV ribozyme sequence between the 3′-end of 5S rRNA and 5′-end of sgRNA was obtained. To verify efficiency of the selected promoters, A. niger G1 (amdS-, ΔglaA, ΔpepA) strain was transformed with the plasmid containing the optimized SpCas9 gene and plasmids with the sgRNA and the protospacer against the alba gene under control of the tested promoters. The knockout of the albA gene results in the disruption of melanin synthesis and formation of colorless colonies. The authors found that the efficiency of the PhU6 and PanU6 promoters was low (20% or less of knockouts from the total number of grown transformants), while all the constructs containing the 5S rRNA promoter have generated knockout in 100% of the transformed colonies. Presence of the 5S rRNA not only failed to decrease knockout efficiency, but even made the procedure more efficient, which could be explained by protection of the 5′-end of sgRNA from RNases.
Next two examples demonstrate possibility of the CRISPR in vivo expression from one autonomously replicating plasmid [108, 131]. In [108], the CRISPR/Cas9 system was adapted for editing of the A. niger genome in order to prevent possible consequences of DSBs. The authors developed a series of constructs with the modified Cas9 and cytidine deaminase (enzyme converting cytidine into thymine without DSB induction). Two phenotype-associated genes, pyrG and fwnA, were inactivated with the efficiency of 47.36-100%; the non-phenotype-associated prtT gene was inactivated with 60% efficiency [108].
Another case is biotechnological application of the CRISPR/Cas9 system for modification of the recombinant A. niger producer strain synthesizing human erythropoietin [131]. Four genes coding for proteolytic enzymes in the A. niger rHuEPO strain were knocked-out (vps, prtT, algC, and och1) to yield the strain producing no proteases, which significantly (41-fold) increased the level of erythropoietin secretion. The components of the CRISPR/Cas9 system were delivered to the cells in the composition of the same plasmid containing the AMA1 sequence, which was developed previously for the Aspergillus fungi [61].
A. oryzae also belongs to the cellulase-producing industrial filamentous fungi. In Japan, A. oryzae is the main microbial producer of homologous and heterologous enzymes. In 2016, Katayama et al. [92] adapted the CRISPR/Cas9 system for A. oryzae mutagenesis (10% efficiency). Later, the same authors used the plasmid containing the autonomous replication AMA1 sequence to increase editing efficiency in A. oryzae to 50-100% [134]. Several copies of the AMA1-containing plasmid were present in the transformed strains, thereby increasing the level of Cas9 and sgRNA expression and, hence, providing more efficient mutagenesis.
In 2015, Fuller et al. [114] adapted the CRISPR/Cas system for A. fumigatis [114] and verified its efficiency for the deletion of the pksP gene coding for polyketide synthase responsible for the conidia black color. The CRISPR/Cas system was integrated into the fungal genome as a single cassette; the Cas9 gene was optimized by using H. sapiens codons and linked with the hygromycin resistance gene hph. The editing efficiency was low (25-35%), which might be explained by the use of NHEJ prone to generation of random insertions [114].
The authors of [91] attempted to develop a highly efficient (95-100%) CRISPR/Cas system for precise sequence integration into the reading frame (with or without the marker) using very short (~35 bp) flanking regions. This method was called the microhomology-mediated end joining (MMEJ). Using this system, the authors achieved efficient and precise integration of the exogenous GFP into the targeted site without marker insertion. The pksP gene providing melanin synthesis in the conidia (see [114]) was used for the clone selection; in vivo expression was carried out using the AMA-based cassette developed in [61].
Genome editing of Penicillium fungi. Filamentous fungus Penicillium chrysogenum is a known producer of β-lactam antibiotics and various secondary metabolites. Successful CRISPR/Cas genome editing of this species was described in [90]. The use of the CRISPR/Cas system was verified using the polyketide synthase gene (pks17), knockout of which disrupts biosynthesis of the green pigment in the spores and can be used for the clone selection. The protoplasts were transformed with the in vitro synthesized Cas9/sgRNA complex and donor DNA with the acetamidase gene (amdS) with 1-kb flanks homologous to the pks17 gene. The strain with disrupted hdfA gene to promote HR was used as a recipient. Correct insertion of the amdS gene was confirmed by sequencing. Similarly, the pks17 and lovF genes coding for diketide synthase involved in lovastatin synthesis were knocked out with 50% efficiency. It was also demonstrated that the 60-bp length of homologous shoulders is sufficient for the successful CRISPR/Cas-mediated incorporation of donor DNA in the P. chrysogenum strain with disrupted nonhomologous recombination. The authors also transformed the protoplasts with linear DNA molecules, one of which contained the amdS marker and the other – the AMA1 sequence, Cas9, and sgRNA. They suggested that these molecules would undergo in vivo recombination due to the existence of homologous ends with the formation of autonomously replicating plasmid with the following selection on acetamide. The Cas9 gene was placed under the xylose-induced xlnA promoter, while sgRNA transcription was studied using three RNA polymerase III promoters (PtRAN-Met, PtRNA-Leu, and PU6). All three promoters provided sgRNA synthesis sufficient for genome editing. However, some clones retained acetamide resistance after several passages on the acetamide-free medium, which suggested incorporation of the marker gene into the fungal genome. The authors concluded based on comparison of the procedures of P. chrysogenum genome editing involving the in vitro assembled complex and expression of its components from the AMA1-carrying plasmid that the first method minimized the off-target effects, while the second method provided higher editing efficiency [90].
A good example of the CRISPR/Cas application in the reverse genetics is the study by Nielsen et al. [110] on modification of biosynthetic pathways for the secondary metabolites in the filamentous fungus Talaromyces atroroseus. T. atroroseus produces talaroconvolutin A and analogue of the talaroconvolutin B, a compound named ZG-1494α, which is inhibitor of the platelet acetyltransferase activating factor. Since the T. atroroseus genome had been sequenced, the authors applied the CRISPR/Cas system earlier developed for Aspergillus [116]. They used the donor plasmid carrying the hygromycin resistance gene for HR at the DSB site and three different plasmids with the AMA1 sequence, Cas9, and sgRNA gene, which differed in the protospacer sequences. The UA08_00425 gene presumably coding for the polyketide synthase (enzyme ensuring gray color of the conidia) was used as a target. The authors were able to obtain transformants with colorless conidia, but the efficiency of editing with different protospacers differed by an order of magnitude. Next, the protospacers were selected to knockout the UA08_04451 gene, which encodes an ortholog of the ccsA gene required for the synthesis of compounds structurally similar to talaroconvolutin A and ZG-1494α in other fungi. As a result, the clones with the deleted UA08_04451 gene also demonstrated disrupted synthesis of talaroconvolutin A and ZG-1494α [110].
Two studies published in 2020 reported genome editing of Penicillium subrubescens [111] and Talaromyces pinophilus [130]. In both cases, the authors used the scheme developed for Aspergillus [83]. The knockout of the ku70 gene in P. subrubescens increased probability of HR in the absence of phenotypic changes. Although the ku70 knockout was verified, no increase in the HR frequency was confirmed [111].
The CRISPR/Cas9 was also adapted for the use in Talaromyces pinophilus, which is a known producer of carbohydrases. The knockout of the seb1 gene resulted in formation of the morphologically altered hyphae (shorter and more branched compared to the wild type) and increased enzymes secretion by 10-15%. The cultural liquid of the mutant strains was characterized by higher (by 20-40%) specific activity of cellulases toward filter paper [130].
Genome editing of filamentous fungi from other genera. As is well known, other filamentous fungi are also widely used in biotechnology, for example, Acremonium chrysogenum is an industrial producer of cephalosporin. There are also multiple parasitic fungi (Alternaria alternate, Magnaporthe oryzae, Fusarium graminearum) fight against which requires the use of modern methods of molecular genetics. Neurospora crassa is a convenient object for studying fungal physiology. There is enough knowledge and experience accumulated in the genome editing in fungi of the Neurospora, Phytophthora, Mucor, Fusarium, and other genera [135].
In the study [88], the CRISPR/Cas technology was used to replace the inducible clr2 promoter (regulator of cellulase biosynthesis) by the constitutive β-tubulin promoter in the genome of N. crassa in order to increase the level of cellulase expression. Cas9 and sgRNA were delivered to the cells in two different plasmids. sgRNA was transcribed using the snr52 promoter and the sup4 terminator; transcription of the humanized Cas9 gene was controlled by the trpC gene promoter and terminator. The donor DNA was added to replace the clr2 gene promoter region with the components of the editing complex. The editing efficiency was high (up to 90%) [88].
The CRISPR/Cas was also used for the genome editing in F. graminearum, a common crop pathogen. The target gene was FgOs1, which codes for the osmosensing histidine kinase. The knockout of this gene results in the resistance to phenylpyrrole and dicarboximide. The components of the CRISPR/Cas system were integrated into the fungal genome as a single cassette; sgRNA transcription was controlled by the tef1 promoter and gpdA terminator separated from the sgRNA sequence by the ribozymes. Transcription of the optimized Cas9 gene was regulated by the gpdA gene promoter and the TRI4 terminator [132].
Another pathogenic basidiomycete, Ustilago maydis, that causes smut on corn and teosinte, was also subjected to genome editing.
The possibility of the CRISPR/Cas system application in this fungus was verified by knocking out the bE1 and bW2 genes. The components of the CRISPR/Cas system were transiently expressed in the fungal cells after cell transformation with the plasmid; sgRNA expression was regulated by the U6 promoter, while the Cas9 gene was controlled by the tef1 promoter. No donor DNA was used, and the editing efficiency was 70-100%.
A. chrysogenum C10 is used in the industrial production of cephalosporin C. However, regulation of the cephalosporin biosynthesis is still poorly understood, mostly because of the technical difficulties of genetic manipulations of the industrial strain. In their recent work, Chen et al. [136] constructed an original CRISPR-Cas9 system based on the chimeric U6/tRNA promoter. The editing efficiency with this system reached 90%. The authors used donor repair DNAs. The system was tested for the removal of large (up to 31.5 bp) DNA fragments involved in the biosynthesis of yellow compound sorbicillinoid, which allowed easy selection of the recombinant clones based on the color of culture liquid. Interestingly, the knockout strain also exhibited increased synthesis of cephalosporin.
CONCLUSIONS
The CRISPR/Cas system has been successfully adapted and is now commonly used for the genome editing in many filamentous fungi. In the last five years, starting from the first case of genome editing in T. reesei, dozens of genomes have been modified in fungal species widely used in industrial biotechnology and agriculture as sources of enzymes, enzyme preparations, secondary metabolites, and other compounds.
One of the major problems in the work with filamentous fungi is the absence of known RNA polymerase III promoters in them. However, this disadvantage can be solved by using either RNA polymerase II promoter and ribozymes or 5S rRNA promoter. It should also be mentioned that the problem of the off-target effects is less urgent in filamentous fungi, since these microorganisms are used for the production of specific compounds. If the off-targets effects do not reduce industrial value of the modified fungus, they can be neglected.
At present, the CRISPR/Cas system still remains an advanced genome editing technology. It is the most universal tool for improvement of physiological and biochemical properties of micro- and macroorganisms. The CRISPR/Cas system is rapidly developing; however, there are still problems related to the editing efficiency, off-target effects, as well as ethical issues associated with genetic modifications. In the nearest future, application of the CRISPR/Cas system in genomics and cell biology will significantly expand the knowledge that has been acquired by using other DNA technologies.
Abbreviations
- CRISPR/Cas:
-
clustered regularly interspaced short palindromic repeats and CRISPR-associated protein
- DSB:
-
double-strand break
- HDV:
-
hepatitis D virus
- HH:
-
hammerhead
- HR:
-
homologous recombination
- NHEJ:
-
non-homologous end joining
- NLS:
-
nuclear localization sequence
- sgRNA:
-
single guide RNA
- tracrRNA:
-
trans-activating CRISPR RNA
References
Punt, P. J., van Biezen, N., Conesa, A., Albers, A., Mangnus, J., and van den Hondel, C. (2002) Filamentous fungi as cell factories for heterologous protein production, Trends Biotechnol., 20, 200-206.
Ward, O. P. (2012) Production of recombinant proteins by filamentous fungi, Biotechnol. Adv., 30, 1119-1139, https://doi.org/10.1016/j.biotechadv.2011.09.012.
Lubertozzi, D., and Keasling, J. D. (2008) Developing Aspergillus as a host for heterologous expression, Biotechnol. Adv., 27, 53-57, https://doi.org/10.1016/j.biotechadv.2008.09.001.
Meyer, V. (2008) Genetic engineering of filamentous fungi – progress, obstacles, expression, Biotechnol. Adv., 26, 177-185, https://doi.org/10.1016/j.biotechadv.2007.12.001.
De Vries, R. (2003) Regulation of Aspergillus genes encoding plant cell wall degrading enzymes: relevance for industrial production, Appl. Microbiol. Biotechnol., 61, 10-20, https://doi.org/10.1007/s00253-002-1171-9.
Schuster, A., and Schmoll, M. (2010) Biology and biotechnology of Trichoderma, Appl. Microbiol. Biotechnol., 87, 787-799.
Kumar, R., Singh, S., Singh, O. V. (2008) Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives, J. Industr. Microbiol. Biotechnol., 35, 377-391.
Sinitsyn, A. P., and Rozhkova, A. M. (2015) Penicillium canescens host as the platform for development of new recombinant strains producers of carbohydrases, in Microbiology Monographs, “Microorganisms in Biorefineries” (Kamm, B., ed.) Springer, USA, pp. 1-19.
Sinitsyn, A. P., Sinitsyna, O. A., and Rozhkova, A. M. (2020) Production of industrially important enzyme using Penicillium verruculosum expression system, Biotechnologiya, 36, 17-34.
Harris, D. M, Westerlaken, I., Schipper, D., van der Krogt, Z. A., Gombert, A. K., and Sutherland, J. (2009) Engineering of Penicillium chrysogenum for fermentative production of a novel carbamoylated cephem antibiotic precursor, Metabol. Engin., 11, 125-137.
Ozcengiz, G., and Demain, A. L. (2013) Recent advances in the biosynthesis of penicillins, cephalosporins and clavams and its regulation, Biotechnol. Adv., 31, 287-311.
Corrêa, R. C. G, Rhoden, S. A., Mota, T. R., Azevedo, J. L., Pamphile, J. A., et al. (2014) Endophytic fungi: expanding the arsenal of industrial enzyme producers, J. Ind. Microbiol. Biotechnol., 41, 1467-1478, https://doi.org/10.1007/s10295-014-1496-2.
Toghueo, R. M. K., and Boyom, F. F. (2020) Endophytic Penicillium species nd their agricultural, biotechnological and pharmaceutical application, 3 Biotech, 10, 1-35, https://doi.org/10.1007/s13205-020-2081-1.
Dashtban, M., Schraft, H., and Qin, W. (2009) Fungal bioconversion of lignocellulosic residues; opportunities and perspectives, Int. J. Biol. Sci., 5, 6, 578-595, https://doi.org/10.7150/ijbs.5.578.
Raveendran, S., Parameswaran, B., Ummalyma, S. B., Abraham, A., Mathew, A. K., et al. (2018) Applications of microbial enzymes in food industry, Food Technol. Biotechnol., 56, 16-30, https://doi.org/10.17113/ftb.56.01.18.5491.
MacKenzie, D. A., Jeenes, D. J., and Archer, D. B. (2004) Filamentous Fungi as Expression Systems for Heterologous Proteins, Genetics and Biotechnology (2nd Edn.) Springer-Verlag Berlin-Heidelberg, pp. 289-315.
Kluge, J., Terfehr, D., and Kuck, U. (2018) Inducible promoters and functional genomic approaches for genetic engineering of filamentous fungi, Appl. Microbiol. Biotechnol., 102, 6357-6372, https://doi.org/10.1007/s00253-018-9115-1.
Penttilä, M. (1998) Heterologous protein production in Trichoderma, in Trichoderma and Gliocladium (Kubicek, C. P., and Harman, G. E., eds) Taylor and Francis Ltd., London.
Siedenberg, D., Mestric, S., Ganzlin, M., Schmidt, M., Punt, P. J., et al. (1999) GlaA promoter controlled production of a mutant green fluorescent protein (S65T) by recombinant Aspergillus niger during growth on defined medium in batch and fed-batch cultures, Biotechnol. Progr., 15, 43-50, https://doi.org/10.1021/bp980105u.
Bulakhov, A. G., Volkov, P. V., Rozhkova, A. M., Gusakov, A. V., Nemashkalov, V. A., et al. (2017) Using an inducible promoter of a gene encoding Penicillium verruculosum glucoamylase for production of enzyme preparations with enhanced cellulase performance, PLoS One, 12, e0170404, https://doi.org/10.1371/journal.pone.0170404.
Toews, M. W., Warmbold, J., Konzack, S., Rischitor, P., Veith, D., and Vienken, K. (2004) Establishment of mRFP1 as a fluorescent marker in Aspergillus nidulans and construction of expression vectors for high-throughput protein tagging using recombination in vitro (GATEWAY), Curr. Genet., 45, 383-389.
Dotsenko, G. S., Gusakov, A. V., Rozhkova, A. M., Korotkova, O. G., and Sinitsyn, A. P. (2015) Heterologous beta-glucosidase in a fungal cellulase system: comparison of different methods for development of multienzyme cocktails, Process Biochem., 50, 1258-1263, https://doi.org/10.1016/j.procbio.2015.05.008.
Landowski, C. P., Huuskonen, A., Wahl, R., Westerholm-Parvinen, A., Kanerva, A., et al. (2015) Enabling cost biopharmaceuticals: a systematic approach to delete proteases from a well-known protein production host Trichoderma reesei, PLoS One, 10, e0134723.
Gallo, A., Bruno, K. S., Solfrizzo, M., Perrone, G., Mule, G., Visconti, A., and Baker, S. (2012) New insight into the ochratoxin A biosynthetic pathway through deletion of a nonribosomal peptide synthetase gene in Aspergillus carbonarius, Appl. Environ. Microbiol., 78, 8208-8218, https://doi.org/10.1128/AEM.02508-12.
Ling, S. O. S., Storms, R., Zheng, Yu., Rodzi, M. R. M., Mahadi, N. M., et al. (2013) Development of a pyrG mutant of Aspergillus oryzae strain S1 as a host for the production of heterologous proteins, Sci. World J., 2013, 634317, https://doi.org/10.1155/2013/634317.
Tanaka, M., Ichinose, S., Shintani, T., and Gomi, K. (2018) Nuclear export-dependent degradation of the carbon catabolite repressor CreA is regulated by a region located near the C-terminus in Aspergillus oryzae, Mol. Microbiol., 110, 176-190, https://doi.org/10.1111/mmi.14072.
Todd, R., Lockington, R., and Kelly, J. (2000) The Aspergillus nidulans creC gene involved in carbon catabolite repression encodes a WD40 repeat protein, Mol. Genet. Genom., 263, 561-570, https://doi.org/10.1007/s004380051202.
Tamayo-Ramos, J. A., and Orejas, M. (2014) Enhanced glycosyl hydrolase production in Aspergillus nidulans using transcription factor engineering approaches, Biotechnol. Biofuels, 7, 103, https://doi.org/10.1186/1754-6834-7-103.
Tani, S., Katsuyama, Y., Hayashi, T., Suzuki, H., Kato, M., et al. (2001) Characterization of the amyR gene encoding a transcriptional activator for the amylase genes in Aspergillus nidulans, Curr. Genet., 39, 10-15, https://doi.org/10.1007/s002940000175.
Liu, L., Liu, J., Qiu, R. X., Zhu, X. G., Dong, Z. Y., and Tang, G. M. (2003) Improving heterologous gene expression in Aspergillus niger by introducing multiple copies of protein-binding sequence containing CCAAT to the promoter, Lett. Appl. Microbiol., 36, 358-361.
Zou, G., Shi, S., Jiang, Y., van den Brink, J., de Vries, R. P., et al. (2012) Construction of a cellulase hyper-expression system in Trichoderma reesei by promoter and enzyme engineering, Microb. Cell Factories, 11, 21.
Sun, X., Zhang, X., Huang, H., Wang, Y., Tu, T., et al. (2020) Engineering the cbh1 promoter of Trichoderma reesei for enhanced protein production by replacing the binding sites of a transcription repressor ACE1 to those of the activators, J. Agricult. Food Chem., 68, 1337-1346, https://doi.org/10.1021/acs.jafc.9b05452.
Weld, R. J., Plummer, K. M., Carpenter, M. A., and Ridgway, H. J. (2006) Approaches to functional genomics in filamentous fungi, Cell Res., 16, 31-44, https://doi.org/10.1038/sj.cr.7310006.
Krappmann, S., Sasse, C., and Braus, G. H. (2006) Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end-joining-deficient genetic background, Eukaryot. Cell, 5, 212-215.
Meyer, V., Arentshorst, M., El-Ghezal, A., Drews, A. C., Kooistra, R., and van den Hondel, C. A. (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant, J. Biotechnol., 128, 770-775.
Ninomiya, Y., Suzuki, K., Ishii, C., and Inoue, H. (2004) Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining, Proc. Natl. Acad. Sci. USA, 101, 12248-12253.
Liu, G., Zhang, J., and Bao, J. (2016) Cost evaluation of cellulase enzyme for industrial-scale cellulosic ethanol production based on rigorous Aspen Plus modeling, Bioproc. Biosyst. Engin., 39, 133-140.
Hu, Y., and Zhu, B. (2016) Study on genetic engineering of Acremonium chrysogenum, the cephalosporin C producer, Synth. Syst. Biotechnol., 1, 3143-3149, https://doi.org/10.1016/j.synbio.2016.09.002.
Peterson, R., and Nevalainen, H. (2012) Trichoderma reesei RUT-C30 – thirty years of strain improvement, Microbiology, 158, 58-68, https://doi.org/10.1099/mic.0.054031-0.
Wu, I., and Arnold, F. (2013) Engineered thermostable fungal Cel6A and Cel7A cellobiohydrolases hydrolase cellulase efficiently at elevated temperatures, Biotechnol. Bioengin., 110, 1874-1883, https://doi.org/10.1002/bit.24864.
Bornscheuer, U. T., Hauer, B., Jaeger, K. E., and Schwaneberg, U. (2019) Directed evolution empowered redesign of natural proteins for the sustainable production of chemicals and pharmaceuticals, Angewande Chemie Int. Edn., 58, 36-40.
Markel, U., Essani, K. D., Besirlioglu, V., Schiels, J., Streit, W. R., and Schwaneberg, U. (2020) Advances in ultrahigh-throughput screening for directed enzyme evolution, Chem. Soc. Rev., 49, 233-262.
Zhang, F., Li, J.-X., Champreda, V., Liu, C.-G., Bai, F.-W., and Zhao, X.-Q. (2020) Global reprogramming of gene transcription in Trichoderma reesei by overexpressing an artificial transcription factor for improved cellulase production and identification of Ypr1 as an associated regulator, Front. Bioengin. Biotechnol., 8, 649, https://doi.org/10.3389/fbioe.2020.00649.
Künkel, W., Berger, D., Risch, S., and Wittmann-Bresinsky, B. (1992) Genetic instability of industrial strains of Penicillium chrysogenum, Appl. Microbiol. Biotechnol., 36, 499-502.
Contreras, F., Thiele, M. J., Pramanik, S., Rozhkova, A., Dotsenko, A. S., et al. (2020) KnowVolution of GH5 Cellulase from Penicillium verruculosum to improve thermal stability for biomass degradation, ACS Sustain. Chem. Engin., 8, 12388-12399, https://doi.org/10.1021/acssuschemeng.0c02465.
Contreras, F., Pramanik, S., Rozhkova, A. M., Zorov, I. N., Korotkova, O. G., et al. (2020) Engineering robust cellulases for tailored lignocellulosic degradation cocktails, Int. J. Mol. Sci., 21, 1589, https://doi.org/10.3390/ijms21051589.
Larue, K., Melgar, M., and Martin, V. J. (2016) Directed evolution of a fungal beta-glucosidase in Saccharomyces cerevisiae, Biotechnol. Biofuels, 9, 52.
Hardiman, E., Gibbs, M., Reeves, R., and Bergquist, P. (2010) Directed evolution of a thermophilic beta-glucosidase for cellulosic bioethanol production, Appl. Biochem. Biotechnol., 161, 301-312.
Sontheimer, E. J., and Barrangou, R. (2015) Origins of the CRISPR genome-editing revolution, Hum. Gene Ther., 26, 413-424, https://doi.org/10.1089/hum.2015.091.
Chen, L., Tang, L., Xiang, H., Jin, L., Li, Q., Dong, Y., et al. (2014) Advances in genome editing technology and its promising application in evolutionary and ecological studies, Gigasciences, 3, 24, https://doi.org/10.1186/2047-217X-3-24.
Liu, W., An, Ch., Sgu, X., Meng, X., Yao, Y., et al. (2020) A dual-plasmid CRISPR/Cas System for mycotoxin elimination in polykaryotic industrial fungi, ACS Public., 9, 2087-2095, https://doi.org/10.1021/acssynbio.0c00178.
Tyagi, S., Kumar, R., Das, A., Won, S.-Y., and Shukla, P. (2020) CRISPR-Cas9 system: a genome-editing tool with endless possibilities, J. Biotechnol., 319, 36-53, https://doi.org/10.1016/j.jbiotec.2020.05.008.
Mali, P, Esvelt, K. M., and Church, G. M. (2013) Cas9 as a versatile tool for engineering biology, Nat. Methods, 10, 957-963, https://doi.org/10.1038/nmeth.2694.
Basset, A. R., and Liu, J. L. (2014) CRISPR/Cas9 and genome editing in Drosophila, J. Genet. Genom., 41, 7-19, https://doi.org/10.1016/j.jgg.20013.12.004.
Blackburn, P. R, Campbell, J. M, Clark, K. J., and Ekker, S. C. (2013) The CRISPR system-keeping zebrafish gene targeting fresh, Zebrafish, 10, 116-118, https://doi.org/10.1089/zeb.2013.9999.
Ebina, H., Misawa, N., Kanemura, Y., and Koyanagi, Y. (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus, Sci. Rep., 3, 2510, https://doi.org/10.1038/srep02510.
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., and Nakata, A. (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase conversation in Escherichia coli, and identification of the gene product, J. Bacteriol., 169, 5429-5433.
Mojico, F. J., Diez-Villasenor, C., Soria, E., and Juez, G. (2000) Biological significans of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria, Mol. Microbiol., 36, 244-246.
Jansen, R., van Embden, J. D. A., Gaastra, W., and Schlous, L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes, Mol. Microbiol., 43, 1565-1575.
Barrangou, R., Fremax, C., Deveau, H., Richards, M., Boyaval, P., et al. (2007) CRISPR provides acquired resistance against viruses in prokaryotes, Science, 315, 1709-1712.
Nødvig, C. S., Nielsen, J. B., Kogle, M. E., and Mortensen, U. H. (2015) A CRISPR-Cas9 system for genetic engineering of filamentous fungi, PLoS One, 10, e0133085, https://doi.org/10.1371/journal.pone.0133085.
DiCarlo, J. E., Norville, J. E., Mali, P., Rios, X., Aach, J., and Church, G. M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR/Cas systems, Nucleic Acids Res., 41, 4336-4343, https://doi.org/10.1093/nar/gkt135.
Horwitz, A. A., Walter, J. M., Schubert, M. G., and Kung, S. H. (2015) Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas, Cell Systems, 1, 88-96, https://doi.org/10.1016/j.cels.2015.02.001.
Curran, K. A., Crook, N., Karim, A., Gupta, A., Wagman, A., and Alpe, H. (2014) Design of synthetic yeast promoters via tuning of nucleosome architecture, Nat. Commun., 5, 4002, https://doi.org/10.1038/ncomms5002.
Doudna, J. A., and Charpentier, E. (2014) The new frontier of genome engineering with CRISPR-Cas9, Science, 346, 1077-1087, https://doi.org/10.1126/science.1258096.
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2013) Tha basic building blocks and evoluation of CRISPR/CAS systems, Biochem. Soc. Trans., 41, 1392-1400, https://doi.org/10.1042/BST20130038.
Wang, S., Chen, H., Tang, X., Zhang, H., Chen, W., and Chen, Y. Q. (2017) Molecular tools for gene manipulation in filamentous fungi, Appl. Microbiol. Biotechnol., 101, 8063-8075, https://doi.org/10.1007/s00253-017-8486-z.
Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enchanced genome editing specificity, Cell, 154, 1380-1389, https://doi.org/10.1016/j.cell.2013.08.021.
Hsu, P. D., Lander, E. S., and Zhang, F. (2014) Development and applications of CRISPR-Cas9 for genome engineering, Cell, 157, 1262-1278, https://doi.org/10.1016/j.cell.2014.05.010.
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., et al. (2011) Evolution and classification of the CRISPR/Cas systems, Nat. Rev. Microbiol., 9, 467-477.
Haft, D. H., Selengut, J., Mongodin, E. F., and Nelson, K. E. (2005) A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR-Cas subtypes exist in prokaryotic genomes, PLoS Computat. Biol., 1, e60.
Chylinsky, K., Le, R. A., and Charpentier, E. (2013) The tracrRNAand Cas9 families of type II CRISPR-Cas immunity systems, RNA Biol., 10, 726-737.
Jinek, M., Chulinski, K., Fonfara, I., Hauer, M., Doudna, J., and Charpentier, E. A. (2012) Programmable dual-RNA-guided DNA endonuclease immunity, Science, 337, 816-821.
Kuscu, C., Arslan, S., Singh, R., Thorpe, J., and Adli, M. (2014) Genome wide analysis reveals characteristics of off-target sites bound by Cas9 endonuclease, Nat. Biotechnol., 32, 677-683.
Cong, L., Ran, F. A., Cox, D., Lin, S., Barreto, R., and Habib, N. (2013) Multiplex genome engineering using CRISPR/Cas systems, Science, 339, 197-217.
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., and Dicarlo, J. E. (2013) RNA-guided human genome engineering via Cas9, Science, 339, 823-826.
Jiang, F., Zhou, K., Ma, L., Gressel, S., and Doudna, J. A. (2015) A Cas9-guide RNA complex preorganaized for target DNA recognition, Science, 348, 1477-1481.
Huai, C., Li, G., Yao, R., Zhang, Y., Cao, M., et al. (2017) Structural insights into DNA cleavage activation of CRISPR-Cas9 system, Nat. Commun., 8, 1375.
Stenberg, S. H., Redding, S., Jinek, M., Greene, E. C., and Doudna, J. A. (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9, Nature, 507, 62-67.
Rutkauskas, M., Sinkunas, T., Songailiene, I., Tikhomirova, M. S., Siksnys, V., and Seidel, R. (2015) Directional R-loop formation by the CRISPR/Cas surveillance complex Cascade provides efficient off-target site rejection, Cell Rep., 10, 1534-1543.
Palermo, G., Miao, Y., Walker, R. C., Jinek, M., and McCammon, J. A. (2016) Striking plasticity of CRISPR-Cas9 and key role of non-target DNA, as revealed by molecular simulations, ACS Cent. Sci., 2, 756-763.
Sternberg, S. H., LaFrance, B., Kaplan, M., and Doudna, J. A. (2015) Conformational control of DNA target cleavage by CRISPR-Cas, Nature, 527, 110-113.
Nødvig, C. S., Hoof, J. B., Kogle, M. E., Jarczynska, Z. D., Lehmbeck, J., et al. (2018) Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli, Fungal Genet. Biol., 115, 78-89, https://doi.org/10.1016/j.fgb.2018.01.004.
Wang, Q., Cobine, P. A., and Coleman, J. J. (2018) Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes, Fungal Genet. Biol., 117, 21-29, https://doi.org/10.1016/j.fgb.2018.05.003.
Chen, J., Lai, Y., Wang, L., Zhai, S., Zou, G., et al. (2017) CRISPR/Cas9-mediated efficient genome editing via blastospore-based transformation in entomopathogenic fungus Beauveria bassiana, Sci. Rep., 8, 45763, https://doi.org/10.1038/srep45763.
Chen, B. X., Wei, T., Ye, Z. W., Yun, F., Kang, L. Z., et al. (2018) Efficient CRISPR-Cas9 gene disruption system in edible-medicinal mushroom Cordyceps militaris, Front. Microbiol., 9, 1157, https://doi.org/10.3389/fmicb.2018.01157.
Kislitsin, V. Yu., Chulkin, A. M., Sinel’nikov, I. G., Sinitsin, A. P., and Rozhkova, A. M. (2020) Expression of CAS9 complex of the CRISPR/CAS system for the genome editing of the filamentous fungus Penicillium verruculosum, Vestn. Mosk. Univ. Ser. 2, 61, 47-54.
Matsu-Ura, T., Baek, M., Kwon, J., and Hong, C. (2015) Efficient gene editing in Neurospora crassa with CRISPR technology, Fungal Biol. Biotechnol., 2, 1-7, https://doi.org/10.1186/s40694-015-0015-1.
Sarkari, P., Marx, H., Blumhoff, M. L., Mattanovich, D., Sauer, M., and Steiger, M. G. (2017) An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger, Bioresour. Technol., 245, 1327-1333, https://doi.org/10.1016/j.biortech.2017.05.004.
Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., and Nygard, Y. (2016) CRISPR/Cas9 based genome editing of Penicillium chrysogenum, ACS Synthet. Biol., 5, 754-764, https://doi.org/10.1021/acssynbio.6b00082.
Zhang, C., Meng, X., Wei, X., and Lu, L. (2016) Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus, Fungal Genet. Biol., 86, 47-57, https://doi.org/10.1016/j.fgb.2015.12.007.
Katayama, T., Tanaka, Y., Okabe, T., Nakamura, H., Fujii, W., and Kitamoto, K. (2016) Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungus Aspergillus oryzae, Biotechnol. Lett., 38, 637-642, https://doi.org/10.1007/s10529-015-2015-x.
Schuster, M., Schweizer, G., and Kahmann, R. (2018) Comparative analyses of secreted proteins in plant pathogenic smut fungi and related basidiomycetes, Fungal Genet. Biol., 112, 21-30, https://doi.org/10.1016/j.fgb.2016.12.003.
Schuster, M., Schweizer, G., Reissmann, S., and Kahmann, R. (2016) Genome editing in Ustilago maydis using the CRISPR/Cas system, Fungal Genet. Biol., 89, 3-9, https://doi.org/10.1016/j.fgb.2015.09.001.
Deng, H., Gao, R., Liao, X., and Cai, Y. (2017) Characterization of a major facilitator superfamily transporter in Shiraia bambusicola, Res. Microbiol., 168, 664-672, https://doi.org/10.1016/j.resmic.2017.05.002.
Deng, H., Gao, R., Liao, X., and Cai, Y. (2017) Genome editing in Shiraia bambusicola using CRISPR-Cas9 system, J. Biotechnol., 259, 228-234, https://doi.org/10.1016/j.jbiotec.2017.06.1204.
Lee, C. M., Cradick, T. J., and Bao, G. (2016) The Neisseria meningitidis CRISPR/Cas System enables specific genome editing in mammalian cells, Mol. Ther., 24, 645-654.
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., et al. (2015) Cpf1 is a single RNA-guided endonuclease of class 3 CRISPR/Cas system, Cell, 163, 759-771.
Swiat, M. A., Dashko, S., den Ridder, M., Wijsman, M., van der Oost, J., et al. (2017) FnCpf1: a novel and efficient genome editing tool for Saccaromyces cerevisiae, Nucleic Acid Res., 45, 12585-12598.
Liu, L., Li, X., Ma, J. Z., Li, Y., Wang, L., Wang, J., et al. (2017) The molecular architecture for RNA-Guided RNA cleavage by Cas13a, Cell, 170, 714-726.
Gao, Y., and Zhao, Y. (2014) Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing, J. Integr. Plant Biol., 56, 343-349, https://doi.org/10.1111/jipb.12152.
Liang, Y., Han, Y., Wang, C., Jiang, C., and Xu, J. R. (2018) Targeted deletion of the USTA and UvSLT2 genes efficiently in Ustilaginoidea virens with the CRISPR-Cas9 system, Front. Plant Sci., 9, 699, https://doi.org/10.3389/fpls.2018.00699.
Zheng, X., Zheng, P., Zhang, K., Cairns, T. C., Meyer, V., Sun, J., and Ma, Y. (2018) 5S rRNA promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger, ACS Synthet. Biol., 8, 1568-1574.
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P., and Lu, T. K. (2014) Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells, Mol. Cell, 54, 698-710, https://doi.org/10.1016/j.molcel.2014.04.022.
Wang, J., Li, X., Zhao, Y., Li, J., Zhou, Q., and Liu, Z. (2015) Generation of cell-type-specific gene mutations by expressing the sgRNA of the CRISPR system from the RNA polymerase II promoters, Protein Cell, 6, 689-692, https://doi.org/10.1007/s13238-015-0169-x.
Xue, W., Chen, S., Yin, H., Tammela, T., Papagiannakopoulos, T., et al. (2014) CRISPR-mediated direct mutation of cancer genes in the mouse liver, Nature, 514, 380-384, https://doi.org/10.1038/nature13589.
Shi, T.-Q., Liu, G.-N., Ji, R.-Yu., Shi, K., Song, P., et al. (2017) CRISPR/Cas9-based genome editing of filamentous fungi: the state of the art, Appl. Microbiol. Biotechnol., 101, 7435-7443.
Huang, L., Dong, H., Zheng, J., Wang, B., and Pan, L. (2019) Highly efficient single base editing in Aspergillus niger with CRISPR/Cas9 cytidine deaminase fusion, Microbiol. Res., 223, 44-50, https://doi.org/10.1016/j.micres.2019.03.007.
Liu, R., Chen, L., Jiang, Y., Zhou, Z., and Zou, G. (2015) Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system, Cell Discov., 1, 15007, https://doi.org/10.1038/celldisc.2015.7.
Nielsen, M. L., Isbrandt, T., Rasmussen, K. B., Thrane, U., Hoof, J. B., and Larsen, T. O. (2017) Genes linked to production of secondary metabolites in Talaromyces atroroseus revealed using CRISPR-Cas9, PLoS One, 12, e0169712, https://doi.org/10.1371/journal.pone.0169712.
Salazar-Cerezo, S., Kun, R. S., de Vries, R. P., and Garrigues, S. (2020) CRISPR/Cas9 technology enables the development of the filamentous ascomycete fungus Penicillium subrubescens as a new industrial enzyme producer, Enzyme Microb. Technol., 133, 109463, https://doi.org/10.1016/j.enzmictec.2019.109463.
Yamato, T., Handa, A., Arazoe, T., Kuroki, M., Nozaka, A., et al. (2019) Single crossover-mediated targeted nucleotide substitution and knock-in strategies with CRISPR/Cas9 system in the rice blast fungus, Sci. Rep., 9, 1-8, https://doi.org/10.1038/s41598-019-43913-0.
Liu, Q., Gao, R., Li, J., Lin, L., Zhao, J., et al. (2017) Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering, Biotechnol. Biofuels, 10, 1-14, https://doi.org/10.1186/s13068-016-0693-9.
Fuller, K. K., Chen, S., Loros, J. J., and Dunlap, J. C. (2015) Development of the CRISPR/Cas9 system for targeted gene disruption in Aspergillus fumigatus, Eukaryot. Cell, 14, 1073-1080, https://doi.org/10.1128/EC.00107-15.
Fang, Y., and Tyler, B. M. (2016) Efficient disruption and replacement of an effector gene in the oomycete Phytophthora sojae using CRISPR/Cas9, Mol. Plant Pathol., 17, 127-139.
Kuivanen, J., Wang, Y.-M. J., and Richard, P. (2016) Engineering Aspergillus niger for galactaric acid production: elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9, Microb. Cell Fact., 15, 210.
Weyda, I., Yang, L., Vang, J., Ahring, B. K., Lubeck, M., and Lubeck, P. S. (2017) A comparison of Agrobacterium-mediated transformation and protoplast-mediated transformation with CRISPR-Cas9 and bipartite gene targeting substrates, as effective gene targeting tools for Aspergillus carbonarius, J. Microbiol. Methods, 135, 26-34.
Wenderoth, M., Pinecker, C., Voß, B., and Fischer, R. (2017) Establishment of CRISPR/Cas9 in Alternaria alternate, Fungal Genet. Biol., 101, 55-60.
Weber, J., Valiante, V., Nødvig, C. S., Mattern, D. J., Slotkowski, R. A., and Mortensen, U. H. (2017) Functional reconstitution of a fungal natural product gene cluster by advanced genome editing, ACS Synthet. Biol., 6, 62-68, https://doi.org/10.1021/acssynbio.6b00203.
Wu, C., Chen, Y., Qiu, Y., Niu, X., Zhu, N., et al. (2020) A simple approach to mediate genome editing in the filamentous fungus Trichoderma reesei by CRISPR/Cas9-coupled in vivo gRNA transcription, Biotechnol. Lett., 42, 1203-1210, https://doi.org/10.1007/s10529-020-02887-0.
Zhang, L., Zhao, X., Zhang, G., Zhang, J., Wang, X., et al. (2016) Light-inducible genetic engineering and control of non-homologous end-joining in industrial eukaryotic microorganisms: LML 3.0 and OFN 1.0, Sci. Rep., 6, 20761.
Kocak, D. D., Josephs, E. A., Bhandarkar, V., Adkar, Sh., Kwon, J., and Gersbach, Ch. (2019) Increasing the specificity of CRISPR systems with engineered RNA secondary structures, Nat. Biotechnol., 37, 657-666, https://doi.org/10.1038/s41587-019-0095-1.
Cui, Y., Xu, J., Cheng, M., Liao, X., and Peng, S. (2018) Review of CRISPR/Cas9 sgRNA design tools, Interdiscip. Sci. Computational Life Sci., 10, 455-465.
Kuivanen, J., Arvas, M., and Richard, P. (2017) Clustered genes encoding 2-ketol-gulonate reductase and l-idonate 5-dehydrogenase in the novel fungal d-glucuronic acid pathway, Front. Microbiol., 8, 225, https://doi.org/10.3389/fmicb.2017.00225.
Li, J., Zhang, Y., Zhang, Y., Yu, P. L., Pan, H., and Rollins, J. A. (2018) Introduction of large sequence inserts by CRISPR-Cas9 to create pathogenicity mutants in the multinucleate filamentous pathogen Sclerotinia sclerotiorum, MBio, 9, e00567-e00518, https://doi.org/10.1128/mBio.00567-18.
Miao, J., Li, X., Lin, D., Liu, X., and Tyler, B. M. (2018) Oxysterol-binding protein-related protein 2 is not essential for Phytophthora sojae based on CRISPR/Cas9 deletions, Environ. Microbiol. Rep., 10, 293-298, https://doi.org/10.1111/1758-2229.12638.
Rantasalo, A., Vitikainen, M., Paasikallio, T., Jäntti, J., Landowski, C. P., and Mojzita, D. (2019) Novel genetic tools that enable highly pure protein production in Trichoderma reesei, Sci. Rep., 9, 5032, https://doi.org/10.1038/s41598-019-41573-8.
Kuivanen, J., Korja, V., Holmström, S., and Richard, P. (2019) Development of microtiter plate scale CRISPR/Cas9 transformation method for Aspergillus niger based on in vitro assembled ribonucleoprotein complexes, Fungal Biol. Biotechnol., 6, 3, https://doi.org/10.1186/s40694-019-0066-9.
Dong, L., Lin, X., Yu, D., Huang, L., Wang, B., and Pan, L. (2020) High-level expression of highly active and thermostable trehalase from Myceliophthora thermophila in Aspergillus niger by using the CRISPR/Cas9 tool and its application in ethanol fermentation, J. Industr. Microbiol. Biotechnol., 47, 133-144, https://doi.org/10.1007/s10295-019-02252-9.
Manglekar, R. R., and Geng, A. (2020) CRISPR-Cas9-mediated seb1 disruption in Talaromyces pinophilus EMU for its enhanced cellulase production, Enzyme Microb. Technol., 140, 109646, https://doi.org/10.1016/j.enzmictec.2020.109646.
Rojas-Sánchez, U., López-Calleja, A. C., Millán-Chiu, B. E., Fernández, F., Loske, M., Gómez-Lim, M. A. (2020) Enhancing the yield of human erythropoietin in Aspergillus niger by introns and CRISPR-Cas9, Protein Express. Purif., 168, 105570, https://doi.org/10.1016/j.pep.2020.105570.
Gardiner, D. M., and Kazan, K. (2018) Selection is required for efficient Cas9-mediated genome editing in Fusarium graminearum, Fungal Biol., 122, 131-137, https://doi.org/10.1016/j.funbio.2017.11.006.
Liu, R., Chen, L., Jiang, Y., Zou, G., and Zhou, Z. (2017) A novel transcription factor specifically regulates GH11 xylanase genes in Trichoderma reesei, Biotechnol. Biofuels, 10, 194.
Katayama, T., Nakamura, H., Zhang, Y., Pascal, A., Fujii, W., and Maruyama, J. I. (2019) Forced recycling of an AMA1-based genome-editing plasmid allows for efficient multiple gene deletion/integration in the industrial filamentous fungus Aspergillus oryzae, Appl. Environ. Microbiol., 85, e01896-e01818, https://doi.org/10.1128/AEM.01896-18.
Song, R., Zhai, Q., Sun, L., Huang, E., Zhang, Y., et al. (2019) CRISPR/Cas9 genome editing technology in filamentous fungi: progress and perspective, Appl. Microbiol. Biotechnol., 103, 6919-6932.
Chen, C., Liu, J., Dua, C., Pan, Y., and Liu, G. (2020) Improvement of the CRISPR-Cas9 mediated gene disruption and large DNA fragment deletion based on a chimeric promoter in Acremonium chrysogenum, Fungal Genet. Biol., 134, 103279, https://doi.org/10.1016/j.fgb.2019.103279.
Funding
This work was partially supported by the Russian Foundation for Basic Research (project no. 18-29-07070).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This article does not contain description of studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Rozhkova, A.M., Kislitsin, V.Y. CRISPR/Cas Genome Editing in Filamentous Fungi. Biochemistry Moscow 86 (Suppl 1), S120–S139 (2021). https://doi.org/10.1134/S0006297921140091
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297921140091