
Abstract
The advent of fluorescent proteins as vital dyes had a major impact in many research fields. Different green fluorescent protein (GFP) variants were established in prokaryotic and eukaryotic organisms within the past 10 years, and other fluorescent proteins were discovered and applied. We expressed the Discosoma red fluorescent protein, DsRed (T4), the improved monomeric red fluorescent protein (mRFP1) and the blue fluorescent protein (BFP) in the filamentous fungus Aspergillus nidulans. Whereas DsRed requires tetramer formation for fluorescence, mRFP1 functions as monomer. We used sGFP, DsRed (T4), mRFP1 and BFP for nuclear and/or mitochondrial labelling. To facilitate gene tagging, we established a number of cloning vectors for the efficient, simultaneous fusion of any protein with mRFP1, BFP and sGFP or the haemagglutinin epitope, 3×HA. A PCR-amplified gene of interest can be inserted into the expression vectors without cloning but using homologous recombination in vitro (GATEWAY). The vectors contain the argB gene as a selection marker for A. nidulans and the inducible alcA promoter for control of expression. The system allows labelling of a protein with several tags in one recombination reaction. Both the nutritional marker gene and the promoter are frequently used in other fungi, suggesting that this set of expression vectors will be very useful tools for gene analysis on a genome-wide scale.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungi are widely used as model organisms to study the control of cell cycle, organelle movement, protein secretion, fungus-host interactions, etc. In addition, the biology of fungi is intensely studied to unravel the principles of fungal growth, adaptation to environmental conditions, metabolic capacities and the regulation or development of reproductive structures, etc. Gene function analyses comprise mainly the study of loss-of-function or gain-of-function mutations and the monitoring of expression levels or subcellular localisation of proteins. The advent of the Aequoria victoria green fluorescent protein (GFP) had a great impact on fungal molecular biology (Cormack 1998). After the initial application of this technology in Escherichia coli (Chalfie et al. 1994) and Saccharomyces cerevisiae (Niedenthal et al. 1996), GFP has been used in a variety of fungi, such as Ustilago maydis (Spellig et al. 1996), Aspergillus nidulans (Fernandez-Abalos et al. 1998; Suelmann et al. 1997), Schizophyllum commune (Lugones et al. 1999) and Neurospora crassa (Fuchs et al. 2002). Within the past 10 years, a variety of GFP variants has been developed, which show increased sensitivity, faster folding of the protein or altered spectroscopic properties (variants with yellow fluorescence or blue fluorescence; Lippincott-Schwartz and Patterson 2003). The proteins with altered spectral properties are especially useful for co-localisation of two given proteins. In addition, new fluorescent proteins have been characterised and introduced into different organisms. However, in the filamentous fungi, mainly GFP and its derivatives have been used so far (Cormack 1998; Pöggeler et al. 2003). Recently, the Discosoma red fluorescent protein, DsRed, was applied in Penicillium paxilli, Trichoderma species (Mikkelsen et al. 2003) and A. nidulans (Dou et al. 2003). However, DsRed requires tetramer formation for the development of fluorescence (Baird et al. 2000). This may hamper the application of DsRed for protein fusions, because forced tetramerisation of the corresponding fusion proteins is likely to disturb the cellular function of the original polypeptide. In addition, the original DsRed isolate required several days for maturation of the fluorescent properties, which is inappropriate for many applications (Baird et al. 2000). To improve the folding properties, several derivatives were engineered, one of which was DsRed (T4). This protein displays a half-time for maturation of 0.71 h and still has a relative brightness of 0.38, in comparison with the slow-folding version (Bevis and Glick 2002). The additional problem of tetramer formation in DsRed was also solved recently with the monomeric red fluorescent protein derivative (mRFP1; Campbell et al. 2002). mRFP1 functions as a monomer and matures quickly. In addition, the excitation and emission peaks, 584 nm and 607 nm, are about 25 nm red-shifted in comparison with the engineered and improved red fluorescent proteins DsRed (T4) and mRFP1 and the blue fluorescent protein (BFP) in the filamentous fungus A. nidulans. All three proteins were used for organelle-labelling. In addition, we introduce a series of vectors for the efficient cloning of tagged expression constructs.
Materials and methods
Strains, plasmids and culture conditions
Supplemented minimal and complete media for A. nidulans were prepared as described by (Käfer 1977) and standard strain construction procedures were used. Standard laboratory Escherichia coli strains (XL-1 blue, Top 10 F’) were used. The A. nidulans strains used were: RMS011 (pabaA1, yA2; ΔargB::trpCΔB; trpC801, veA1; Stringer et al. 1991), SRF200 (pyrG89; ΔargB::trpCΔB; pyroA4; veA1; Karos and Fischer 1999), SDM1004 (RMS011 transformed with pJH19 and pRS54), SDM25 (RMS011 transformed with pRF280 and pSK700) and SSK90 (RMS011 transformed with pJH19).
Molecular techniques
Standard DNA transformation procedures were used for A. nidulans (Yelton et al. 1984) and E. coli (Sambrook and Russel 1999). For PCR experiments, standard protocols were applied, using a capillary rapid cycler (Idaho Technology, Idaho Falls, USA) for the reaction cycles. DNA sequencing was done commercially (MWG Biotech, Ebersberg). Western blot analysis was performed as described by the supplier of the Hybond membranes and the Western blot kit (Amersham Pharmacia, Freiburg and Roche, Mannheim).
Plasmids
For nuclear labelling, the plasmids used were: pRF280 [gpdA(p)::sgfp::stuA(NLS), argB in pBluescript; a derivative of pRS31; Suelmann et al. 1997], pRF281 [gpdA(p)::sgfp::stuA(NLS), pyr4 in pBluescript], pJW18 [alcA(p)::DsRed (T4)::stuA(NLS), argB], and pJH19 [alcA(p) in pJW18 substituted by the gpd promoter]. For mitochondrial labelling, the plasmids used were: pRS54 [gpdA(p)::citrate synthase N-term::sgfp in pBluescript; Suelmann and Fischer 2000], pSK800 (sgfp in pRS54 substituted by mRFP1) and pSK700 [sgfp in pRS54 substituted by DsRed (T4)].
Destination vectors
For pMT-OvE and pMT-3×HA, the vector pBluescript KS-Δ was used, with argB cloned into NotI and alcA(p) cloned into BamHI. The suicide ccdB box [containing the ccdB gene (Bernard and Couturier 1992), the chloramphenicol cat gene and attR sites] was amplified with pDEST 14 (Invitrogen) as template (primers 5’-CTC GAG ATA GGG AGA CCA CAA CGG-3’, 5’-CTC GAG CAG CTT CCT TTC GGG C-3’) and cloned into XhoI downstream of the alcA promoter. 3×HA was cloned as a KpnI fragment downstream of the ccdB box. For pMT-sGFP, pMT-BFP and pMT-mRFP1, the vector pSNi11 (Schier and Fischer 2002) including argB and alcA(p) was used to insert a commercially available ccdB box-containing fragment (Invitrogen) blunt-ended into a SmaI site, as described by the supplier. The tags were cloned into EcoRI and KpnI. The sGFP gene was amplified with the primer combination 5’-GAA TTC ATG GTG AGC AAG GGC GAG-3’ and 5’-GGT ACC CTA TTT GTA CAG CTC GTC-3’, the BFP gene with the same primers as for sGFP and mRFP1 with the primers 5’-GAA TTC ATG GCC TCC TCC GAG G-3’ and 5’-GGT ACC TTA GGC GCC GGT GGA G-3’. The template for mRFP1 was obtained from Dr. Prastio (University of San Diego, USA) and the template for BFP from Dr. Ram (Leiden University, The Netherlands). For the amplification of all destination vectors, ccdB gene-resistant E. coli cells (Library Efficiency DB3.1 competent cells; Invitrogen) were used. Entry vectors pMT-veA and pMT-stuA(NLS) were based on the vector pENTR/D-TOPO (Invitrogen). The vector pMT-veA included the PCR-amplified veA gene (primers 5’-CAC CGC AAC AAG TCT TCT AGA GC-3’, recombination was performed with the LR clonase enzyme mix (Invitrogen), as described by the supplier. In each reaction, 300 ng destination vector and 300 ng entry vector were used. If more than one destination vector were used in a single reaction, the different destination vectors were used in equal amounts, so that the total amount of destination vectors was always 300 ng per reaction. The same applied for the use of several entry vectors in a single LR reaction. The LR recombination reaction was transformed into E. coli, as described by the supplier.
Fluorescence microscopy
Fluorescent proteins were visualised with appropriate filter combinations (no. 15 for red fluorescence, no. 9 for green fluorescence; Zeiss, Jena, Germany), using an Axiophot microscope (Zeiss). Images were captured with a high-resolution Orca ER camera (Hamamatsu, Munich, Germany). Alternatively, we used a TCSSp2 confocal microscope (Leica).
Results and discussion
Expression of DsRed (T4) and mRFP1 in A. nidulans
In previous work, we fused sGFP to the C-terminal domain containing the nuclear localisation signals (NLS) of the developmental transcription factor StuA and expressed the construct under the control of the constitutive gpd promoter (Suelmann et al. 1997). A similar construct (pJH19) was established with the DsRed (T4) gene instead of the sgfp gene. The construct was introduced into wild-type A. nidulans (RMS011) and stable transformants were analysed for red fluorescence. Microscopic inspection revealed that nuclei were brightly labelled. In previous experiments, we found that the sGFP fusion protein diffuses out of the nucleus during mitosis (Suelmann et al. 1997). This result was surprising, because in fungi the nuclear envelope remains intact during nuclear division. However, small proteins could diffuse through the nuclear pore complex and, since the sGFP-StuA(NLS) fusion protein has a predicted molecular mass of about 45 kDa, it could leak out of the nucleus. In comparison, fluorescent proteins fused to DNA-binding proteins remain in the nucleus during mitosis (Fernandez-Martinez et al. 2003; unpublished data from our laboratory). Since the DsRed protein needs to form a tetramer for fluorescence, the nuclear-targeted protein should have a molecular mass of about 190 kDa. To test whether this fusion protein would remain in the nucleus, we did a time-lapse analysis of mitosis and found that the sGFP-StuA and the DsRed-StuA fusion proteins behaved identically (Fig. 1). These observations suggest that diffusion of the fluorescent proteins out of the nucleus during mitosis is not dependent on the molecular mass of the proteins but rather depends on DNA interaction. Since the StuA(NLS) portion of the StuA protein only comprises the putative NLS but probably not the DNA-interacting domain, it is unlikely that the fusion proteins bind to DNA. Our results could be explained in two ways: (1) the nuclear envelope does not remain intact during mitosis and becomes largely leaky or (2) the nuclear envelope remains intact but the nuclear import machinery is not active.

Localisation of DsRed (T4) in the nuclei of hyphae of A. nidulans before (a), during (b) and after (c) mitosis. Plasmid pJH19 was transformed into RMS011
To demonstrate that the red fluorescent protein can be used for double-labelling experiments, we combined the red nuclear label [pJW19, DsRed (T4)] with sGFP-labelled mitochondria (pRS54; Fig. 2a). We also expressed a fusion between the mRFP1 targeted to mitochondria (pSK700) in combination with green-labelled nuclei (Fig. 2b).

Double-labelling of nuclei and mitochondria with DsRed (T4) and sGFP. a, c Phase contrast image of germlings. b, d Fluorescence picture of the same germlings as in a, c. a, b Labelling of nuclei with DsRed (T4) and mitochondria with sGFP in strain SDM1004. c, d Labelling of nuclei with sGFP and mitochondria with DsRed (T4) in strain SDM25
Construction of expression vectors for over-expression and protein tagging with mRFP1, BFP, sGFP or 3×HA
The tagging of proteins by conventional cloning usually involves several time-consuming steps, and sometimes it is hard to achieve because of the lack of unique restriction sites in the vectors and/or the gene of interest. This problem has been solved by the introduction of a vector system based on recombination in vitro (Landy 1989). After cloning of the gene of interest into a vector flanked by attL sites (pENTR/D-TOPO, kanamycin resistance), the gene is transferred to the destination vector by in vitro recombination. Due to the recombination event, the suicide ccdB box (Bernard and Couturier 1992) is replaced by the gene of interest. The obtained expression vectors confer ampicillin resistance to the recipient E. coli strains, which are not resistant to the ccdB box (XL-1 blue, Top 10 F’) and thus only plasmids with successful recombination are able to amplify on ampicillin-containing media. Meanwhile, a great variety of expression vectors are commercially available from Invitrogen and have been adapted for use in plants (Curtis and Grossniklaus 2003; Karimi et al. 2002). However, those vectors are not useful for filamentous fungi, due to the lack of a promoter and a fungus-specific selection marker. Therefore, we designed a number of constructs which allow the tagging of proteins with sGFP, mRFP1, BFP and the haemagglutinin epitope, 3×HA (Fig. 3). To test the functionality of the system, we tagged part of the StuA transcription factor, containing the NLS sequences. The in vitro LR-recombination reaction with the entry vector pMT-stuA(NLS) and destination vectors pMT-BFP and pMT-mRFP1 should result in two different expression vectors: stuA(NLS) tagged with BFP and stuA(NLS) tagged with mRFP1. After transformation of the recombination reaction in E. coli, 24 colonies were analysed. Five of the plasmids contained the stuA(NLS) tagged with mRFP1 and 13 plasmids contained the stuA(NLS) tagged with BFP (Fig. 4). All inserts were in the correct orientation. StuA(NLS) was also tagged with sGFP. The nuclei of the corresponding transformants (SRF200) harboured red, green or blue nuclei, respectively (Fig. 5a). The fluorescence of the BFP is very weak and it is not recommendable for standard use. In another example, we used pMT-veA as the entry vector with three different destination vectors (including pMT-3×HA). Seven from 14 tested E. coli colonies contained the tagged veA gene and three of these were fused to 3×HA. The function of the VeA-3×HA expression vector in A. nidulans (SRF200) was shown by Western blot (Fig. 5b). It was also possible to use several entry vectors with only one destination vector in a single in vitro LR-recombination reaction. Using this strategy, one can transfer several genes of interest into one destination vector (data not shown). Meanwhile, the system was successfully used to label other cellular proteins in our laboratory. Hence, the introduced vectors are very useful tools for quick and efficient protein-tagging in filamentous fungi. High-throughput analyses will be of increasing importance with the increasing number of full fungal genome sequences available.

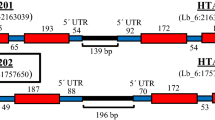
Scheme of the constructed vectors. The sequences and details of these vectors are available at http://www.uni-marburg.de/mpi/fischer/fischer.html. The web site will be updated by adding new vectors (with relevant information) as they are constructed in our laboratory. Restriction enzyme sites: B BamHI, Bg BglII, E EcoRI, F FspI, K KpnI, N NotI, S SmaI, X XhoI

Scheme of an example of the recombination reaction between one entry-clone and two destination vectors (a) and the analysis of plasmids obtained after the recombination (b). a The location of the primers for the amplification of the gene of interest is indicated. The forward primer should contain CACC upstream of the ATG and the reverse primer should end just before the stop codon. b Plasmids were digested with FspI and separated on a 1% agarose gel. A total of 13 clones were derived from recombination with pMT-BFP (e.g. lane 1) and five from pMT-mRFP1 (e.g. lane 2). The other six cannot be explained by the recombination event. Lambda DNA digested with Eco1301 was used as a size marker

Analysis of SRF200 strains transformed with plasmids created by recombination between pMT-stuA(NLS) and the destination vectors for labelling with fluorescent proteins or the veA gene in the entry vector pMT-veA and the hemagglutinin (HA)-tagging plasmid as destination vector, as described in Fig. 3. a Fluorescence analysis. b Western blot analysis of protein extracts from a control strain expressing the cyclin PclA fused to 3×HA epitopes (left lane; Schier et al. 2001), the wild type without any HA-tagged protein (middle lane) and a strain expressing the transcription factor VeA fused to 3×HA (right lane). The arrows indicate PclA:3×HA and VeA:3×HA. One hundred micrograms of total protein were loaded and epitope-tagged proteins detected with an anti-HA antibody and peroxidase-coupled secondary antibodies
References
Baird GS, Zacharias DA, Tsien RY (2000) Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc Natl Acad Sci USA 97:11984–11989
Bernard P, Couturier M (1992) Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J Mol Biol 226:735–745
Bevis BJ, Glick BS (2002) Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat Biotechnol 20:83–87
Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird BS, Zacharias DA, Tsien RY (2002) A monomeric red fluorescent protein. Proc Natl Acad Sci USA 99:7877–7882
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994) Green fluorescent protein as a marker for gene expression. Science 263:802–805
Cormack B (1998) Green fluorescent protein as a reporter of transcription and protein localization in fungi. Curr Opin Microbiol 1:406–410
Curtis MD, Grossniklaus U (2003) A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133:462–469
Dou X, Wu D, An W, Davies J, Hashmi SB, Ukil L, Osmani SA (2003) The PHOA and PHOB cyclin-dependent kinases perform an essential function in Aspergillus nidulans. Genetics 165:1105–1115
Fernandez-Abalos JM, Fox H, Pitt C, Wells B, Doonan JH (1998) Plant-adapted green fluorescent protein is a versatile vital reporter for gene expression, protein localization and mitosis in the filamentous fungus Aspergillus nidulans. Mol Microbiol 27:121–130
Fernandez-Martinez J, Brown CV, Diez E, Tilburn J, Arst HN, Penalva MA, Espeso EA (2003) Overlap of nuclear localisation signal and specific DNA-binding residues within the zinc finger domain of PacC. J Mol Biol 334:667–684
Fuchs F, Prokisch H, Neupert W, Westermann B (2002) Interaction of mitochondria with microtubules in the filamentous fungus Neurospora crassa. J Cell Sci 115:1931–1937
Käfer E (1977) Meiotic and mitotic recombination in Aspergillus and its chromosomal aberrations. Adv Genet 19:33–131
Karimi M, Inzé D, Depicker A (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7:193–195
Karos M, Fischer R (1999) Molecular characterization of HymA, an evolutionarily highly conserved and highly expressed protein of Aspergillus nidulans. Mol Genet Genomics 260:510–521
Landy A (1989) Dynamic, structural and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem 58:913–949
Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living cells. Science 300:87–91
Lugones LG, Scholtmeijer K, Klootwijk R, Wessels JG (1999) Introns are necessary for mRNA accumulation in Schizophyllum commune. Mol Microbiol 32:681–689
Mikkelsen L, Sarrocco S, Lubeck M, Jensen DF (2003) Expression of the red fluorescent protein DsRed-Express in filamentous ascomycete fungi. FEMS Microbiol Lett 223:135–139
Niedenthal RK, Riles L, Johnston M, Hegemann JH (1996) Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast 12:773–786
Pöggeler S, Masloff S, Hoff B, Mayrhofer S, Kück U (2003) Versatile EGFP reporter plasmids for cellular localization of recombinant gene products in filamentous fungi. Curr Genet 43:54–61
Sambrook J, Russel DW (1999) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Schier N, Fischer R (2002) The Aspergillus nidulans cyclin PclA accumulates in the nucleus and interacts with the central cell cycle regulator NimX(Cdc2). FEBS Lett 523:143–146
Schier N, Liese R, Fischer R (2001) A pcl-like cyclin of Aspergillus nidulans is transcriptionally activated by developmental regulators and is involved in sporulation. Mol Cell Biol 21:4075–4088
Spellig T, Bottin A, Kahmann R (1996) Green fluorescent protein (GFP) as a new vital marker in the phytopathogenic fungus Ustilago maydis. Mol Genet Genomics 252:503–509
Stringer MA, Dean RA, Sewall TC, Timberlake WE (1991) Rodletless, a new Aspergillus developmental mutant induced by directed gene inactivation. Genes Dev 5:1161–1171
Suelmann R, Fischer R (2000) Mitochondrial movement and morphology depend on an intact actin cytoskeleton in Aspergillus nidulans. Cell Motil Cytoskeleton 45:42–50
Suelmann R, Sievers N, Fischer R (1997) Nuclear traffic in fungal hyphae: in vivo study of nuclear migration and positioning in Aspergillus nidulans. Mol Microbiol 25:757–769
Yelton MM, Hamer JE, Timberlake WE (1984) Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci USA 81:1470–1474
Acknowledgements
We thank Dr. Glick (University of Chicago, USA) for sending us DsRed (T4), Dr. Prastio (University of San Diego, USA) for sending us the mRFP1 and Dr. Ram (Leiden University, The Netherlands) for BFP. We are grateful to Jochen Scheld for excellent technical assistance and to Anne Blumenstein and Evelyn Vollmeister. This work was supported by the Max-Planck-Institute for terrestrial microbiology, the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (DFG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by U. Kück
Rights and permissions
About this article
Cite this article
Toews, M.W., Warmbold, J., Konzack, S. et al. Establishment of mRFP1 as a fluorescent marker in Aspergillus nidulans and construction of expression vectors for high-throughput protein tagging using recombination in vitro (GATEWAY). Curr Genet 45, 383–389 (2004). https://doi.org/10.1007/s00294-004-0495-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-004-0495-7