
Abstract
Therapy-related myeloid neoplasms (t-MN) are aggressive leukemia that develops as a complication of prior exposure to DNA-damaging agents. Clonal cytopenia of undetermined significance (CCUS) is a precursor of de novo myeloid neoplasms. Characteristics of CCUS that develop following cytotoxic therapies (therapy-related clonal cytopenia, t-CC) and outcomes following t-CC have not been described. We identified 33 patients with t-CC and compared to a cohort of the WHO-defined t-MN (n = 309). t-CC had a distinct genetic and cytogenetic profile: pathogenic variants (PV) in TET2 and SRSF2 were enriched in t-CC, whereas TP53 PV was more common in t-MN. Ten (30%) t-CC patients developed a subsequent t-MN, with a cumulative incidence of 13%, 23%, and 50% at 6 months, 1, and 5 years, respectively. At t-MN progression, 44% of evaluable patients had identifiable clonal evolution. The median survival following t-CC was significantly superior compared all t-MN phenotype including t-MDS with <5% bone marrow blasts (124.5 vs. 16.3 months, P < 0.001) respectively. The presence of cytogenetic abnormality and the absence of variants in DNMT3A, TET2, or ASXL1 (DTA-genes) were associated with a higher likelihood of developing a subsequent t-MN and an inferior survival. We describe a putative precursor entity of t-MN with distinct features and outcomes.
Similar content being viewed by others


Introduction
With the widespread adaptation of sequencing techniques, precursor states of myeloid neoplasms such as clonal hematopoiesis of indeterminate potential (CHIP), idiopathic cytopenia of undetermined significance (ICUS) or clonal cytopenia of undetermined significance (CCUS) are recognized. CHIP is defined as the presence of expanded clonal blood cells carrying one or more somatic mutations, in the absence of any detectable hematological abnormalities. CCUS is characterized by unexplained cytopenia of cytopenia in the context of clonal hematopoiesis, but in the absence of known hematological malignancies. Finally, the presence of unexplained cytopenia without the evidence of clonality is defined as ICUS. While all these entities are associated with a higher risk of subsequent hematological malignancies, their malignant potential is diverse [1, 2].
Therapy-related myeloid neoplasms (t-MN) develop as a complication of prior DNA-damaging therapies including chemotherapy, radiation, stem cell transplantation (SCT), or immunosuppressive therapies (IST) for autoimmune diseases (AID). t-MN are aggressive neoplasms with overall survival of approximately 1 year from diagnosis, regardless of the therapies employed [3, 4].
In the context of DNA-damaging therapy, the impact of coexistent clonal hematopoiesis (CH) is context dependent. For example, in lymphoma and solid tumors patients, CH increases the risk of future t-MN [5, 6]. In contrast, the presence of CH did not predict a higher risk of t-MN in multiple myeloma patients undergoing autologous SCT [7]. Moreover, the therapeutic modality as well as different therapeutic classes have a distinct pattern of CH [8]. Combined how these host-related and external forces shape the clonal evolution leading to leukemic transformation is not known.
While studying t-MN patients, we encountered patients who had received DNA-damaging therapy and developed unexplained cytopenia with clonal abnormality, without morphological evidence of a myeloid neoplasm (i.e., CCUS). As a vast majority of patients in the CHIP, ICUS, or CCUS cohorts did not receive prior DNA-damaging therapies [2, 9,10,11], the significance of CCUS following such therapies and its outcomes is not known. We hypothesized that clonal cytopenia following DNA-damaging therapy (therapy-related clonal cytopenia or t-CC) is a distinct entity from the WHO-defined t-MN. To test this hypothesis, we analyzed clinicopathological characteristics and outcomes of t-CC as well as the risk factors for developing a subsequent t-MN.
The emergence of any cytogenetic abnormalities following DNA-damaging therapy, raises the concern for a t-MN. In a cohort of patients exposed to prior DNA-damaging therapy, 46% patients with the deletion of chromosome 7q (del 7q) progressed to t-MN. Thus, while del 7q was associated with a very high risk of progression to t-MN; it—by itself—did not define t-MN [12]. This is in contrast to the de novo context, wherein unexplained cytopenia in the presence of MDS-defining cytogenetic abnormality would be diagnosed as myelodysplastic syndrome, unclassifiable (MDS-U) [13, 14]. Therefore, we studied outcomes of following the emergence of MDS-defining cytogenetic abnormalities and compared with t-CC.
Methods
Patient cohort
Following the institutional review board approval, we conducted a retrospective review of all adult patients treated at Mayo Clinic. After obtaining informed consent, we defined t-CC using the following criteria: (i) history of exposure to DNA-damaging agents in the form of chemotherapy, radiation, autologous SCT for non-myeloid diseases, or IST; (ii) unexplained cytopenia persisting ≥4 months; (iii) evidence of clonality using cytogenetic analysis or next-generation sequencing (NGS); and (iv) no morphological evidence of a myeloid neoplasm. t-MN was defined using the WHO guideline [13]. t-CC patients that subsequently developed t-MN were included in the t-CC cohort for the purpose of this analysis. Finally, we identified patients with prior exposure DNA-damaging therapy and unexplained cytopenia that had no morphological evidence of a myeloid neoplasm but had at least one MDS-defining cytogenetic abnormality (referred to as t-MDS (cyto). According to the WHO guidelines [13], these patients were classified as t-MN and their outcome was compared to t-CC and other t-MN phenotypes.
Clinicopathological characteristics
Demographic and clinical characteristics including age at the time of primary condition, sex, DNA-damaging therapies received, and hematological parameters were abstracted. All available bone marrow biopsies were re-reviewed by 2 hematopathologists independently (R.H. and D.C.) to exclude t-MN. Diagnostic and therapeutic decisions were made per treating physician’s discretion.
Next-generation sequencing
DNA was extracted from fresh bone marrow aspirates and next-generation sequencing (NGS) testing was performed using a targeted next-generation sequencing (NGS) panel that included 42 genes commonly mutated in myeloid neoplasms: ANKRD26, ASXL1, BCOR, CALR, CBL, CEBPA, CSF3R, DDX41, DNMT3A, ELANE, ETNK1, ETV6, EZH2, FLT3, GATA1, GATA2, IDH1, IDH2, JAK2, KDM6A, KIT, KRAS, MPL, NPM1, NRAS, PHF6, PTPN11, RAD21, RUNX1, SETBP1, SH2B3, SF3B1, SRP72, SMC3, SRSF2, STAG2,TERT, TET2, TP53, U2AF1, WT1, and ZRSR2. The library preparation, sequencing, and data analysis were performed as described [15]. Briefly, libraries were prepared using the Agilent SureSelect‐XT Target Enrichment Kit (SureSelectXT, Agilent, Santa Clara, CA). and sequencing was performed on MiSeq or HiSeq platforms (Illumina, San Diego, CA) at the Mayo Clinic Clinical Genome Sequencing Laboratory. Pathogenic and likely pathogenic variants calling was performed as described [16]. Only the variants at the sites with a total read depth >100, supported by more than five alternate variant reads and a variant allele frequency (VAF) ≥ 5%, were retained for further analysis.
Statistical analysis
Univariate analysis was performed using logistic regression for nominal characteristics and Cox proportional hazard for time-to-event endpoints. Myeloid neoplasm-free survival (MNFS) was defined as interval from t-CC diagnosis to t-MN progression or the last follow-up. Progression-free survival (PFS) was defined as interval from t-CC diagnosis to t-MN progression or death. Finally, overall survival (OS) was calculated from t-CC diagnosis to last follow-up or death, whichever occurred first. Kruskal–Wallis test for continuous variables and Fisher Exact test for categorical variables were used with a significance level of 5% or less (P-value ≤0.05). Statistical analysis was performed using BlueSky Statistics (Chicago, IL) and figures were generated using GraphPad (v9, San Diego, USA). Oncoplot was prepared as described [17, 18].
Results
Clinical and pathological characteristics
We identified 90 patients who developed unexplained cytopenia following cytotoxic therapy. Of these, 36 were excluded for reasons as shown (Fig. 1). Twelve patients with no morphologic evidence of a myeloid neoplasm but the presence of MDS-defining cytogenetic abnormalities [referred to as t-MDS (cyto)] were classified as t-MN according to the WHO guideline [13]. The remaining 42 cases were re-reviewed, of which 9 (21.4%) cases were determined to have t-MN, while 33 patients were determined to have t-CC. Detailed pathological features of t-CC patients are described in Table 1 and serial bone marrow examinations from a representative case are shown (Fig. 2). We also identified 309 WHO-defined t-MN patients and compared clinicopathological features of the two cohorts (Supplementary Table 1). The interval from the primary diagnosis to t-CC was shorter compared to t-MN (34.4 vs. 79.8 months, P < 0.001). t-CC patients were more likely to have received IST (30.3% vs. 8.7%, P = 0.001); whereas a higher proportion of t-MN patients had received chemotherapy (82.5% vs. 63.6%, P = 0.018). Presentation as t-MN was associated with a higher degree of anemia (9 vs. 10.9 g/dL, P < 0.001), absolute neutropenia (1.1 vs. 1.6, P = 0.022), and thrombocytopenia (63 vs. 101, P = 0.037); though the white blood cell (WBC) count did not differ between the two cohorts. t-CC patients had a lower likelihood of abnormal cytogenetics (24.2% vs. 84.9%, P < 0.001), complex karyotype (CK, none vs. 52.3%, P < 0.001), and monosomal karyotype (MK, none vs. 50.3%, P < 0.001) compared to t-MN. Finally, we compared the clinicopathological features of t-CC, t-MDS (cyto), and t-MN separately and found that t-MDS (cyto) had distinct clinicopathological features (Supplementary Table 2 and Supplementary Fig. 1).

(i) CONSORT diagram; and (ii) Venn diagram showing the relation between therapy-related clonal cytopenia (t-CC) and therapy-related myeloid neoplasm (t-MN) patients. *1 patient did not have NGS analysis performed. CCUS clonal cytopenia of undetermined significance. MDS myelodysplastic syndrome.

A Bone marrow aspirate smear (600x) and B bone marrow biopsy (400x) at the time of t-CC diagnosis. Six months later, C bone marrow aspirate smear (600x) and D bone marrow biopsy (400x) showed dysplastic changes in the megakaryocytes (*) and increase in CD34+ myeloblasts (# and inset), consistent with the diagnosis of t-MN.
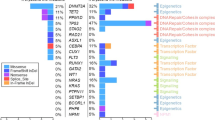
The proportion of patients with abnormal NGS was similar between t-CC and t-MN (96.3% vs. 90.3%, P = 0.477). Clinicopathological features and its correlation with NGS findings are shown in Fig. 3 and Supplementary Table 3. Median variance allele frequency (VAF) at t-CCUS was 35% (range 5–76%). The median VAF was not different between the DTA and non-DTA genes (33% vs. 39%, P = 0.45). The proportion of patients with a pathogenic variant (PV) in TP53 was significantly higher in t-MN compared to t-CC (40.9% vs. 7.4%, P < 0.001). On the other hand, a higher proportion of t-CC patients had PV in TET2 (55.6% vs. 10%, P < 0.001), and SRSF2 (23.1% vs. 8.1%, P = 0.028) compared to t-MN. The most common PV in t-CC were in TET2 19 (37%, Fig. 3), DNMT3A 7(14%), SRSF2 66(12%), RUNX1 4 (8%), and TP53 3 (6%). In contrast, the most common PVs in CCUS [9] were TET2 (21%), SRSF2 (12%), DNMT3A (7%), ZRSR2 (7%), and U2AF1 (6%) and the most common PV in t-MN were TP53 (26%), TET2 (8%), ASXL1 (7%), DNMT3A (6%), SRSF2 (4%) and IDH1 (4%). Thus, the genetic landscape of t-CC is distinct from t-MN as well as compared to the recently described cohort of CCUS patients [9].

(i) Clinical and genetic characteristics of t-CC patients; and (ii) the distribution of pathogenic variants in clonal cytopenia of undetermined significance (CCUS), therapy-related clonal cytopenia (t-CC), and therapy-related myeloid neoplasm (t-MN). AID autoimmune disease, Heme primary hematological malignancy, Solid primary solid tumor, Chemo chemotherapy, Rad radiation, Auto SCT autologous stem cell transplant, IST immunosuppressive therapy, Neutro neutropenia, Thrombo thrombocytopenia, cyto cytogenetics.
Risk of t-MN progression
At the last follow-up, 10 (30.3%) patients progressed to t-MN. We compared the characteristics of t-CC patients that subsequently developed t-MN compared to those who did not (Table 2). The distribution of the primary condition was different between the two cohorts: a higher proportion of patients that developed t-MN had hematological malignancy, whereas none of the 5 patients with AID developed t-MN. A higher degree of thrombocytopenia (platelets 46 vs. 11, P = 0.012), the presence of bone marrow blasts (1 vs. 0, P = 0.021), and nucleated red blood cells (0.6 vs. 0, P < 0.001) were associated with a higher likelihood of a subsequent t-MN. Similarly, the presence of a cytogenetic abnormalities (P = 0.004) and the absence of DTA variants (P = 0.015) were associated with a higher likelihood of a subsequent t-MN. Regardless of the presence of a non-DTA variants, the presence of at least one DTA variant was associated with a low risk of a subsequent t-MN: none of the 12 patients with DTA variants only developed t-MN, whereas 1 of the 8 patients with combined DTA and non-DTA variants progressed to t-MN.
The cumulative incidence of t-MN at 6 months, 1 year, and 5 years was 13%, 23%, and 50% respectively (Fig. 4). The presence of cytogenetic abnormalities at the time of t-CC diagnosis was associated with a statistically shorter MNFS (20.4 months vs. not reached, P = 0.02, Supplementary Table 4). Similarly, the absence of DTA variants was also associated with a statistically shorter MNFS (33.1 vs. 48 months, P = 0.02).

(i) Cumulative incidence (CI) of t-MN in patients with therapy-related clonal cytopenia (t-CC); (ii) Myeloid neoplasm free survival (MNFS) stratified by the presence of abnormal cytogenetics and (iii) the presence of pathogenic variants in DNTM3A, TET2, or ASXL1 (DTA) genes. Abnl abnormal, cyto. cytogenetics, DTA DNMT3A, TET2, and ASXL1 genes, PV pathogenic variant, NR not reached.
Clonal evolution at the time of t-MN progression
Paired cytogenetics and NGS at t-CC and t-MN were available for 9 and 4 patients, respectively. Combined, 4 (44.4%) of 9 evaluable patients had identifiable clonal evolution (Table 3) at t-MN progression. Among those with paired cytogenetic analyses, 3 patients acquired additional cytogenetic abnormalities (deletion of chromosome 13q, del 7q/trisomy 21, and a complex clone). Among those with paired NGS, one patient each noted the acquisition of NRAS and CEBPA/TP53, whereas in one patient each, DNMT3A/RUNX1 and TP53 were undetectable. Thus, leukemic transformation was associated with diverse biological mechanisms.
Outcomes following t-CC
Most common management strategy for t-CC was observation in 19 patients. The rest were treated with growth factor support (n = 7), chemotherapy (n = 1), IST (n = 1), intravenous immunoglobulin (n = 1), or a clinical trial (n = 1). One patient was diagnosed as MDS at an outside institution and underwent allogeneic SCT. At the last follow up, 11 (33.3%) deaths were noted. Primary causes of death included: t-MN (n = 4), infection (n = 2), cardiac complications (n = 2), primary malignancy (n = 1), progressive cytopenia without MN (n = 1), and undetermined (n = 1). Thus, 4 of 11 deaths were noted in patients who did not develop a subsequent t-MN.
Median PFS and OS for the entire cohort were 33 and 124.5 months respectively (Fig. 5). The presence of cytogenetic abnormality (9.5 vs. 47.7 months, P = 0.01) and the absence of DTA variants (20.6 vs. 47.7 months, P = 0.03) were associated with a shorter PFS. Similarly, the presence of cytogenetic abnormality (17.9 months vs. not reached, P = 0.02) was associated with inferior OS and there was a trend towards an inferior survival in the absence of DTA variants (29.9 months vs. not reached, P = 0.07). Other factors associated with shorter PFS were the history of autologous SCT (HR 5.02, P = 0.03, Supplementary Table 5), the presence of thrombocytopenia (HR 9.1, P = 0.03), or the presence of anemia (HR 8.58, P = 0.04) at t-CC diagnosis. Similarly, history of autologous stem cell transplant (HR 8.82, P = 0.01, Supplementary Table 6) was associated with an inferior OS.

(i) Progression-free survival (PFS), and (ii) overall survival (OS) of the entire AQ11 cohort. (iii) PFS and (iv) OS stratified by the presence of abnormal cytogenetics.(v) PFS and OS (vi) stratified by the presence of pathogenic variants in DNTM3A, TET2, or ASXL1 (DTA) genes. Abnl abnormal, cyto. cytogenetics, DTA DNMT3A, TET2, and ASXL1 genes, PV pathogenic variant, NR not reached.
Outcomes following t-CC compared to t-MN
We next assessed the impact of the development of a subsequent t-MN on survival. Patients who developed t-MN (n = 10) had a statistically significantly inferior survival compared to those who did not (17.1 months vs. not reached, P = 0.03, Fig. 6). Comparing the overall survival following t-CC to that with various phenotypic subclassifications of t-MN: t-MDS with <5% bone marrow blasts, t-MDS with excess blasts (t-MDS-EB), t-MDS/MPN, and t-AML showed that t-CC patients had a significantly superior survival (124.5 months) compared to t-MDS with <5% blasts (16.3 months), t-MDS-EB (14 months), and t-AML (13 months).

(i) Overall survival (OS) in therapy-related clonal cytopenia (t-CC) as stratified by the subsequent development of therapy-related myeloid neoplasm (t-MN); (ii) t-CC has significantly superior OS compared to t-MN, including compared to t-MDS (<5% bone marrow blasts). t-MDS (cyto)—therapy-related myelodysplastic syndrome based on the presence of MDS-defining cytogenetics; t-MDS (<5%)—t-MDS with <5% blasts at the time of presentation; t-MDS (EB)—t-MDS with excess blasts (5–19%); t-AML—therapy-related acute myeloid leukemia.
The significance of MDS-defining cytogenetic abnormalities
Of the 12 t-MDS (cyto) patients, 9 (75%) developed the morphological evidence of a myeloid neoplasm at a median of 10.5 months, while 3 (25%) patients did not. One patient (UPIN 2048) died within 1 month due to sepsis. UPIN 2041 died of progressive lymphoma 12 months without developing morphological evidence of a myeloid neoplasm. Finally, UPIN 1036 was alive at 44 months despite harboring the deletion of chromosome 5q, as well as PVs in ASXL1 and TP53. Time to develop morphological evidence of t-MN was significantly shorter for those with t-MDS(cyto) compared to t-CC (Supplementary Table 7, Supplementary Fig. 2). Importantly, median OS of the patients with MDS-defining cytogenetic abnormalities was significantly inferior compared to t-CC patients (13.2 vs. 124 months, P = 0.01), but was comparable to the other t-MN phenotypes (13–16.3 months, Fig. 5).
Discussion
Therapy-related myeloid neoplasms are one of the most aggressive malignancies with no effective therapies and an exceedingly poor survival [20]. Therefore, there is an urgent need to predict and prevent future t-MN.
The evidence of clonality in patients with otherwise unexplained cytopenia, impart a 17–27% risk of developing a subsequent myeloid neoplasm [9,10,11]. However, these studies did not assess the impact of prior DNA-damaging therapies, which is one of the strongest known risk factors for myeloid neoplasms [21].
We comprehensively characterized CCUS developing after the exposure to DNA-damaging therapies and found that the cumulative incidence of t-MN was 50% at 5 years. The interval from the diagnosis of primary malignancy/AID to t-CC diagnosis was shorter than t-MN. In addition, t-MN was accompanied by a higher degree of anemia, absolute neutropenia, and thrombocytopenia. A significantly smaller proportion of t-CC patients had TP53, CK, and MK—features known to predict inferior outcomes [3]. This raises two possibilities—the first is that patients presenting as t-CC, including those progressed to t-MN, represent a distinct subset of t-MN. Another possibility is that the leukemic transformation is a distinct biological event characterized by the acquisition of high-risk features including the acquisition of PV in TP53 as well as chromosomal instability. While limited by a small cohort size, the paired cytogenetic/genetic analysis at t-CC and t-MN showed complex clonal evolution seen at the time of leukemic transformation. Using a lower VAF threshold (≥2%), a recent study found that the leukemic transformation was associated with acquisition of additional somatic mutations, including chromosomal aneuploidies or mutations in genes in 91% of cases [8]. Combined, these observations raise the possibility that a prompt and accurate diagnosis of t-CC may allow for interventions which may not be feasible in t-MN patients.
Designing optimized surveillance strategies and counseling will require an accurate identification of patients at a higher risk of t-MN. We noted that the presence of cytogenetic abnormalities was associated with inferior outcomes, whereas the presence of DTA variants—regardless of the co-existing non-DTA variants—were associated with superior outcomes. The mechanism underlying this observation remains unclear. A possible explanation is that the presence of DTA variants may denote ‘true’ CHIP-like clone, whereas the presence of cytogenetic abnormalities and/or non-DTA variants may represent a therapy-related clone.
Long-term follow up following t-CC revealed 2 interesting themes: more than a third of patients died without developing t-MN. These findings are commensurate with CHIP patients who experience increased morbidity and mortality [1, 19]. On the other hand, the survival of t-CC patients was significantly superior compared to t-MN patients. In the de novo context, an argument can be made that the clear distinction between CCUS and low-risk MDS (LR-MDS) may be of little importance; as observation is the preferred option for both entities [1], and there is no difference in survival [9]. This is in stark contrast with our findings as t-CC appears to have superior survival compared to all t-MN phenotypes, including t-MDS with no increase in blasts. In addition, once diagnosed with t-MN, very few patients would be watched without interventions. Thus, we argue that the distinction between t-CC and t-MN is clinically meaningful.
Finally, we assessed if, in the context of prior DNA-damaging therapy, the presence of MDS-defining cytogenetic abnormalities carries similar prognostic significance as in the de novo context [12, 13]. Survival of t-MDS(cyto) patients was no different compared to t-MN, but significantly superior to t-CC—supporting the assertion that even in the absence of morphological evidence of a myeloid neoplasm, these cases should be considered as t-MN. However, the interval from the emergence of cytogenetic abnormality to morphological progression varied greatly (0.9–81 months) and 2 patients did not develop morphological evidence of t-MN despite 12 and 44 months of follow-up. Goswami et al. followed patients with prior DNA-damaging therapy who developed isolated deletion of 7q—an MDS-defining cytogenetic anomaly [12]. While these patients fulfilled the WHO-defined t-MN diagnostic criteria, less than half actually progressed to t-MN. Collectively, these results underline the heterogeneity of the cohort and the need for larger studies that will help more accurate risk stratification of this cohort.
A limitation of our study was that the samples obtained prior to DNA-damaging therapy were not available and that a subset of patients did not have paired NGS performed at t-CC and t-MN. Therefore, whether a clone strictly represented CHIP, or the effect of the prior DNA-damaging therapy could not be established. Second, the absolute and relative risks of t-CC or t-MN following chemotherapy could not be inferred. Third, PPM1D that has a well-known association with t-MN; [22] as well as CUX1 [23], recently described to be a gatekeeper in t-MN pathogenesis, were not assessed. Fourth, with regards to the paired sequencing, the inability to identify a corresponding t-MN clone at the time of t-CC does not necessarily denote the absence of such a clone. It is possible that low-level clone is present below the detection threshold of NGS and cytogenetics technique. Finally, given that t-CC, at least in some cases, acted as a precursor to t-MN, whether the difference in outcomes reflects the differences in the biology of the 2 entities or the lead-time bias could not be ascertained [24]. Therefore, a larger prospective study of all patients undergoing cytotoxic therapies with a longer follow-up will be needed to answer these questions.
In summary, we describe t-CC as a putative precursor entity of t-MN and identify the risk factors for poor outcomes including the progression to t-MN. t-CC had a distinct clinical and genetic profile as well as overall superior survival compared to t-MN. The presence of cytogenetic abnormality and the absence of variants in DNMT3A, TET2, or ASXL1 genes were associated with a higher likelihood of progressing to t-MN and an inferior survival. Paired analysis at t-CC and t-MN as well as comparative analyses of the t-CC and t-MN cohorts suggest that the leukemic transformation as an event characterized by acquisition of morphologic evidence of dysplasia as well as cytogenetic and genetic abnormalities. Given that half of the patients develop t-MN over the next 5 years, a wider recognition of t-CC may allow to individualize counseling, optimize surveillance, and design prevention studies—ultimately improving outcomes in t-MN.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
DeZern AE, Malcovati L, Ebert BL. CHIP, CCUS, and other acronyms: definition, implications, and impact on practice. Am Soc Clin Oncol Educ Book. 2019;39:400–10.
Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126:2355–61.
McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:513.
Nadiminti K, Sidiqi MH, Meleveedu K, Alkhateeb HB, Hogan WJ, Litzow M, et al. Characteristics and outcomes of therapy-related myeloid neoplasms following autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2021;11:63.
Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35:1598–605.
Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–82.e4.
Mouhieddine TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun. 2020;11:1–9.
Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52:1219–26.
Li M, Binder M, Lasho T, Ferrer A, Gangat N, Al-Kali A, et al. Clinical, molecular, and prognostic comparisons between CCUS and lower-risk MDS: a study of 187 molecularly annotated patients. Blood Adv. 2021;5:2272–8.
Malcovati L, Gallì A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129:3371–8.
Gallì A, Todisco G, Catamo E, Sala C, Elena C, Pozzi S, et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood. 2021;138:965–76.
Goswami RS, Wang SA, DiNardo C, Tang Z, Li Y, Zuo W, et al. Newly emerged isolated Del(7q) in patients with prior cytotoxic therapies may not always be associated with therapy-related myeloid neoplasms. Mod Pathol. 2016;29:727–34.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Cazzola M. Myelodysplastic syndromes. N. Engl J Med. 2020;383:1358–74.
Mehta N, He R, Viswanatha DS. Internal standardization of the interpretation and reporting of sequence variants in hematologic neoplasms. Mol Diagnosis Ther. 2021;25:517–26.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1–pl.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl J Med. 2017;377:111–21.
Higgins A, Shah MV. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes 2020;11:749.
Morton LM, Dores GM, Schonfeld SJ, Linet MS, Sigel BS, Lam CJK, et al. Association of chemotherapy for solid tumors with development of therapy-related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5:318–25.
Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23:700–13.e6.
Imgruet MK, Lutze J, An N, Hu B, Khan S, Kurkewich J, et al. Loss of a 7q gene, CUX1, disrupts epigenetically driven DNA repair and drives therapy-related myeloid neoplasms. Blood. 2021;138:790–805.
Duffy SW, Nagtegaal ID, Wallis M, Cafferty FH, Houssami N, Warwick J, et al. Correcting for lead time and length bias in estimating the effect of screen detection on cancer survival. Am J Epidemiol. 2008;168:98–104.
Acknowledgements
The authors are grateful to our patients and their families. M.V.S. was supported by K12, Leukemia Research Foundation New Investigator Award, and Bridget Kiely Clinician Career Development in Transplant Research.
Author information
Authors and Affiliations
Contributions
M.V.S. designed the study, collected data, performed statistical analysis, and wrote the draft of the manuscript; M.V.S., A.A., K.H.B., H.B.A. M.A.E., W.J.H., M.R.L., K.M., A.T., N.G. M.M.P., and A.A. contributed patients; PG performed cytogenetic evaluation and acquired samples for N.G.S. analysis; R.H. analyzed N.G.S. results; D.C. and R.H. performed independent rereview of pathology. All authors agree to the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Patnaik—Membership on an entity’s Board of Directors or advisory committees (Stemline Therapeutics) and Research funding (Kura Oncology). Al-Kali—Research funding (Novartis), and Research support to institution (Astex). Litzow—Research funding (AbbVie, Astellas, Amgen, Actinium, Pluristem); Advisory board (Jaz, Omeros); Data monitoring committee (BioSight). All other authors disclose no conflicts.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A portion of these data was presented at the 63rd annual meeting of the American Society of Hematology, Atlanta, GA, USA (2021).
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, M.V., Mangaonkar, A.A., Begna, K.H. et al. Therapy-related clonal cytopenia as a precursor to therapy-related myeloid neoplasms. Blood Cancer J. 12, 106 (2022). https://doi.org/10.1038/s41408-022-00703-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00703-8
- Springer Nature Limited