
Abstract
Therapy-related myeloid neoplasms (T-MN) are poorly characterized secondary hematological malignancies following chemotherapy/radiotherapy exposure. We compared the clinical and mutational characteristics of T-MN (n = 129) and primary myelodysplastic syndrome (P-MDS, n = 108) patients. Although the somatic mutation frequency was similar between T-MN and P-MDS patients (93% in both groups), the pattern was distinct. TP53 mutations were more frequent in T-MN (29.5 vs. 7%), while spliceosomal complex mutations were more common in P-MDS (56.5 vs. 25.6%). In contrast to P-MDS, the ring sideroblasts (RS) phenotype was not associated with better survival in T-MN, most probably due to genetic association with TP53 mutations. SF3B1 was mutated in 96% of P-MDS with ≥15% RS, but in only 32% T-MN. TP53 mutations were detected in 92% T-MN with ≥15% RS and SF3B1 wild-type cases. Interestingly, T-MN and P-MDS patients with “Very low” or “Low” Revised International Prognostic Scoring System (IPSS-R) showed similar biological and clinical characteristics. In a Cox regression analysis, TP53 mutation was a poor prognostic factor in T-MN, independent of IPSS-R cytogenetics, disease-modifying therapy, and NRAS mutation. Our data have direct implications for T-MN management and provide evidence that, in addition to conventional disease parameters, mutational analysis should be incorporated in T-MN risk stratification.
Similar content being viewed by others

Introduction
Therapy-related myeloid neoplasms (T-MNs) are secondary malignancies that occur following chemotherapy (CT) and/or radiotherapy (RT) exposure for the treatment of malignant or non-malignant diseases [1]. They account for 5–10% of patients newly diagnosed with MNs. The World Health Organization (WHO) included therapy-related myelodysplastic syndrome (T-MDS), myelodysplastic/myeloproliferative neoplasm (T-MDS/MPN), and acute myeloid leukemia (T-AML) within the single clinical group of T-MN, due to the perceived similarity between their biological characteristics and outcome. The incidence of these clinically aggressive malignancies varies from <1 to 24% [2] depending on the type of primary disease (non-malignant vs. malignant), the type and intensity of genotoxic exposure, and the length of follow-up.
The prognosis of T-MN is generally poor, with a median survival <12 months, which is attributed to high-risk cytogenetic changes. Unlike primary MDS (P-MDS), which has validated prognostic classifications (e.g., Revised International Prognostic Scoring System [IPSS-R]), there is no universal standard to prognosticate T-MN. Recently, a modified version of IPSS-R was applied to segregate T-MDS and T-AML (<30% bone marrow [BM] blasts) into different prognostic groups [3]. Intriguingly, the overall survival (OS) between T-MN and P-MDS was different, even when matched for IPSS-R. This indicates a possible contribution of sub-chromosomal genetic lesions to the pathogenesis and dismal outcome of this disease.
Although comprehensive mutation profiling of P-MDS has been reported on large patient cohorts (157–2250 patients) [4,5,6,7], limited data are available for T-MN patients. Mutational frequency in T-MN was reported as highly variable and lower than in P-MDS (33–59 vs. 78–90%, respectively) [8, 9]. This is likely due to relatively small sample size and limited repertoire of genes analyzed. Importantly, very few studies have compared the mutation frequency of T-MN and P-MDS using a uniform bioinformatics pipeline. This study analyzes the somatic mutation profile of T-MN and P-MDS patients registered in the South Australian-MDS Registry.
Materials and methods
Patient samples
The South Australian-MDS (SA-MDS) Registry is a statewide registry enrolling patient diagnosed with P-MDS, AML with ≤30% BM blasts, MDS/MPN overlap, or T-MN. The demographic, clinical, laboratory, treatment, and follow-up data of enrolled patients were analyzed.
The study was conducted in accordance with the Declaration of Helsinki and was approved by ethics committees of participating institutions. Diagnosis was from peripheral blood/BM smear examination by expert hemato-pathologists. Perl’s stain was used to characterize BM iron stores and ring sideroblasts (RS). The WHO classification [1] was employed to further classify the MNs. Where applicable, the IPSS-R classification [10] was used for prognostic purposes in both P-MDS and T-MN. Patients were categorized as receiving “best supportive care only” or “disease-modifying therapy” (DMT). DMT was further specified as hypomethylating agent, intensive CT, allogeneic hematopoietic stem cell transplant (allo-HSCT), investigational therapy, or any combination of these.
Mutational profiling
Genomic DNA collected at diagnosis from 108 P-MDS and 129 T-MN patients was subjected to sequencing and mutational analysis. Patients with samples obtained only at time-points other than diagnosis were excluded from both cohorts (n = 37 and 15, respectively). Paired germline samples (hair follicle or BM mesenchymal stromal cell DNA) were available for 31/108 (29%) P-MDS and 58/129 (45%) T-MN samples. The genomic profile of T-MN and P-MDS samples (BM or germline) were analyzed using either custom-designed 217-gene NimbleGen Capture Platform (Roche NimbleGen Inc., Madison, WI, USA), Ion AmpliSeq (Thermo Fisher Scientific Inc., Waltham, MA, USA), or Fluidigm Access Array (Fluidigm Inc., San Francisco, CA, USA) platforms (see Supplementary Information for details). Sequence reads were aligned to the GRCh37 human reference genome (Supplementary method).
Forty-three genes known to be recurrently mutated in myeloid malignancies were selected for further analysis (Table S1). Only variants at sites with a total read depth >100, supported by more than five alternate variant reads and a variant allele frequency (VAF) ≥3%, were retained for further analysis. Exceptions to this were ASXL1 (c.1934dupG) and SRSF2 (P95) variants (see Supplementary Information). Figure S1 summarizes the criteria used for selecting somatic mutations. Comparison was performed with paired germline samples when available. All selected variants were manually curated for functional deleteriousness.
Statistical analysis
Comparison of patient characteristics was performed using Mann–Whitney, Fisher’s exact, or χ2 tests, as appropriate. Overall survival (OS) was calculated from the date of diagnosis to that of last contact or death. Approximately 21% P-MDS and 39% T-MN patients in our cohort received DMT reflecting current MDS management. Hence, we adjusted the OS using a time-varying covariate to account for DMT exposure. Patients alive at the last contact date or lost to follow-up were censored. Survival analyses were performed using the Kaplan–Meier method and curves were compared using the log-rank test in univariate analysis. These factors were further analyzed in a multivariate Cox regression model with time-varying covariate. The statistical analyses were performed using GraphPad Prism (version 6.07 for Windows, GraphPad Software) and R (https://www.r-project.org). All tests were two sided, unless specified, and differences were considered significant if p < 0.05.
Results
Clinical characteristics
The demographic, clinical, and laboratory characteristics of 108 P-MDS and 129 T-MN patients in our cohort are shown in Table 1. T-MN patients were younger than P-MDS (71.1 vs. 75.3 years, p < 0.001) at diagnosis of their MN.
The most common primary disease in T-MN patients requiring cytotoxic therapy was lymphoproliferative disease (36.4%), followed by breast (14.7%) and prostate (11.6%) cancers (Table S2). Approximately 10% T-MN received cytotoxic therapy for autoimmune diseases. Patients received CT (45.7%), RT (21.7%), or both (combined modality treatment [CMT], 32.6%) to treat their primary disease. Notably, 36.8% of lymphoproliferative disease and myeloma patients developing T-MN had autologous HSCT.
Most T-MN patients presented in MDS phase (74%) and fewer with AML (≥20% blasts, 26%). Two patients had therapy-related acute promyelocytic leukemia at diagnosis. Therapy-related chronic myelomonocytic leukemia (T-CMML) and T-MDS/MPN were infrequently seen in our cohort (7 and 4 cases, respectively). The morphological MDS subtypes were not different between P-MDS and T-MN groups (Table 1).
Comparing T-MN patients with or without leukemic presentation (T-AML vs. T-MDS), T-AML showed a higher frequency of 11q23 translocations (p = 0.004). The OS of T-AML was poorer than that of T-MDS (3.5 vs. 13.2 months, p < 0.0001), despite a higher proportion of T-AML patients being treated with induction CT (41.2 vs. 9.5%) and allo-HSCT (17.7 vs. 7.4%; Table S3).
Poor risk cytogenetic abnormalities and high IPSS-R scores are common in T-MN
A higher proportion of T-MN patients had IPSS-R Poor and Very poor risk cytogenetic abnormalities. Conversely, Very low and Low risk cytogenetics were more common in P-MDS (p < 0.0001; Table 1). Consequently, the IPSS-R distribution was skewed towards High and Very high categories in T-MN and Very low and Low in P-MDS groups (Table 1).
Cytogenetic abnormalities, including del7/7q (18.4 vs. 6.5%, p = 0.009), complex karyotype (≥3 abnormalities, 28.8 vs. 8.3%; p < 0.0001), chromosomal translocations (39.2 vs. 10.2%; p < 0.0001), monosomal karyotype (26.4 vs. 4.6%; p < 0.0001), and marker chromosomes (20.8 vs. 1.9%; p < 0.0001), were more common in T-MN compared to P-MDS (Table 1). The 11q23 translocations (MLL or KMT2A fusions) were only seen in T-MN patients and were not associated with topoisomerase exposure.
T-MN and P-MDS have a similar overall mutation frequency, but the mutation pattern is distinct
The mutational spectrum of T-MN is depicted in Fig. 1a, b and Tables S4 and S5. In P-MDS, 323 nonsynonymous somatic mutations were detected in 100/108 (92.6%) patients in 35 of the 43 genes tested (Fig. S2 and Tables S4 and S5). Similarly, 347 nonsynonymous mutations were detected in 34 genes in 120/129 (93.0%) T-MN patients. Mutations in >1 gene were detected in 73 and 66% of P-MDS and T-MN, respectively (Fig. 1 and Fig. S2). The mutation burden was similar between P-MDS and T-MN (median, 3 mutations). Of the 43 genes analyzed, no mutations were seen in MYD88, PDGFRA, and SRP72 in either P-MDS or T-MN (Table S4).

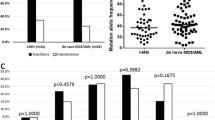
Frequency and distribution of mutations in therapy-related myeloid neoplasms (T-MN) and primary MDS (P-MDS) cases: a Distribution of mutations in 129 T-MN patients. b Frequency of mutations in T-MN (solid fill) and P-MDS (hashed fill). TP53 is the most commonly mutated gene in T-MN, while TET2 followed by SF3B1 are the most commonly mutated genes in P-MDS. In TP53-mutated cases, mutations in other genes are less frequent compared to non-TP53-mutated cases (p < 0.0001). The antecedent condition leading to T-MN and the therapy received are listed on the bottom rows. c Distribution of mutations in TP53 according to key functional domains in P-MDS and T-MN. PD – primary disease; Others – other cancers; AID – autoimmune disease; Ca Colon – colon cancer; MM – multiple myeloma; Ca Prostate – prostate cancer; Ca Breast – breast cancer; LPD – lymphoproliferative disorders; CT – chemotherapy; RT – radiotherapy; CMT – combined modality treatment (CT and RT)
Although the overall mutation frequency was similar, the mutation pattern was different between P-MDS and T-MN. TP53 mutations were more frequent in T-MN than in P-MDS (29.5 vs. 7.0%; p < 0.0001) (Fig. 1 and Table S4). A total of 56 TP53 mutations (41 missense, 7 nonsense, 5 splice-site, and 3 frameshift) were detected in 38 T-MN patients (Fig. 1c). Multiple TP53 mutations were detected in a higher proportion of T-MN (10.8%) than P-MDS (3.0%; p = 0.03). Two TP53 mutations were detected in 11 T-MN patients, while 3 and 4 TP53 mutations were detected in 2 and 1 T-MN patients, respectively. Considering the highest VAF in patients with multiple mutations, the median VAF of TP53 mutations was high (39% in 38 patients).
The presence of TP53 mutation was strongly associated with complex karyotype (p < 0.0001; Fig. S3A). Additionally, patients with TP53 VAF ≥20% (25/30; 83%) had a higher frequency of complex karyotype compared to patients with VAF <20% (3/7, 43%; p = 0.04). The median number of mutations in genes other than TP53 in patients with mutated TP53 (TP53mut) was much lower compared to that in patients with wild-type TP53 (TP53wt; 0 vs. 3 mutations/sample, p < 0.0001; Fig. 1b). The BM blast count was <5% in half of the patients at the time of T-MN diagnosis. Conspicuously, no TP53mut patient had BM morphology consistent with T-CMML. The frequency of primary hematological malignancy (55.3 vs. 40.6%) or CT exposure (81.6 vs. 76.9%) was not different between T-MN with TP53mut and TP53wt.
We also assessed the impact of TP53 VAF on the OS of T-MN patients. There was no difference in this parameter when T-MN were segregated by a TP53 VAF median value of 39%. However, patients with a VAF of <20% showed longer OS than patients with ≥20% (10.4 vs. 7.7 months, p = 0.03; Fig. S3B). There was no difference in the proportion of patients with High or Very high IPSS-R scores or AML between these two groups.
Spliceosome mutations were more frequent in P-MDS compared to T-MN (56.5 vs. 25.6%; p < 0.0001; Table S4 and Fig. 1a). Within the spliceosomal complex, SF3B1 (25.9 vs. 6.2%; p < 0.0001) and U2AF1 mutations (7.4 vs. 1.6%; p = 0.04) were detected at higher frequency in P-MDS compared to T-MN, while there was no difference in the frequency of SRSF2 or ZRSR2 mutations. As expected, most of the spliceosome mutations were mutually exclusive, except in one patient (PID 1355) harboring two SF3B1 variants (K666M; VAF 27.5% and K666N; VAF 10.4%) with each likely being in separate sub-clones.
In the other major myeloid genes, TET2, ASXL1, DNMT3A, RUNX1, and EZH2, the mutation frequency was similar between P-MDS and T-MN (Fig. 1a). SETBP1 mutations were seen in 6.2% of T-MN patients, of which 6 (75%) had del7/7q abnormalities. The primary disease type and its treatment did not influence the mutation pattern in T-MN.
Additionally, the frequency and median number of mutations were not different between T-MDS and T-AML. SETBP1, SF3B1, and ZRSR2 mutations were exclusively seen in T-MDS, while the mutation pattern in other genes was not different between the two groups (Table S6).
The frequency of RS is similar in T-MN and P-MDS, but the genetic association is distinct
RS were seen upon Perl’s stain in P-MDS (29.6%) and T-MN (24.8%). In 21.3% P-MDS and 14.7% T-MN, RS were present in ≥15% of erythroid cells. Spliceosomal complex mutations were seen in all P-MDS, but only in 37% T-MN with ≥15% RS (p < 0.0001), with SF3B1 mutations being the most frequent (96 vs. 32%; p < 0.0001; Fig. 2a and Table S7). Interestingly, TP53 mutations were detected in 92% T-MN with ≥15% RS and wild-type SF3B1 (SF3B1wt), compared to 29.5% in the entire T-MN cohort. Notably, the only P-MDS sample with ≥15% RS and SF3B1wt lacked TP53 mutation. A similar correlation was seen for SF3B1 mutation frequency with any number of RS (Table S7, footnote and Fig. S4A).

Comparison of the genetic association of ring sideroblasts (RS) and survival in therapy-related myeloid neoplasms (T-MN) and primary MDS (P-MDS): a Distribution of SF3B1 and TP53 mutations in patients with ≥15% or more RS on bone marrow morphology. SF3B1 mutation is most common in P-MDS, while TP53 is most common in T-MN. b The overall survival of T-MN patients with ≥15% RS is poorer than P-MDS patients. c Poor OS in T-MN with ≥15% RS is most probably due to association with TP53 mutation in T-MN cases
Survival of P-MDS with ≥15% RS was better than those with <15% RS (37.5 vs. 27.7 months, p = 0.008), most probably due to the strong association of RS with favorable SF3B1 mutations in P-MDS. The OS of P-MDS patients with SF3B1 mutations was better than that of wild-type patients (38.2 vs. 26.8 months, p = 0.004). However, the OS in T-MN with ≥15% RS was not different from that of those patients with <15% RS (12 vs. 11 months; p = 0.33). In addition, the OS of T-MN with ≥15% RS was worse than that of P-MDS with ≥15% RS (12 vs. 38 months, p < 0.0001; Fig. 2b), likely due to higher prevalence of TP53 mutations (Fig. 2c) in these T-MN cases. Similarly, when RS ≥1% were considered, the OS of T-MN was worse than that of P-MDS (Fig. S4B).
Overall, although the frequency of ≥15% RS was similar in P-MDS and T-MN, it was not associated with better survival in T-MN, most likely due to poor correlation with SF3B1 mutation and stronger association with TP53 mutations in SF3B1wt cases.
T-MN and P-MDS with Very low or Low IPSS-R score share clinical and biological characteristics and have similar outcomes
T-MN patients with Very low IPSS-R score had a median survival of 106 months, which worsened with an increase in the score, with median survival in Very high IPSS-R group being only 7.8 months (p < 0.0001). The survival of T-MN cohort was significantly poor as compared to P-MDS cohort (10.6 vs. 31.4 months, p < 0.0001; Fig. 3a) and the difference between the two groups was maintained when the analysis was restricted to patients with ≤30% blasts (12.8 vs. 32.4 months; p < 0.0001; Fig. S5). T-MN with Intermediate IPSS-R score had poorer OS compared to Intermediate score P-MDS (14 vs. 35 months, p = 0.01; Table S8), while it was equally poor in T-MN and P-MDS with High or Very high IPSS-R scores (8.8 vs. 13 months, p = 0.09).

Survival in Revised International Prognostic Scoring System (IPSS-R) Very low and Low risk therapy-related myeloid neoplasms (T-MN) is not different from the primary MDS (P-MDS) counterpart: a The overall survival (OS) of T-MN patients is poorer than P-MDS. b In contrast, the OS of patients with Low or Very low IPSS-R is similar in T-MN and P-MDS. c Cox proportional hazard model for OS in T-MN. d Cox proportional hazard model for OS in P-MDS. UCL – upper confidence limit; LCL – lower confidence limit
Interestingly, survival of T-MN with Very low or Low risk was not different from P-MDS (64 vs. 45 months, p = 0.9; Fig. 3b). Hence, we compared the clinical and genetic characteristics of P-MDS (n = 52) and T-MN (n = 22) patients with Very low or Low IPSS-R score (Table 2). There was no difference between their demographics and MDS subtypes. The majority of patients had normal karyotype (79 vs. 82%), and no poor-risk chromosomal abnormalities were seen in the remainder. The median number of mutations was not different (2 vs. 3 mutations). Importantly, the mutation profile of both groups was also remarkably similar. We did not find any difference in the frequency of mutations in either individual genes or pathways (Table 2).
Predictors of survival in T-MN
The OS (adjusted for DMT) was poor in T-MN patients compared to P-MDS patients (10.6 vs. 31.4 months, p < 0.0001), despite a higher proportion of T-MN patients receiving intensive CT (17.8 vs. 7.4%) and allo-HSCT (10.1 vs. 1.9%). The OS was compared between P-MDS and T-MN patients with respect to the mutational status of genes that were mutated in five or more patients. In univariate analysis, TET2 and SF3B1 mutations were associated with better OS, while ASXL1, CBL, U2AF1, TP53, and NRAS mutations were associated with poorer outcome in P-MDS. In T-MN, TP53, NRAS, WT1, and BCOR mutations were associated with a poorer OS, while ASXL1 and IDH2 mutations were favorable. There was no relationship between OS and the number of mutations.
In a multivariate Cox regression analysis (Fig. 3c), the presence of ≥20% BM blasts at diagnosis (hazard ratio (HR) 5.05, 95% confidence interval (CI) 2.82–9.06, p < 0.0001), IPSS-R cytogenetic category (HR 1.57, 95% CI 1.24–1.99, p = 0.0002), DMT (HR 0.35, 95% CI 0.20–0.60, p = 0.0002), and NRAS (HR 4.51, 95% CI 2.06–9.87, p = 0.0002) and TP53 (HR 2.05, 95% CI 1.16–3.62, p = 0.0131) mutations were independent predictors of OS in the T-MN cohort. In P-MDS, in addition to IPSS-R cytogenetic and DMT categories, age, male gender, and mutations in ASXL1, NRAS, RUNX1, and CBL were independent factors associated with poor survival. Conversely, TET2 mutations were associated with improved OS in P-MDS (Fig. 3d).
Discussion
T-MN represents an aggressive malignancy with extremely poor long-term outlook compared to de novo MDS/AML. Here, we present the clinical, laboratory, and genetic profiles of 129 well-characterized T-MN patients, compared with 108 P-MDS patients. Our results show that mutation burden in T-MN is as high as P-MDS but the pattern is different. Further, the strong genetic association of SF3B1 mutation with RS is absent in T-MN. Also, the survival outcome between T-MN and P-MDS with Low or Very low IPSS-R score was not significantly different.
Unlike in P-MDS and AML, the cytogenetic categories are still not well defined in T-MN. Previous studies on T-MN (T-MDS and T-AML) have used various classification systems, including European LeukaemiaNet (ELN) [11], UK Medical Research Council (UK-MRC) [12], IPSS [13], and others [14]. Interestingly, the IPSS-R is one of the best predictive tools in T-MDS [15]; hence, we used the IPSS-R to stratify cytogenetics in our patients with ≤30% blasts. Of 21 T-AML patients with >30% blasts, 17 did not have complex, del7 or other chromosomal abnormalities, and would be categorized similarly by the IPSS-R, ELN, or UK-MRC classifications. The only difference would be for four patients with 11q23 abnormality, which is considered of intermediate risk in IPSS-R, but adverse in ELN or UK-MRC. Consistent with previous studies, our T-MN cohort showed earlier age of onset, higher frequency of poor-risk cytogenetic abnormalities, higher IPSS-R scores, and poorer OS in comparison to P-MDS [3, 8, 14, 16].
Frequency and pattern of mutations in our P-MDS cohort were similar to published literature [4,5,6]. We detected mutations in 93% of P-MDS patients, most commonly in TET2, ASXL1, SF3B1, SRSF2, and DNMT3A. Spliceosomal complex mutations were mutually exclusive, with SF3B1-mutated status being associated with RS and good prognosis. Notably, our T-MN cohort showed a high mutation frequency (93% of all T-MN) similar to P-MDS. Hence, on a gene level, T-MN is as heterogeneous as P-MDS [5, 6] or AML [17, 18], and this heterogeneity is not offset by an increased frequency of karyotypic abnormalities, a key feature of T-MN.
The frequency of mutations in our T-MN cohort was substantially different from previously published studies [8, 9, 19,20,21,22] for a number of reasons. Firstly, analyses employing low-sensitivity sequencing methods (e.g., Sanger [19, 20, 22]) reported generally lower mutation frequency than next-generation sequencing (NGS)-based studies. Secondly, most reports have focused only on mutational hotspots for some genes [8, 19, 23] such that mutations elsewhere in the gene have been overlooked. For example, in a large T-MN cohort (n = 77) that screened only for the DNMT3A hotspot by Sanger sequencing [19], 5.2% of cases were mutated, which was much lower than the 26% reported in an separate T-AML cohort sequenced on an NGS platform [23]. In closer agreement with the latter study, we found DNMT3A mutations in 27% T-AML and 16% T-MDS. Additionally, genes of considerable importance in the pathogenesis of MNs have been excluded in most studies. For example, spliceosomal complex genes, ASXL1, SETBP1, WT1, and TET2, were not included in panels selected by Ok et al. [8] or the “Copenhagen Series” [20]. Similarly, important genes such as SF3B1, SRSF2, JAK2, DDX41, MPL, GNAS, PTEN, and SETBP1 were not included in another study on T-AML [9]. Lastly, studies performing deep sequencing on the most myeloid-relevant genes are exclusively on T-AML [9, 23]. These factors have led to inconsistencies in reporting and understanding the mutational profile of the spectrum of T-MN. Notably, in comparisons between different T-AML cohorts, the mutation frequency ranged from 33 to 97% of patients [8, 9, 21, 23]. Our cohort is a spectrum of T-MDS and T-AML sequenced using a panel of genes covering most of the important genes/pathways. Importantly, our results are in line with comparable P-MDS and AML studies, which also show a much higher and comparable mutation frequency, with 80–90% or more showing at least one mutation [5, 17, 18].
TP53 was the most commonly mutated gene in T-MN, followed by the spliceosomal complex. Other groups have also reported a similar high prevalence of TP53 mutations in T-MN patients [8]. One study on a smaller pediatric T-MDS cohort found that TP53 mutations mostly occur as an isolated genetic event, similarly to our findings [24]. The observed high frequency of TP53 mutations in T-MN could be due to preferential selection and expansion of rare hematopoietic progenitor cells (HSPC) carrying TP53 mutations following CT exposure, possibly due to their innate chemo-resistance nature. TP53 mutations are known to be associated with complex cytogenetics. This was recapitulated in our T-MN cohort, with the association being stronger than that seen in P-MDS. Despite its strong association with complex cytogenetics, the TP53 mutational status was independently associated with poor OS in a multivariate model. The OS of TP53mut T-MN further decreased as the mutated TP53 allele load increased above 20%, as reported in P-MDS [25].
A detailed spectrum of spliceosomal complex mutations has not been previously reported in T-MN. These mutations occurred in up to a quarter of T-MN patients and were mutually exclusive. In contrast to P-MDS, however, we found that SRSF2 mutations were the dominant spliceosomal mutation in T-MN (60%) instead of SF3B1. Both SF3B1 and SRSF2 are heterozygous mutations limited to specific codons resembling oncogenes and seen in normal individuals (clonal hematopoiesis) [26, 27]. It has been recently shown in a pre-clinical mouse model that HSPC carrying SRSF2 mutations are able to survive CT-induced pro-mutagenic genomic insults and tolerate DNA damage accumulation [28]. It is possible that some age-related mutations (e.g., SF3B1) are not as resistant to such insults and are wiped out as hematopoiesis continues under the stress of mutagen exposure. This may explain the higher frequency of SRSF2 in T-MN.
Interestingly, we find that the frequency of patients with RS is similar between P-MDS and T-MN. As published previously [29], 96% of P-MDS with ≥15% RS harbored SF3B1 mutations, but only 32% of T-MN with RS had SF3B1 mutations. Furthermore, >90% of SF3B1wt T-MN cases with RS had TP53 mutation. This finding has not been previously reported in T-MN, although such an observation has been seen in P-MDS with RS, where 10% of SF3B1wt P-MDS cases harbored TP53 mutations [29]. The exact biological mechanism behind this is not clear. However, TP53 is known to play an important role in mitochondrial homeostasis via multiple mechanisms, which ultimately impact respiratory and oxidative phosphorylative functions [30]. It is possible that TP53 mutations occurring in undifferentiated CD34+ HSPC may downregulate genes involved in the mitochondrial ribosome and in the electron transport chain, leading to later appearance of RS [31]. Further studies are warranted to examine this phenomenon.
In agreement with recent findings [15], the OS, clinical and genetic characteristics of IPSS-R Very low and Low risk T-MN and P-MDS were not significantly different. However, study was underpowered for subgroup analysis and validation in a larger independent cohort is required.
Despite almost seven decades since T-MN were first described [32, 33], prognostic factors still remain to be firmly ascertained. Multiple studies show that karyotype and previous CT/RT exposure (i.e., being therapy related) are important prognostic factors [11, 12, 34,35,36]. With the BM blasts, the prognostic value was found to be more impactful at a cut-off level of 20% [12, 37]. Overall, the discriminatory power of traditional MDS prognostic classifications like IPSS, IPSS-R, WPSS (WHO classification-based Prognostic Scoring System), and others does not seem to be as robust in T-MN as for P-MDS [3, 15], and requires modification from the original schema [3]. This suggests that factors beyond the conventional parameters are operative in T-MN prognosis. In multivariable analysis, we found that patients with T-AML had the highest risk of death, followed by patients carrying TP53 and NRAS mutations. Our findings are supported by studies showing poor prognosis of TP53 and the RAS mutations in MDS patients treated with DMT or/and allo-HSCT [24, 38,39,40,41]. Interestingly, TP53 VAF also predicted poor survival; T-MN patients with high TP53 VAF (>20%) had poorer survival. This is in agreement with findings in P-MDS, albeit at a different clone size cut-off [25]. In our cohort, treatment with DMT was associated with improved OS, but the study is underpowered to draw a definite conclusion.
In conclusion, we show similarities and differences in mutational signatures and clinical outcomes between T-MN and P-MDS. The mutation frequency is similar in T-MN and P-MDS, but the mutation pattern is different. The T-MN mutational landscape is dominated by TP53 and SRSF2, probably due to selection and expansion of HSPC harboring these mutations under genotoxic stress. The frequency of RS is similar in P-MDS and T-MN, but is not associated with better survival in T-MN, most likely due to fewer SF3B1 mutations and the presence of TP53 mutations in SF3B1 wild-type cases. Our results suggest that, in addition to conventional disease parameters, mutational analysis provides added prognostic markers and should be incorporated in T-MN risk stratification.
Data deposition
The sequence data have been deposited at the European Genome-Phenome Archive, which is hosted by the European Bioinformatics Institute, under accession #EGAS00001003547.
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Sem Oncol. 2013;40:666–75.
Ok CY, Hasserjian RP, Fox PS, Stingo F, Zuo Z, Young KH, et al. Application of the international prognostic scoring system-revised in therapy-related myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Leukemia. 2014;28:185–9.
Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–82.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–47.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27.
Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2016;49:204–12.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–55.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Luana F, Livio P, Alfonso P, Anna C, Gianluca G, Massimo B, et al. Characteristics and outcome of therapy-related myeloid neoplasms: report from the Italian network on secondary leukemias. Am J Hematol. 2015;90:E80–E85.
Bacher U, Haferlach C, Alpermann T, Schnittger S, Kern W, Haferlach T. Patients with therapy-related myelodysplastic syndromes and acute myeloid leukemia share genetic features but can be separated by blast counts and cytogenetic risk profiles into prognostically relevant subgroups. Leuk Lymphoma. 2013;54:639–42.
Hiwase DK, Singhal D, Strupp C, Chhetri R, Kutyna MM, Wee LA, et al. Dynamic assessment of RBC-transfusion dependency improves the prognostic value of the revised-IPSS in MDS patients. Am J Hematol. 2017;92:508–14.
Schoch C, Kern W, Schnittger S, Hiddemann W, Haferlach T. Karyotype is an independent prognostic parameter in therapy-related acute myeloid leukemia (t-AML): an analysis of 93 patients with t-AML in comparison to 1091 patients with de novo AML. Leukemia. 2003;18:120–25.
Zeidan AM, Al Ali N, Barnard J, Padron E, Lancet JE, Sekeres MA, et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia. 2017;31:1391.
Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686–98.
Voso MT, Fabiani E, Fianchi L, Falconi G, Criscuolo M, Santangelo R, et al. Mutations of epigenetic regulators and of the spliceosome machinery in therapy-related myeloid neoplasms and in acute leukemias evolved from chronic myeloproliferative diseases. Leukemia. 2013;27:982–5.
Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22:240–8.
Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908–12.
Fabiani E, Falconi G, Fianchi L, Criscuolo M, Leone G, Voso MT. SETBP1 mutations in 106 patients with therapy-related myeloid neoplasms. Haematologica. 2014;99:e152–e53.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Strahm B, Wlodarski MW, Pastor VB, Przychodzien B, Catala A, Dworzak M, et al. Impact of somatic mutations on the outcome of children and adolescents with therapy-related myelodysplastic syndrome. Blood. 2016;128:3162.
Sallman DA, Komrokji R, Vaupel C, Cluzeau T, Geyer SM, McGraw KL, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2015;30:666–73.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Alfonso Pierola A, Marchesini M, Takahashi K, Gañán-Gómez I, Fiorini E, Ogoti Y, et al. The role of Chip-related DNA damage response dysfunction in therapy-related myeloid neoplasms. Blood. 2016;128:958.
Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jädersten M, Jansson M, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233–41.
Kamp WM, Wang P-y, Hwang PM. TP53 mutation, mitochondria and cancer. Curr Opin Genet Dev. 2016;38:16–22.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95.
Crosby WH. Acute granulocytic leukemia, a complication of therapy in Hodgkin’s disease? Clin Res. 1969;17:463.
Steinberg MH, Geary CG, Crosby WH. Acute granulocytic leukemia complicating hodgkin’s disease. Arch Intern Med. 1970;125:496–98.
Kayser S, Dohner K, Krauter J, Kohne CH, Horst HA, Held G, et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117:2137–45.
Kern W, Haferlach T, Schnittger S, Hiddemann W, Schoch C. Prognosis in therapy-related acute myeloid leukemia and impact of karyotype. J Clin Oncol. 2004;22:2510–11.
Armand P, Kim HT, DeAngelo DJ, Ho VT, Cutler CS, Stone RM, et al. Impact of cytogenetics on outcome of de novo and therapy-related AML and MDS after allogeneic transplantation. Biol Blood Marrow Transplant. 2007;13:655–64.
Singh ZN, Huo D, Anastasi J, Smith SM, Karrison T, Le Beau MM, et al. Therapy-related myelodysplastic syndrome morphologic subclassification may not be clinically relevant. Am J Clin Pathol. 2007;127:197–205.
Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28:78–87.
Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376:536–47.
Della Porta MG, Alessandrino EP, Bacigalupo A, van Lint MT, Malcovati L, Pascutto C, et al. Predictive factors for the outcome of allogeneic transplantation in patients with MDS stratified according to the revised IPSS-R. Blood. 2014;123:2333–42.
Acknowledgements
This work was supported by the Royal Adelaide Hospital Research Committee, Contributing Hematologists Committee, and Royal Adelaide Hospital.
Author contributions
DS: planned and performed mutational analysis, analyzed data, and wrote the manuscript. LYAW: processed samples, analyzed data, and edited the manuscript. MMK and RC: analyzed data and edited the manuscript. JG, AWS, JF, and PP-SW: analyzed bioinformatics data and provided critical comments. MB, WTP, and SH: processed samples and analyzed data. SE: provided statistical advice. SM and PA: analyzed data and provided critical comments. SB and ALB: provided critical comments and edited the manuscript. NS and RG: provided clinical information and critical comments. TK, LBT, PGB, IDL, RJD: provided critical comments. JPM: provided critical comments and edited the manuscript. HSS: developed the project, analyzed data, and edited the manuscript. CNH: developed the project, analyzed data, and edited the manuscript. DKH: developed the project, analyzed data, and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SB received research funding and honoraria and served on advisory committees from Novartis and Bristol-Myers Squibb, and consultancy fees and honoraria from Qiagen and Cepheid. HSS received honoraria from Celgene. The other authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Singhal, D., Wee, L.Y.A., Kutyna, M.M. et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia 33, 2842–2853 (2019). https://doi.org/10.1038/s41375-019-0479-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-019-0479-8
- Springer Nature Limited
This article is cited by
-
Comparison of the 2022 world health organization classification and international consensus classification in myelodysplastic syndromes/neoplasms
Blood Cancer Journal (2024)
-
Analysis of clinical and genomic profiles of therapy-related myeloid neoplasm in Korea
Human Genomics (2023)
-
Factors predicting survival following alloSCT in patients with therapy-related AML and MDS: a multicenter study
Bone Marrow Transplantation (2023)
-
TP53 mutation variant allele frequency of ≥10% is associated with poor prognosis in therapy-related myeloid neoplasms
Blood Cancer Journal (2023)
-
Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes defined by international consensus classification
Blood Cancer Journal (2023)