
Abstract
Given its chronicity, contribution to disability and morbidity, and prevalence of more than 2%, the effective treatment, and prevention of bipolar disorder represents an area of significant unmet medical need. While more than half a century has passed since the introduction of lithium into widespread use at the birth of modern psychopharmacology, that medication remains a mainstay for the acute treatment and prevention of recurrent mania/hypomania and depression that characterize bipolar disorder. However, the continued limited understanding of how lithium modulates affective behavior and lack of validated cellular and animal models have resulted in obstacles to discovering more effective mood stabilizers with fewer adverse side effects. In particular, while there has been progress in developing new pharmacotherapy for mania, developing effective treatments for acute bipolar depression remain inadequate. Recent large-scale human genetic studies have confirmed the complex, polygenic nature of the risk architecture of bipolar disorder, and its overlap with other major neuropsychiatric disorders. Such discoveries have begun to shed light on the pathophysiology of bipolar disorder. Coupled with broader advances in human neurobiology, neuropharmacology, noninvasive neuromodulation, and clinical trial design, we can envision novel therapeutic strategies informed by defined molecular mechanisms and neural circuits and targeted to the root cause of the pathophysiology. Here, we review recent advances toward the goal of better treatments for bipolar disorder, and we outline major challenges for the field of translational neuroscience that necessitate continued focus on fundamental research and discovery.
Similar content being viewed by others


“I believe the brain, like any other organ, can get sick and it can also heal.”
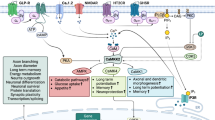
Inadequate nature of current treatments for bipolar disorder
With a lifetime prevalence estimate of more than 2% [3], BPD is a common neuropsychiatric disorder representing one of the leading causes of disability worldwide, impacting individuals, their families, and society as a whole [4, 5]. While episodes of mania in BPD are increasingly effectively managed through a number of medications, including antipsychotics, recurrent major depressive episodes continue to represent a significant challenge in clinical practice. Longitudinal studies indicate that some individuals with bipolar I and II disorder are burdened by significant depressive symptoms, syndromal or subsyndromal, for much of their course despite standard treatments [6, 7]. These symptoms contribute to the substantial morbidity and mortality observed in BPD, including persistent functional impairment [8] as well as suicide [9]. National and international treatment guidelines recognize the challenges in treating BPD, particularly depression, as existing mood stabilizers are only effective at reducing depressive symptoms in ~1/3 of patients [10,11,12,13]. Standard antidepressants have repeatedly failed to show benefit in randomized, placebo-controlled trials [13, 14]. Lithium [1], considered a gold standard treatment in preventing recurrence by all major guidelines, nonetheless does not consistently show superiority to placebo for treatment of depression. Lamotrigine also failed to separate consistently from placebo in large acute depression monotherapy trials [15]. While some atypical antidepressants have demonstrated efficacy for BPD, they benefit only a subset of individuals [16,17,18,19], while metabolic risks are substantial [20] and rates of discontinuation are high [21]. Taken together, there is a clear need to advance the discovery of treatments for BPD, for acute episodes as well as prevention of recurrence.
Challenge of modeling & target identification for bipolar disorder
The development of therapeutics for BPD continues to be a challenge due to the limited understanding of the mechanism of action of existing therapeutics, their polypharmacology, and the lack of validated cellular and animal models that are etiologically based [4, 22, 23]. While recognizing no nonhuman model will be able to fully capture the complexity and full range of symptoms relevant to BPD [22], advances in three main areas seeking to develop innovative new model systems to investigate the pathophysiology of BPD and to discover novel therapeutic targets have begun to turn the tide on this historical challenge for the field of psychopharmacology.
The first is based upon the application of reprogramming technology that now enables the generation of genetically accurate, patient-derived, induced pluripotent stem cells (iPSC) as novel types of “ex vivo” cellular models [24,25,26,27,28,29,30,31,32,33]. In general, 2-dimensional iPSC-derived cultures are well suited to the identification of cell autonomous phenotypes that depend less on cell-cell interactions, but success in measuring network-level electrophysiological properties, including lithium-sensitive hyperexcitability phenotypes of BPD patients has been reported [28, 32, 34]. Recent advances in the development of protocols for 3-dimensional cultures of brain organoids have the potential to allow greater investigation of cell nonautonomous phenotypes related to higher-order properties due to the greater cell-cell interactions inherent to tissue-like architectures [35]. These models bring their own challenges regarding variability and heterogeneity, but standardization of protocols has significantly improved these features and will continue to in the future [36]. A major limitation to date of the studies published to date is the relatively small number of patients that have been characterized, particularly given the recognition that line-to-line variability can be substantial. Multiple efforts are underway in the community to expand to much larger sample sizes in the hundreds. For example, one publicly-available biobank includes more than 400 patient-derived iPSC lines (among them ~80 individuals with bipolar disorder) [37], including neurocognitive testing and self-report battery capturing NIMH Research Domain Criteria features [38, 39]. In conjunction with performing detailed clinicopathological phenotyping, this generation of a “living library” of BPD patient cells opens up new avenues to elucidate the underlying neurobiology of BPD and to aid in the discovery of novel therapeutic targets. Moreover, understanding the ex vivo response of patients as assessed using iPSC-derived, or additionally or alternatively other peripheral cells such as primary lymphocytes, ultimately at the single cell level [40], in advance of in vivo testing has the potential order to reveal stratified, subpopulations of individuals likely to respond differentially and thereby facilitate clinical investigation [41].
Second, based upon an emerging genetic understanding of risk factors for BPD, new rodent models have begun to be characterized with lithium and related pharmacological agents. An excellent example of this advance that provides a direct link to the potential etiological basis of BPD as supported by large-scale genome-wide association studies (GWAS) comes from studies of the ankyrin 3 gene (ANK3) encoding the adaptor protein Ankyrin-G (AnkG). While the precise mechanisms of the genetic association remain unclear, BPD-associated SNPs have elevated brain-specific expression patterns and there is emerging evidence that rare loss-of-function, splice-site SNPs may be protective for BPD [42,43,44]. Studies of AnkG in mouse models have begun to elucidate its potential role in disease pathophysiology, connections to key pathways regulating neurotransmission and neuroplasticity, and ability of lithium to normalize behavioral abnormalities detected upon AnkG loss of function [45,46,47,48,49,50,51].
Finally, as a higher-throughput and lower cost alternative to rodents, zebrafish are being used as vertebrate animal model system for characterization of existing and novel agents for BPD through both quantitative behavioral [52] and functional imaging studies that combined the generation of large-scale brain activity maps through the use of transgenic zebrafish with a genetically encoded calcium indicator, utilizing machine learning for predictive analysis [53].
Lithium & the GSK3 hypothesis: pathways & probes
A long-standing hypothesis concerning the mechanism of action of lithium relevant to the treatment of BPD is that of the inhibition of glycogen synthase kinase-3 (GSK3) family of serine/threonine protein kinase comprised of the two paralogs GSK3α and GSK3β [54,55,56]. GSK3 activity is dynamically regulated through the combined effects of activating autophosphorylation of Tyr279/216 (α and β isoforms respectively), its inhibitory phosphorylation on Ser21/9 (α and β isoforms, respectively) on its N-terminal tail, and the activity of protein phosphatase that counteract its phospho-inhibition.
At the biochemical level, lithium has been shown to directly inhibit GSK3 kinase activity through competition for magnesium in the ATP binding site of GSK3 [54, 55, 57]. In addition, lithium indirectly inhibits GSK3 kinase activity through activation of the AKT kinase family of kinases leading to N-terminal, inhibitory phosphorylation and the inhibition of phosphatases that dephosphorylate GSK3 [56, 58,59,60,61]. Outside of cellular and rodent studies, additional support for the relevance of GSK3 comes from studies of peripheral blood of BPD patients treated with lithium that also show elevated N-terminal inhibitory GSK3 phosphorylation levels [62, 63]. Furthermore, multiple structurally distinct antipsychotics and antidepressants indirectly modulate GSK3 activity [56, 64, 65]. Despite this growing knowledge, since the precise mechanisms of GSK3 relevant for regulation of affective behavior remain incompletely understood, the elucidation of lithium-sensitive neural substrates and downstream pathways remain an active area of investigation [56, 59, 66,67,68,69,70,71,72].
WNT/GSK3β/β-catenin signaling pathways
One of the early cellular pathways implicated in lithium’s regulation of neuroplasticity as a result of GSK3 inhibition is that of WNT/GSK3β/β-catenin signaling [54, 55, 67]. Here GSK3 inhibition leads to reduced phosphorylation and attenuated proteasomal degradation of β-catenin, resulting in accumulation of β-catenin in the cytoplasm, its interaction with membrane-localized, cell-adhesion molecules like cadherin [73], as well as the translocation of β-catenin to the nucleus where it acts as an activator of transcription of TCF/LEF-dependent genes that form the basis of canonical WNT signaling pathway [66, 67, 71, 72, 74,75,76]. WNT/GSK3β/β-catenin signaling also plays a fundamental role in memory consolidation [77] and regulating neurogenesis [75, 76, 78,79,80]. Besides GSK3 inhibition pharmacologically or genetically, the overexpression of β-catenin itself has been shown to cause an antidepressant-like phenotype in the forced swim test of behavioral despair in mice [70]. However, adult hippocampal neurogenesis, which occurs over a much longer time frame, is not necessary for the response to lithium in mouse behavioral assays like the forced swim test in which acute lithium administration is effective [81]. In the treatment of BPD though, the onset of clinical benefit of lithium generally requires weeks of administration leaving open the possibility of the contribution of neurogenesis and other adaptive changes. Finally, besides evidence that lithium can modulate WNT/GSK3β/β-catenin signaling that may be of relevance for its therapeutic effects in BPD, a number of rare genetic risk factors for neuropsychiatric disorders are also known to regulate or be regulated by WNT/GSK3β/β-catenin signaling [82,83,84]. In particular, loss of function mutations in the CTNNB1 gene encoding β-catenin are now recognized as a frequent cause of intellectual disability and autism spectrum disorder [85, 86], which further points to the fundamentally important role of β-catenin in the neurocircuits important for brain development and cognition.
β-Arrestin-2/AKT/PP2A-GSK3 signaling
In addition to lithium’s regulation of WNT/GSK3β/β-catenin, elegant studies from the Caron and Beaulieu laboratories have shown that multiple lithium-sensitive behaviors, in particular amphetamine-induced hyperactivity, requires the scaffolding protein β-arrestin 2, which forms a complex with protein phosphatase 2 A (PP2A) and AKT leading to AKT dephosphorylation and inactivation in response to dopamine D2 receptor activation in a manner that can be antagonized by lithium treatment [66, 71, 87]. Building off these studies, Klein and colleagues have now shown using both pharmacological inhibitors and in vivo genetic approaches involving the overexpression or loss of one copy of the GSK3β gene, that GSK3 activity is a critical regulator of the stability of the β-arrestin-2/AKT/PP2A complex with lithium disrupting this complex and GSK3β overexpression restoring the complex in lithium treated mice [88]. Since the activity of PP2A-mediated dephosphorylation and inactivation of AKT can in turn lead to elevated GSK3 activity due to a loss of reduced inhibitory Ser9/21 N-terminal tail phosphorylation, the disruption of the stability β-arrestin-2/AKT/PP2A complex by lithium would serve to amplify its direct inhibitory effects on GSK3. In line with these findings, using acute administration of a pharmacological inhibitor of AKT, Pan et al. [72] have demonstrated that AKT kinase activity is required for lithium to regulate inhibitory GSK3 phosphorylation and affective behaviors in mice with the viral-mediated expression of activated AKT shown conversely to confer lithium responsively both to cultured cell lines and a strain of mice that does not normally respond behaviorally to lithium. In contrast, when GSK3 was inhibited directly using the selective, ATP-competitive inhibitor CHIR099021, a requirement for AKT activation was no longer observed for the behavioral effects of lithium in either lithium responsive or non-responsive mouse strains [72]. Collectively, while the precise mechanisms remain uncertain, the findings implicating a key role for AKT activity and the disruption of β-arrestin-2/AKT/PP2A complex in response to lithium could possibly explain how such a weak direct inhibitor of GSK3 as lithium has sufficiently robust pharmacodynamic effects to control affective behavior in the context of BPD. Such interwoven and specific mechanisms suggest potentially important aspects of lithium’s specificity at the level of neurocircuits since not all cell types appear to form β-arrestin-2/AKT/PP2A complexes [71, 88]. In support of the latter notion, studies using cell-type selective Cre-recombinase expressing mice and floxed GSK3 alleles [89, 90], as well as with CRISPR/Cas9-mediated genome engineering [91], have demonstrated that the hyperlocomotory response to amphetamine is dependent on GSK3β activity specifically in dopamine D2 receptor-expressing medium spiny neurons (MSNs). These MSNs mediate neurotransmission within the indirect pathway of the basal ganglia circuit, separate from dopamine D1 receptor-expressing MSNs within the direct pathway where the loss of GSK3β had no effect on the response to amphetamine. Besides lithium, there is further evidence for the therapeutic relevance of antagonizing β-arrestin-2 complexes coming from studies of antipsychotics targeting the dopamine D2 receptor many of which are commonly used to treat BPD [92]. Finally, to expand this notion of neurocircuits outside those formed by dopamine D2 receptor expressing neurons, similar studies inactivating GSK3β selectively in forebrain, pyramidal neurons of mice have revealed enhanced social interaction and anxiolytic-like effects [90]. These findings pointing to a broader role of GSK3β activity in aspects of neurotransmission that are potentially relevant to mood and anxiety symptoms in BPD. In support of this notion, the anxiolytic-like effects of GSK3β inhibition have been further revealed through CRISPR/Cas9-mediated somatic knockout in medial prefrontal cortex neurons of adult mice [93]. These behavioral effects occurred in concert with reduced glutamatergic neurotransmission through reduction of AMPA-mediated excitatory postsynaptic currents and were mimicked by overexpression of FXR1P (fragile X mental retardation-related protein 1), an RNA binding protein that is a known phospho-substrate regulated by GSK3β [93, 94]. Beyond anxiolytic effects, the cell-type-specific deletion of GSK3β in dopamine D2 receptor-expressing neurons has been shown to have pro-cognitive effects in a test of working memory impairment due to N-methyl-D-aspartate (NMDA) receptor antagonist (MK-801) treatment [95]. In concert, the resistance to cognitive dysfunction was related to alterations of synaptic plasticity in the medial prefrontal cortex with NMDA receptor activity increased in layer V pyramidal neurons, enhanced dopamine-dependent modulation of NMDA receptor currents, and increased dendritic spine density of layer V pyramidal neurons [95]. These changes in NMDA receptor-dependent synaptic plasticity were further related to elevated expression levels of GRIN2A and GRIN2B in a manner correlated with chromatin modifications (elevated histone H3 Lys18 and Lys27 acetylation) in the corresponding promoter regions [95]. Although the precise causal relationship to the pro-cognitive effects of GSK3β ablation in D2 receptor expressing neurons and mechanistic basis is unclear, these changes in epigenetic status of the NMDA receptor subunits were associated with a reduction of total levels and the specific loss at the GRIN2B promoter of HDAC2 (histone deacetylase 2) [95], a major suppressor of synaptic plasticity and target for pro-cognitive and mood stabilizing therapeutic development [96,97,98,99,100,101,102,103,104,105,106,107]. Overall, these findings further emphasize the potential developmental- and cell-type-specific effects of deletion of GSK3β such that the age of the mouse and neurocircuit being targeted are important factors when extrapolating the relevance of mouse model studies to novel therapeutic development.
Advances in pharmacological targeting & imaging of GSK3
To further advance the pharmacological targeting of GSK3 from a neuropsychiatric disease perspective continued efforts have largely focused on the development of GSK3 inhibitors with different levels of potency, selectivity (including between the two isoforms GSK3α and GSK3β), and modes of action [72, 82, 108,109,110,111,112,113]. For example, Wagner et al. have reported highly selective, brain penetrant, and behaviorally active GSK3 inhibitors that have lithium-like effects in cellular assays and behavioral models of psychostimulant-induced hyperactivity [108, 111]. Similarly, in a study addressing neuropsychiatric symptoms of Alzheimer’s disease and related tauopathies Griebel et al. [112] reported on a novel, potent GSK3 inhibitor SAR502250 that reduced the depressive-like state of mice in a chronic mild stress paradigm, attenuated aggression in a mouse defense test and decreased psychostimulant-induced hyperactivity. In addition, using structure-guided drug design and an integrated panel of cell-based assays, including ones assessing GSK3 substrate phosphorylation in human iPSC-derived neuronal cells, Bernard-Gauthier et al. [113] recently described the development of a series of highly potent and selective, brain-penetrant oxazole-4-carboxamide-based inhibitors of GSK3. This included OCM-51, one of the most potent (picomolar IC50) and selective (>tenfold GSK3β vs. GSK3α) GSK3β inhibitor known to date [113]. The achievement of picomolar-selective GSK3β inhibition, as well as demonstrating the feasibility of achieving at least 10-fold β-isozyme specificity, is a significant advance over mostly double-digit nanomolar inhibitors of GSK3 that lacked selectivity. To allow neuroimaging of GSK3, these studies also radiolabeled the lead compound OCM-44, which showed excellent brain exposure in rodent pharmacokinetic studies with equal partitioning between brain tissue and plasma, and performed microdosed positron emission tomography (PET) imaging in nonhuman primates [113]. The continued optimization of this series of GSK3 inhibitors as PET tracers and progression into human studies is ongoing and may provide a critically needed tool to help address in vivo target engagement of GSK3 inhibitors to aid in finding appropriate doses and administration schedules.
While improvement in potency and selectivity along with the synthesis of PET tracers for assessing in vivo target engagement are important developments for the field’s efforts to advance GSK3 inhibitors, a number of other critical hurdles remain to be addressed. While details of multiple GSK3 programs that were initiated but terminated have not been disclosed, a recent summary of studies by AstraZeneca outlined the challenges encountered in progressing efficacious and safe compounds that had been originally being developed for the purpose of treating Alzheimer’s disease starting in 2003 [109, 114]. While highly potent (Ki 5–30 nM) and orally bioactive compounds within different structural classes were identified that showed in vivo efficacy in preclinical models assessing tau phosphorylation and precognitive effects, preclinical toxicological effects on the musculoskeletal system, as well as histopathological changes in the gallbladder and cholecystitis observed in dogs and biliary hyperplasia in rats, required abandoning clinical development of multiple compound series [114]. This included the compound AZD1080 that was demonstrated in a Phase I clinical trial in healthy controls to show GSK3 target engagement in peripheral cells and was reported to be well tolerated, but without a sufficient exposure margin its further development was forced to halt [109].
With these pre-clinical and clinical issues in mind, numerous opportunities still exist to tackle selective GSK3 inhibition with different modes of inhibition, including allosteric inhibitors and substrate competitive inhibitors. An alternative approach can tackle enhancing safety by tuning the pharmacokinetic properties, including enhanced brain to plasma ratios and minimized peripheral target engagement. Understanding the key substrates driving the neurobehavioral effects of GSK3 inhibition provides the potential to guide the clinical development of a new generation of efficacious and safe GSK3 inhibitors by allowing optimization of pharmacokinetic and pharmacodynamic profiles. In addition, since the kinetics of phosphorylation and dephosphorylation of substrates can vary widely, having knowledge of the pharmacodynamics will enable exploration of appropriate alternative dosing regimens to maximize efficacy and enhance safety.
Novel target discovery via lithium mimetics
As an alternative to a direct GSK3 inhibitor, one approach to understanding the therapeutically relevant mechanism of action of lithium is to identify pharmacological “lithium mimetics” that recapitulate the effect of lithium on specific molecular targets in order to determine whether such agents have similar neurochemical and behavioral effects. Such a screening strategy may also yield a lithium ‘enhancer’ that improves the benefit of a lower dose of lithium, which given its narrow therapeutic index could reduce toxicity, or restore lithium responsivity to patients that are refractory or lose sensitivity to lithium, thereby broadening the clinical population that could obtain benefit. While therapeutically lithium represents a gold standard, pharmacologically it poses challenges as a target because of the complexity of its effects— that is, the wide variety of effects in vitro raises the possibility that mirroring its effects on a single target pathway may have no therapeutic relevance. To attempt to refute this hypothesis, a number of innovative approaches have been taken aiming to discover pharmacological probes with potential for clinical translation.
Targeting phospho-CRMP2
In an effort to identify lithium mimetics, Tobe et al. [30] recently demonstrated that p-CRMP2T514, the inactivated form of Collapsin response mediator protein 2 (CRMP2), was elevated in lithium-responsive BPD patient iPSC-derived neuronal models as compared with nonlithium responsive BPD patients, as well as patients with other psychiatric and neurological disorders. This particular phospho-substrate of GSK3 is of interest based upon global quantitative proteomic profiling in human iPSC-derived neuronal cells using stable isotope labeling by amino acids in cell culture (SILAC) methodology that showed it to be one of the most highly regulated phosphoproteins in response to treatment with the selective GSK3 inhibitor CHIR-99021 [115], which has been shown to have lithium-like effects in mouse behavioral models [72]. Previous biochemical and cellular studies have also shown that p-CRMP2T514 is regulated by GSK3β and not GSK3α and that its phosphorylation levels is regulated by lithium and other GSK3 inhibitors in rodent neurons [116, 117]. In addition, using CRISPR/Cas9-mediated genome engineering in mice, a double knockout of GSK3β and CRMP2 in dopamine D2 receptor-expressing MSNs was demonstrated to block the suppression of amphetamine-induced hyperactivity that occurs when GSK3β is knocked out in the same dopamine D2 receptor-expressing MSN in a manner that lead to decreased dendritic branching complexity and spine density [91]. These data further implicate a critical role for GSK3β’s regulation of neurotransmission in MSN within the indirect pathway of the basal ganglia as a critical regulator of affective behavior [89, 91].
Since these collective data strongly suggest that CRMP2 serves as a key neural substrate downstream of GSK3β that regulates the neurocircuitry involved in affective behaviors in mouse models, and since p-CRMP2T514 levels served as robust marker of GSK3β inhibition in human neurons, Zhao et al. [115] performed an unbiased screen of a chemogenomic library for novel regulators of p-CRMP2T514. This screen yielded both known GSK3 inhibitors, many of which were previously shown to have lithium-like effects in the amphetamine-induced hyperactivity model, as well as a series of nondirect GSK3β modulators that decreased p-CRMP2T514 levels, like lithium, both in neural progenitor cells (NPCs) and post-mitotic neurons. This latter class included both FDA-approved drugs, which have the potential for more readily being repurposed for testing in the clinic, as well as novel natural products and bioactive probes not previously shown to regulate CRMP2 activity.
In connecting ex vivo iPSC-derived assay results to in vivo data with intact neurocircuits, systemic administration of a subset, but not all, of the CRMP2-phosphorylation suppressors was found to mimic lithium’s attenuation of amphetamine-induced hyperlocomotion in mice. For example, of the class of non-GSK3β targeting p-CRMP2T514 suppressors that were identified, NNC-711, a reported blocker of GABA uptake via inhibition of SLC6A1 (solute carrier family 6 member 1; (GAT1)), suppressed amphetamine-induced hyperactivity when administered on its own to mice, but showed no additive effect in combination with lithium administration. In contrast, the known D2 receptor antagonist sulpiride, an agent shown in a double-blind comparative study to have equivalent antidepressant activity to amitriptyline in BPD patients with recurrent depression that were being treated with lithium [118], only suppressed hyperlocomotion when administered in combined with lithium. An additional interesting p-CRMP2T514 suppressor identified by this screen was the natural product, and widely consumed nutritional supplement, huperzine A, a sesquiterpene alkaloid, which suppressed locomotion on its own and showed an additive effect with lithium in the amphetamine-induced hyperactivity assay. Mechanistically, huperzine A is known to inhibit acetylcholinesterase and to be an antagonist of NMDA receptors, and has been investigated as a disease-modifying drug for dementia and shown in a series of randomized, controlled trials as an adjunctive treatment to modestly improve neurocognitive function in schizophrenia patients [119]. In preclinical mouse models of Alzheimer’s disease, huperzine A has been shown to enhance cognition and its neuroprotective activities were linked to increased levels of inhibitory GSK3αS21/βS9 phosphorylation [120]. Although the precise mechanism through which huperzine A suppressed p-CRMP2T514 levels in human neurons is unknown, given recent GWAS studies in BPD that have implicated the GRIN2A subunit of the NMDA receptors (see below), these findings potentially link together CRMP2 to a genetically implicated mechanism in BPD as discussed further above in the context of Ankyrin-G (ANK3) [51]. Taken as a whole, further testing of CRMP2 modulating agents may afford a new strategy to mimic the effects of lithium and direct GSK3 inhibitors on affective behavior while minimizing side effects due to the central role that GSK3 plays in multiple levels of cellular physiology.
Targeting inositol monophosphatase
An alternative mechanism to lithium’s GSK3 inhibition that continues to be explored is its inhibition of inositol monophosphatase (IMPase) [121, 122], a key enzyme involved in Gq-protein coupled receptor and other signaling pathways. While selectively targeting IMPase with brain penetrant agents has remained a challenge, recent studies seeking to identify a lithium mimetic identified ebselen as a blood-brain barrier-penetrant IMPase inhibitor [123]. In preclinical mouse models, ebselen preferentially reduced motor impulsivity over choice impulsivity through a mechanism at least in part through inhibition of 5-HT2A receptors [124]. Given the relationship of impulsivity to potential suicide risk, and putative suicide- and attempt-sparing ability of lithium [125], these data support the lithium mimetic activity of ebselen, at least indirectly. In partial support of these pre-clinical data, acute oral ebselen was shown in a double-blind, placebo-controlled trial with healthy participants to reduce brain myo-inositol in cortical regions and to affect emotional processing, to decrease latency time in the acoustic startle paradigm, and to decrease the reinforcement of rewarding stimuli along with decreased slow-wave sleep [126]. Other placebo-controlled randomized studies in healthy controls also showed that ebselen treatment decreased impulsivity and produced a positive bias in emotional processing [127]. Future studies in BPD subjects of ebselen alone or in combination with lithium, with assessment of affective symptoms, will be needed to determine whether the effects translate to meaningful benefit for patients.
Novel targets & mechanisms emerging from human genetics
Twin studies of BPD indicate that it is highly heritable (~80%); children with an affected parent are at increased risk for a variety of symptoms and neurobiological abnormalities [128, 129]. In agreement with twin and family studies, recent data from large-scale GWAS have confirmed the risk for developing BPD is highly polygenic in nature [129]. This polygenicity overlaps incompletely with schizophrenia and other major neuropsychiatric disorders [130]. The incomplete overlap is consistent with the diagnostic challenge of distinguishing these disorders clinically, as well as the differences in prognosis as well as apparent pharmacological specificity. That is, mainstays of treatment in one disorder may be entirely ineffective in a genetically related disorder. Lithium is not an effective antipsychotic for treatment of schizophrenia; standard antidepressants remain largely ineffective as monotherapy for bipolar depression and may even worsen disease course in some individuals.
Simply identifying tens or hundreds of risk loci does not necessarily identify BPD treatment targets. Indeed, even largely monogenic diseases such as Alzheimer’s disease or Huntington’s disease have required substantial work to move from a risk variant to a therapeutic strategy. This work may entail intersecting risk loci with knowledge of changes in the transcriptome, epigenome, and proteome from patient-derived cell models and post-mortem tissue [131] – particularly if only common variants, rather than “smoking gun” functional variants, have been identified. As of the end of 2019, the most recent GWAS of 20,352 cases and 31,358 controls of European descent, with follow-up analysis in an additional 9,412 cases and 137,760 controls identified 30 loci as genome-wide significant [129]. Subsequently pathway analysis identified nine pathways that were significantly enriched for genes with BPD associations, including ones implicated in endocannabinoid signaling, regulation of insulin secretion, and motor control [129]. While any of these represent potential therapeutic targets [23], more work is required to understand the way in which these pathways may contribute either to neurodevelopmental effects, or to acute symptomatology, or both.
When considering formulating a therapeutic hypothesis on the basis of these data, the complex genetic architecture of BPD presents a number of formidable challenges. First, the nature of linkage disequilibrium amongst associated polymorphisms means that the index variant may not be the causal variant. Hence, what gene or set of genes to follow up on remains unclear in many if not all cases. Resolving the causal variant requires additional fine mapping to be performed along with analysis of functional data from transcriptomic studies and epigenomic studies in order to link the variant to specific cell types, transcripts, or genomic regulator elements. On the basis of these data, it may be possible in the future to more clearly assign the directionality of the effect in terms of the “gain” or “loss” of a particular biological function to guide a clear therapeutic hypothesis. Here, inspirational examples come from other areas of medicine where the experiments of nature that genetic studies represent have been highly informative through the elucidation of an allelic series that points to a clear directionality of the effect and informs a therapeutic hypothesis in terms of what the expected physiological result should be by increasing or decreasing the target function by a specified amount [132]. The second major challenge comes from the fact that the effect size of the majority of the associated loci across the genome are small. Recognizing that the effect size is a statistical metric derived from a population level analysis the modulation of the function encoded by single genetic risk factor may not produce sufficient change to a complex biological system when considered in one particular individual’s cells. In addition, given the robustness of biological systems that are often buffered from change and subject to constraints for dynamic change in response to perturbations, the identification of central, highly connected nodes in networks of interaction may be required to ultimately impact the system.
A number of these BPD-associated genes, including GRIN2A, CACNA1C, SCN2A, and HDAC5 [129], have available pharmacological probes, including some of which are FDA-approved drugs with a history of investigation in the context of bipolar disorders. Most notable in this context are the L-type calcium channel antagonists that target the α1c subunit encoded by the CACNA1C gene discussed above as a focus of rodent model systems to investigate BPD pathophysiology. Prior to any of the present genetic observations concerning L-type calcium channels in BPD, as reviewed recently [133, 134], multiple clinical studies of different classes of L-type calcium channel antagonists were performed starting in the late 1980s. These agents were either investigated as monotherapy or in combination with lithium, an antipsychotic or anticonvulsant, with the majority of studies having been performed with verapamil. The overall results have been mixed with challenges to the interpretation of the findings due to the varied doses used and absence of clear data demonstrating in vivo target engagement in the brain, for example from a PET tracer, and mixed pharmacology particularly in the case of verapamil that limits precise conclusions regarding contributions of the L-type channel to efficacy as compared with other mechanisms [134]. For example, despite promising initial results with the dihydropyridine nimodipine, either as a monotherapy or in combination with lithium or the anticonvulsant carbamazepine on mania in BPD [135,136,137,138], the Phase IIa study of MEM-1003, a dihydropyridine of the same class that was defined to exhibit reduced peripheral side effects, especially blood pressure lowering, was found to be ineffective as a monotherapy for acute mania in BPD. A preliminary study with the dihydropyridine isradipine showed promise for this class of pharmacological agents [139].
Recent efforts are seeking to overcome these shortcomings of the earlier studies, for example with the Oxford study of Calcium channel Antagonism, Cognition, Mood instability and Sleep [140], which is a randomized, double-blind, placebo-controlled study on the effect of the dihydropyridine LTCC nicardipine in healthy young adults with mood instability with a battery of psychiatric, cognitive, circadian, physiological, biochemical, and functional magnetic resonance imaging (fMRI) and magnetoencephalography neuroimaging parameters being collected over a 4-week period with subjects stratified by the CACNA1C risk single-nucleotide polymorphism (SNP) rs1006737. While the benefit of such a study is premised upon the directionality of the risk variant in the CACNA1C locus causing increased L-type calcium channel activity, this mechanistic detail remains unclear, with published data from pre-clinical studies supporting both directionalities. Ultimately, ongoing human clinical studies combined with genetic markers stratified based upon risk SNPs within the CACNA1C may provide the most relevant, definitive answer to whether selective L-type calcium channel antagonists versus potentiators are desired since these will address the complex role that such channels play in different cell types of the nervous system.
Another example of a putative risk gene for BPD from the most recent large-scale GWAS studies that is of high-interest is the identification of HDAC5 encoding a member of the class II histone deacetylase family, though at this stage it has more limited pharmacological investigation and there is a need for fine mapping to pinpoint the true causal variant within the locus. In previous functional studies in rodent models, Tsankova et al. [141] had shown that viral-mediated HDAC5 overexpression in the hippocampus blocked imipramine’s ability to reverse depression-like behavior and found that chronic imipramine was associated with a selective downregulation of HDAC5. Studies by Renthal et al. [142] also showed that chronic but not acute exposure to stress decreased HDAC5 function in the nucleus accumbens, a brain region that plays a critical role in brain reward response, and loss of HDAC5 caused hypersensitivity to chronic but not acute stress, suggesting its regulation may play a causal role in the response to stress. Such observations point to the importance of elucidating the particular regions of the brain relevant to BPD pathophysiology that a putative genetic risk factor like HDAC5 may be important for, since the functional consequences of gain and loss of function may be distinct in different brain regions, thereby presenting a challenge for conventional pharmacological modulation that may lack brain region or cell type specificity. These observations, along with preclinical mouse model data looking at both antidepressant-like and antimanic-like activity of selective HDAC inhibitors [96,97,98,99,100,101,102,103,104,105], as well as the advent of the PET radiotracer [11C]Martinostat [143] that allows for “neuroepigenetic” imaging of HDAC levels in the living human brain of psychiatric disease patients [144], support the notion that histone modifications may play a key role in the pathophysiology and future treatment of affective disorders.
Beyond specific targets implicated by the peak SNPs in large-scale BPD GWAS, the implication of the endocannabinoid system as a potentially important pathway determining risk, and most notably the implication of the gene FAAH as one of the key drivers of this association, is of potential relevance for therapeutic development [129]. FAAH encodes fatty acid amide hydrolase, an enzyme that along with monoacylglyercol lipase (MAGL), breaks down endocannabinoids in the brain that serve as endogenous ligands of the principal cannabinoid receptors CB1/CB2. As reviewed in Arimand et al. [145], inhibition of FAAH and MAGL is the focus of ongoing efforts to develop new treatments for pain and neuropsychiatric disorders, including in BPD. Of note, whereas CB1 receptors are localized both pre- and post-synaptically on neurons throughout the nervous system, CB2 receptors are located largely peripherally in immune system cells and microglia with evidence that CB2 receptor agonist may have anti-neuroinflammatory properties through the ability to suppress microglial activation and cytokine release [146]. This has led to the suggestion that potentiating CB2 receptor activity, either directly or through modulating the endocannabinoid system, may produce a novel pharmacotherapy that targets neuroinflammatory dysfunction in BPD [145]. Last, the implication of the regulation of insulin secretion in the pathway analysis with a broad set of potentially key driver genes [129], including calcium channel subunits (CACNA1C, CACNA1D), the dopamine receptor D2 (DRD2) that encodes the primary target of multiple atypical antipsychotics already clinically used to treat BPD, cyclic AMP regulated kinase and regulatory enzymes (PRKCA, ADCY2, PDE3B), and potassium ion channel subunits (KCNB1, KCNC2, KCNS3, KCNG2), is of interest given the existing and potential for novel pharmacological agents targeting these factors. Moreover, previous double-blinded, controlled trials of intranasal insulin on neurocognitive function in euthymic BPD patients suggests the potential for investigating this pathway more systematically in conjunction with the new knowledge of genetic variation that may determine response and thus be used to stratify patients [147].
Novel targets & mechanisms emerging from classical neuropharmacology
Since the findings from human genetics of BPD have only recently begun to resolve themselves into recognizable pathways, and multiple steps are still required to translate these findings into a clear therapeutic hypothesis [132], another path to continue to discover potential new therapies for BPD is to advance human experimental therapeutic trials with novel mechanism of action medications that have emerged from classical neuropharmacology, often interventions that are first FDA-approved for other indications.
A recent example of this effort is that of lurasidone, which was first approved as an antipsychotic for treatment of schizophrenia in 2010 with the desire of minimizing undesired side effects, namely extrapyramidal and metabolic effects, that plague first- and second-generation antipsychotics, respectively. Rather than pursuing mania as an indication, typically the path for preceding antipsychotics, lurasidone was approved in 2013 for the treatment of bipolar depression in adults as a monotherapy and as an adjunct therapy with either lithium or valproate [148] and then most recently in 2018 for bipolar depression in pediatric patients (aged 10–17) [149]. Lurasidone exhibits a mixed pharmacology showing high affinity binding as an antagonist to the dopamine D2, 5-HT2A, 5-HT7, and α2c adrenergic receptors, along with partial agonism of the 5-HT1A receptor [150]. Intriguingly, besides efficacy for treatment of bipolar depression, preliminary, randomized, open-label studies of cognition in euthymic patients with BPD suggest lurasidone is able to ameliorate cognitive impairment [151], and a 3-week, a double-blind, controlled study in schizophrenia patients also reported pro-cognitive effects for lurasidone whereas the atypical antipsychotic ziprasidone did not [152]. These observations, along with preclinical studies in rodent models showing that lurasidone, but not other antipsychotics such as ziprasidone, risperidone, aripiprazole, clozapine and haloperidol can suppress MK-801-induced cognitive deficits and shown other procognitive properties [150, 153, 154], point to potentially unique properties of this compound. Exploration of the underlying molecular and cellular mechanisms of the pro-cognitive effects of lurasidone could open up a critically needed new path to agents for BPD and related neuropsychiatric disorders. To this end, studies in patient-derived iPSC models and related systems are now underway with promising findings pointing to differences in pathways critical for synaptic plasticity (SJH, RK, unpublished observations). More generally, work with lurasidone highlights efforts to find treatments that improve cognition in BPD, a complex set of impairments that may represent trait markers of the disease, compounded by state effects and particularly by other medications [155]. Such impairments contribute to the functional consequences and chronicity of bipolar disorder but have previously been underappreciated in light of the more evident mood symptoms.
Outside of lithium and atypical antipsychotics, BPD is often treated with anticonvulsant agents such as lamotrigine, valproate, and carbamazepine. While the mechanistic rationale for the efficacy of some anticonvulsants but not others remains unclear, the proven clinical efficacy of such anticonvulsant agents suggests the value of continuing to explore the potential for repurposing new mechanism of action anticonvulsants as adjunctive or monotherapy. An exemplar of this strategy is recent studies being pursued in the context of BPD with lacosamide. Here, Cuomo et al. [156] have reported results of an open-label study of more than 100 individuals with acute BPD treated with lacosamide compared with a retrospective analysis of a BPD sample treated with other antiepileptics. While the study design limits firm conclusions, lacosamide was suggested to be well-tolerated and effective in reducing mania, depression, and anxiety and in improving global functioning, at doses of lower than those typically used in epilepsy [156].
Mechanistically, lacosamide is thought to target voltage-gated sodium channels, but unlike other anticonvulsants it does so through a unique mechanism of enhancing the slow inactivation of voltage-gated sodium channels rather than through affecting their fast inactivation. While the mechanism through which this slow inactivation of sodium channels is not fully understood, of potential relevance is the fact that lacosamide has also been recently shown to modulate CRMP2 [157]. As discussed above, CRMP2 has emerged as a neural substrate sensitive to lithium and GSK3β inhibitors and appears to be differentially regulated in BPD iPSC-derived neuronal models [30, 115]. Since CRMP2 can regulate microtubule dynamics [157], which may be involved in the regulation of the slow inactivation of voltage-gated sodium channels, future studies on lacosamide’s mechanism of action and testing of CMRP2 modulating small molecules, along with larger, placebo-controlled randomized trials may yield a new mechanism for BPD pharmacotherapy.
Following a similar line of reasoning, besides lacosamide, additional novel anticonvulsant agents in the category that merit further investigation in BPD for benefit include: (1) rufinamide, an agent approved for treatment of Lennox-Gastaut syndrome whose use in BPD is supported by two case reports of potential mood stabilizing effects with a mechanism of action not conclusively known but may involve antagonism of voltage-gated sodium channels [158]; and (2) ezogabine (retigabine) [159], an opener of voltage-gated KCNQ/Kv7 potassium channels approved as an add‐on medication to treat seizures associated with epilepsy, which has been shown by in an open-label trial to have an acute antimanic effect in a subset of BPD patients [160], as well as demonstrated to have antimanic like activity in both the amphetamine plus chlordiazepoxide-induced hyperactivity model and repeated sensitization amphetamine-induced hyperactivity model [161,162,163].
Targeting trace amine-associated receptors
Outside of ideas for novel BPD pharmacotherapies emerging from consideration of anticonvulsants, recent studies have suggested Trace Amine-Associated Receptor 1 (TAAR1) activation as a potential novel class of antipsychotics [164]. While not dependent on dopamine D2 receptor antagonism for efficacy, mechanistic evidence indicates the activation of TAAR1-D2R heterodimers induces biased signaling through β-arrestin-mediated mechanisms leading to a reduction of GSK3β signaling [165], providing evidence independent from that of lithium for the importance of β-arrestin-mediated regulation of GSK3 [71, 88]. Previous studies in both C. elegans and mice have also suggested that trace amine pathways signaling through TAAR1 are important for clozapine’s behavioral effects [166]. Together, these data suggest that, in addition to their current development as novel mechanism of action antipsychotics, there may be utility in considering TAAR1 agonists as BPD treatments. In particular, combinations of TAAR1 agonists and lithium may yield an effective therapeutic and one that would enable lowering of the lithium dose, along with potential restoration of lithium sensitivity among otherwise non-responsive patients.
Omega-3 fatty acids
Another example of pharmacological agents outside of the anticonvulsants and more traditional ion channel and monoamine receptors that are the targets of current agents and ones in development, an intriguing example of a class of natural product supplements of potential interest for BPD therapeutic development are the long-chain omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Deficits in these omega-3 fatty acids have long been associated with the pathophysiology of BPD [167, 168]. Studies in preclinical animal models support the notion that omega-3 fatty acids promote synaptic maturation and plasticity in developing cortical brain circuits and their deficiency can lead to aberrant network connectivity. For example, a preclinical study in a nonhuman primate model using resting-state functional connectivity MRI demonstrated that lower levels of omega-3 fatty acids were associated with decreased functional connectivity in cortical regions critical for cognitive function [169].
In terms of treatment trials using omega-3 fatty acids as a dietary supplements in BPD, as reviewed in Saunders et al. [168], a total of five nonrandomized and open-label studies of omega-3 supplementation in bipolar disorder that consisted of administration of either of mixture of DHA and EPA or EPA alone found beneficial effects on clinical measures related to mood in four of the five studies. In contrast to these encouraging results, randomized, double-blind, placebo-controlled trials of omega-3 fatty acid supplementation have been more varied with benefit observed in only two of the seven studies. However, multiple methodological differences in potentially critical variables such as the dose of the supplement, the exact nature of the ratio of EPA to DHA (and other fatty acids), concurrent pharmacotherapy, design of trials in addition to heterogeneity of diagnosis make this comparison of studies problematic, thus leaving open the question of the benefit of omega-3 fatty acids.
With these issues in mind, Zhao et al. [170] recently described the results of studies of DHA using human iPSC-derived NPCs and post-mitotic neurons to gain insight into the molecular and cellular mechanisms that may underlie the potential beneficial therapeutic effect of DHA on synaptic plasticity and neuronal connectivity, as well as to develop functional biomarkers that could be used to guide future experimental therapeutic trials in terms of optimal dosing and schedule of administration. These studies demonstrated, for the first time in the context of a living human neuron, that exposure to DHA at physiologically relevant levels led to a dose-dependent, significant upregulation of both CREB and WNT signaling pathways along with a dose-dependent gene-expression signature. Using live-cell imaging, they also showed that DHA treatment enhanced viability of proliferating NPCs and the complexity of axonal and dendritic branching on differentiating iPSC-derived neurons [170]. These studies provided direct experimental evidence that exposure of human neuronal cells to DHA can have a potentially beneficial effect on critical pathways regulating neuroplasticity. With the growing number of approved prescription omega-3 fatty acid products being used to treat hypertriglyceridema [171] it may be possible in the near future to leverage epidemiological data from BPD patients taking these agents and be beneficial to perform trials with purified omega-3 fatty acids, alone or in combination with other agents, rather than more complex mixtures derived from marine sources.
Peroxisome proliferator-activated receptors (PPARs)
An additional target of potential relevance for BPD pharmacotherapy that connects to a different area of neurobiology through metabolism and mitochondrial function is that of the family of peroxisome proliferator-activated receptors (PPARs) and their regulation by peroxisome proliferator-activated receptor gamma coactivator-1 alpha [172]. As PPAR agonists have been developed and are clinically used as anti-diabetic agents in the case of the thiazolidines and anti-triglyceride fibrates (PPAR agonists), this has allowed both open-label and randomized, double-blind, placebo-controlled trials with promising results [173, 174]. An additional proof-of-concept trial of bezafibrate for bipolar depression coupled with resting state fMRI analysis as a marker of neuronal changes is currently being performed [172].
Psilocybin and psychedelics
The use of psilocybin, a functionally selective, serotonin 5-HT2A receptor agonist (with additional effects on other serotonin receptors), which occurs naturally in over 200 known mushroom species through biosynthesis from L-tryptophan, is an area of growing interest in the context of treatment-resistant, unipolar depression, along with a range of other neuropsychiatric conditions, including alcohol dependence and anxiety [175]. Early, albeit still small, studies suggest have shown the safety and rapid efficacy of psilocybin in patients with unipolar treatment-resistant depression [176, 177]. The psychedelic effects of psilocybin have been shown to correlate with 5-HT2A receptor occupancy [178]. Plasma levels of the pharmacologically active psilocin derived from dephosphorylation of the prodrug psilocybin, and blood oxygen-level dependent resting-state functional connectivity measured with fMRI have shown changes in depressed patients upon psilocybin administration [176]. Hence, psilocybin and its derivatives may be of interest to investigate in the context of safety and placebo-controlled, randomized trials in BPD, particularly those with depression. Interestingly, earlier placebo-controlled, randomized studies in acute BPD mania with L-tryptophan [179], the biosynthetic precursor for both serotonin (5-hydroxytryptamine) and psilocybin, suggested a beneficial effect, as did similar studies examining dietary L-tryptophan on affective behavior where decreased depressive and anxiety symptoms were observed [180].
Chronotherapeutic targets regulating biological rhythms
Disturbed sleep appears to be an early symptom of bipolar disorder [181], and growing evidence suggests that changes in sleep and daytime activity that are characteristic features of mania and depressive states are core features of BPD [182,183,184]. This is of clinical relevance as changes in sleep have been shown to be highly predictive of impending affective instability [185]. Although large-scale human genetic studies have failed to date to provide robust support for direct involvement of classical circadian genes (e.g., PER2), these clinical observations, on top of the evidence that lithium can have a beneficial effect on circadian abnormalities in BPD, in particular delaying sleeping-wake phase rhythms, suggests a potentially important area of investigation is the development of “chronotherapeutics” that seek to normalize abnormalities in biological rhythms that may be at the core of the disease pathophysiology [186,187,188].
Support for the notion that stabilizing circadian rhythms may be therapeutically beneficial in BPD has emerged from efforts to target melatonin MT1 and MT2 receptors. In a small, open-label, adjunctive study with the MT1/MT2 agonist agomelatine, within a week of treatment there was a beneficial response to more than 80% of the subjects, although multiple patients over an extended time period of one year experienced adverse effects and 4 patients who were on lithium at the same time experienced manic or hypomanic episodes [189]. These findings have been extended in additional open-label trials with similar beneficial response rates [190]. However, a more recent placebo controlled trial of adjunctive agomelatine in BPD patients with depression on either lithium or valproate failed to show benefit [191], and another randomized controlled trial was unable to demonstrate any efficacy of ramelteon, another M1/M2 agonist, as an adjunctive maintenance therapy for BPD [192]. These mixed results suggest that alternative mechanisms for modulating melatonergic transmission may need to be explored with heterogeneity of the circadian abnormalities in BPD potentially providing a barrier to translation without appropriate patient stratification.
Challenging a purely pharmaceutical-centric view to developing BPD therapeutics, but also potentially highly valuable when considered as an adjunctive treatment or for use with treatment-resistant patients, are recent advances in noninvasive approaches to BPD therapy that have come through consideration of the impact of light on depressive aspects of the disorder [188]. Most notably, exposure to morning bright light treatment for BPD depression, often as an adjunct to mood stabilizers, has been repeatedly shown to be efficacious and safe, including in two recent randomized placebo-controlled trials, as well as with studies that have extended the time period of efficacy of bright light administration the midday period [193, 194]. While there is need for careful consideration to prevent a switch to mania and there are facets that remain to be understood regarding the optimal light intensity, wavelength spectrum, duration of exposure and mechanism of action in terms of effects on the circadian system and specific neurotransmitter pathways, the noninvasive nature of such modality, the rapid onset of benefit of less than a week, and the potential for personalizing the treatment for individuals based upon their personal physiology holds much promise [186, 188].
As an alternative nonpharmacological, chromotherapeutic strategy to stabilize circadian and sleep abnormalities in BPD, efforts are underway to create virtual darkness chronotherapy involving the blocking of blue light at critical periods in the night [187, 195]. Blue light in the wavelength of 400–500 nm has been shown to activate intrinsically photosensitive retinal ganglion cells that respond to blue light due to the expression of the photopigment melanopsin that upon interaction with light signals through G-proteins to activate phospholipase C leading to opening of TRPC-type ion channels resulting in depolarization of the neuron [196]. These neurons project to neurons located within the suprachiasmatic nucleus along with other brain regions [197], and through their activity are able to block the production of melatonin [198]. Thus, following the rationale that augmenting melatonin production at critical periods may normalize the circadian and sleep abnormalities in BPD, and with the pharmacology of the melanopsin receptor encoded by the OPN4 gene at a nascent stage [199], a particularly promising strategy that has been advanced involves simply the wearing of amber colored glasses that block blue light from entering the eye. In early studies with this concept, blue light blocking glasses improved sleep in BPD patients [200]. More recently, in a pioneering study, a randomized trial in BPD patients comparing the adjunctive use of blue-light blocking glasses compared with clear glasses for a 1-week period during the evening hours that entailed use of continuous demonstrated a rapid onset of antimanic effects within 3 days [201]. In addition to the low cost (~$7.00/pair) of the glasses, and the minimal harm that could come from their use, the notion of blue light blocking glasses opens up the possibility of potential new personalized strategy for prevention of BPD through stabilizing an individual’s biological rhythm disturbances, along with other options to reduce one’s exposure to blue light [195].
Reporting summary
There remains much to be learned regarding the neurobiology of BPD, along with a clear and present need for innovation by the next generation of neuroscientist, chemical biologist, biochemists, geneticists, computational scientists, and clinicians to address this challenge. Technological developments on a number of fronts ranging from genome sequencing, patient-derived iPSC models, CRISPR/Cas9 and related genome editing and epigenome editing techniques, and novel PET imaging probes are examples of unprecedented strategies to validate novel neurotherapeutic targets to advance the treatment and prevention of BPD. Balanced against the long-term promise of these approaches is the immediate need for better therapeutic strategies: waiting for future breakthroughs in neuroscience is simply not an option for patients, families, and clinicians seeking treatment today. The elegance of these new approaches cannot distract from the clinical imperative.
When contemplating the future, and specifically in weighing the role that technical versus conceptual innovation will have in revolutionizing the treatment of complex neuropsychiatric disorders like BPD, it is worth pondering lessons from the past in fields outside that of psychiatry. One such case is a field far from psychiatry, but one that underwent the type of revolutionary change that may be needed to seriously transform our understanding and treatment of neuropsychiatric disorders: astronomy. Here, individuals like Galileo Galilei were pivotal figures for the development of the foundations of modern astronomy in embracing the new technology of the telescope to discover a seemingly infinitely expansive groups of stars and a set of planets. The long-term impact of Galileo and his contribution to society at large, however, was not that he continued to map the skies in ever-increasing detail with the latest technical advances in telescopes. Rather, it was Galilelo’s scientific challenge to geocentricism and long-standing superstitions that ultimately were the catalyst for the change in understanding our place in the cosmos. By analogy, researchers in psychiatry should carefully consider the opportunities provided by new ‘telescopes’ to overturn outdated dogma and realize the full promise of precision medicine.
References
Cade JF. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949;2:349–52.
https://enacademic.com/dic.nsf/enwiki/9778 Accessed 1 July 2020.
Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry. 2007;64:543–52.
Schioldann J. History of the introduction of lithium into medicine and psychiatry: birth of modern psychopharmacology. Adelaide: Academic Press; 1949.
Parker G, McCraw S, Hadzi-Pavlovic D, Fletcher K. Costs of the principal mood disorders: a study of comparative direct and indirect costs incurred by those with bipolar I, bipolar II and unipolar disorders. J Affect Disord. 2013;149:46–55.
Judd LL, Akiskal HS, Schettler PJ, Coryell W, Endicott J, Maser JD, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry. 2003;60:261–9.
Judd LL, Akiskal HS, Schettler PJ, Endicott J, Maser J, Solomon DA, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59:530–7.
Tohen M, Hennen J, Zarate CM Jr, Baldessarini RJ, Strakowski SM, Stoll AL, et al. Two-year syndromal and functional recovery in 219 cases of first-episode major affective disorder with psychotic features. Am J Psychiatry. 2000;157:220–8.
Osby U, Brandt L, Correia N, Ekbom A, Sparen P. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58:844–50.
Keck PE, Perlis RH, Otto MW, Carpenter D, Ross R, Docherty JP. Expert Consensus Guideline Series: Treatment of Bipolar Disorder 2004. Postgrad Med. 2004:1–120.
Hirschfeld RA, Bowden CL, Gitlin MJ, Keck PE, Perlis RH, Suppes T et al. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry. 2002;159(Suppl 4):1–11.
Suppes T, Dennehy EB, Hirschfeld RM, Altshuler LL, Bowden CL, Calabrese JR, et al. The Texas implementation of medication algorithms: update to the algorithms for treatment of bipolar I disorder. J Clin Psychiatry. 2005;66:870–86.
Sachs GS, Nierenberg AA, Calabrese JR, Marangell LB, Wisniewski SR, Gyulai L, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711–22.
Nemeroff CB, Evans DL, Gyulai L, Sachs GS, Bowden CL, Gergel IP, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906–12.
Calabrese JR, Huffman RF, White RL, Edwards S, Thompson TR, Ascher JA, et al. Lamotrigine in the acute treatment of bipolar depression: results of five double-blind, placebo-controlled clinical trials. Bipolar Disord. 2008;10:323–33.
Tohen M, Vieta E, Calabrese J, Ketter TA, Sachs G, Bowden C, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079–88.
Thase ME, Macfadden W, Weisler RH, Chang W, Paulsson B, Khan A, et al. Efficacy of quetiapine monotherapy in bipolar I and II depression: a double-blind, placebo-controlled study (the BOLDER II study). J Clin Psychopharmacol. 2006;26:600–9.
Vieta E, Calabrese JR, Goikolea JM, Raines S, Macfadden W. Quetiapine monotherapy in the treatment of patients with bipolar I or II depression and a rapid-cycling disease course: a randomized, double-blind, placebo-controlled study. Bipolar Disord. 2007;9:413–25.
Weisler RH, Calabrese JR, Thase ME, Arvekvist R, Stening G, Paulsson B, et al. Efficacy of quetiapine monotherapy for the treatment of depressive episodes in bipolar I disorder: a post hoc analysis of combined results from 2 double-blind, randomized, placebo-controlled studies. J Clin Psychiatry. 2008;69:769–82.
Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19:1–93.
Scherk H, Pajonk FG, Leucht S. Second-generation antipsychotic agents in the treatment of acute mania: a systematic review and meta-analysis of randomized controlled trials. Arch Gen Psychiatry. 2007;64:442–55.
Cosgrove VE, Kelsoe JR, Suppes T. Toward a valid animal model of bipolar disorder: how the research domain criteria help bridge the clinical-basic science divide. Biol Psychiatry. 2016;79:62–70.
Scolnick EM. The path to new therapies for schizophrenia and bipolar illness. FASEB J. 2017;31:1254–9.
Chen HM, DeLong CJ, Bame M, Rajapakse I, Herron TJ, McInnis MG, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry. 2014;4:e375.
Wang JL, Shamah SM, Sun AX, Waldman ID, Haggarty SJ, Perlis RH. Label-free, live optical imaging of reprogrammed bipolar disorder patient-derived cells reveals a functional correlate of lithium responsiveness. Transl Psychiatry. 2014;4:e428.
Madison JM, Zhou F, Nigam A, Hussain A, Barker DD, Nehme R, et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry. 2015;20:703–17.
Bavamian S, Mellios N, Lalonde J, Fass DM, Wang J, Sheridan SD, et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry. 2015;20:573–84.
Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–99.
Kim KH, Liu J, Sells Galvin RJ, Dage JL, Egeland JA, Smith RC, et al. Transcriptomic analysis of induced pluripotent stem cells derived from patients with bipolar disorder from an old order amish pedigree. PLoS ONE. 2015;10:e0142693.
Tobe BTD, Crain AM, Winquist AM, Calabrese B, Makihara H, Zhao WN, et al. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci USA. 2017;114:E4462–71.
Vizlin-Hodzic D, Zhai Q, Illes S, Sodersten K, Truve K, Parris TZ, et al. Early onset of inflammation during ontogeny of bipolar disorder: the NLRP2 inflammasome gene distinctly differentiates between patients and healthy controls in the transition between iPS cell and neural stem cell stages. Transl Psychiatry. 2017;7:e1010.
Stern S, Santos R, Marchetto MC, Mendes APD, Rouleau GA, Biesmans S, et al. Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients’ responsiveness to lithium. Mol Psychiatry. 2018;23:1453–65.
Haggarty SJ, Silva MC, Cross A, Brandon NJ, Perlis RH. Advancing drug discovery for neuropsychiatric disorders using patient-specific stem cell models. Mol Cell Neurosci. 2016;73:104–15.
Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 2018;23:2509–23.
Marton RM, Pasca SP. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 2019;30:133–43.
Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570:523–7.
NIMH Repository & Genomics Resource (https://www.nimhgenetics.org). 2020.
Insel TR. The NIMH research domain criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171:395–7.
McCoy TH Jr, Castro VM, Hart KL, Pellegrini AM, Yu S, Cai T, et al. Genome-wide association study of dimensional psychopathology using electronic health records. Biol Psychiatry. 2018;83:1005–11.
Lago SG, Tomasik J, van Rees GF, Steeb H, Cox DA, Rustogi N, et al. Drug discovery for psychiatric disorders using high-content single-cell screening of signaling network responses ex vivo. Sci Adv. 2019;5:eaau9093.
Perlis RH. Translating biomarkers to clinical practice. Mol Psychiatry. 2011;16:1076–87.
Rueckert EH, Barker D, Ruderfer D, Bergen SE, O’Dushlaine C, Luce CJ, et al. Cis-acting regulation of brain-specific ANK3 gene expression by a genetic variant associated with bipolar disorder. Mol Psychiatry. 2013;18:922–9.
Hughes T, Hansson L, Sonderby IE, Athanasiu L, Zuber V, Tesli M, et al. A loss-of-function variant in a minor isoform of ANK3 protects against bipolar disorder and schizophrenia. Biol Psychiatry. 2016;80:323–30.
Hughes T, Sonderby IE, Polushina T, Hansson L, Holmgren A, Athanasiu L, et al. Elevated expression of a minor isoform of ANK3 is a risk factor for bipolar disorder. Transl Psychiatry. 2018;8:210.
Leussis MP, Berry-Scott EM, Saito M, Jhuang H, de Haan G, Alkan O, et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol Psychiatry. 2013;73:683–90.
Gottschalk MG, Leussis MP, Ruland T, Gjeluci K, Petryshen TL, Bahn S. Lithium reverses behavioral and axonal transport-related changes associated with ANK3 bipolar disorder gene disruption. Eur Neuropsychopharmacol. 2017;27:274–88.
Zhu S, Cordner ZA, Xiong J, Chiu CT, Artola A, Zuo Y, et al. Genetic disruption of ankyrin-G in adult mouse forebrain causes cortical synapse alteration and behavior reminiscent of bipolar disorder. Proc Natl Acad Sci USA. 2017;114:10479–84.
van der Werf IM, Van Dam D, Missault S, Yalcin B, De Deyn PP, Vandeweyer G, et al. Behavioural characterization of AnkyrinG deficient mice, a model for ANK3 related disorders. Behav Brain Res. 2017;328:218–26.
Lopez AY, Wang X, Xu M, Maheshwari A, Curry D, Lam S, et al. Ankyrin-G isoform imbalance and interneuronopathy link epilepsy and bipolar disorder. Mol Psychiatry. 2017;22:1464–72.
Nelson AD, Caballero-Floran RN, Rodriguez Diaz JC, Hull JM, Yuan Y, Li J et al. Ankyrin-G regulates forebrain connectivity and network synchronization via interaction with GABARAP. Mol Psychiatry. 2018. https://doi.org/10.1038/s41380-018-0308-x.
Garza JC, Qi X, Gjeluci K, Leussis MP, Basu H, Reis SA, et al. Disruption of the psychiatric risk gene Ankyrin 3 enhances microtubule dynamics through GSK3/CRMP2 signaling. Transl Psychiatry. 2018;8:135.
Ellis LD, Soanes KH. A larval zebrafish model of bipolar disorder as a screening platform for neuro-therapeutics. Behav Brain Res. 2012;233:450–7.
Lin X, Duan X, Jacobs C, Ullmann J, Chan CY, Chen S, et al. High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology. Nat Commun. 2018;9:5142.
Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–9.
Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–8.
Freland L, Beaulieu JM. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci. 2012;5:14.
Ryves WJ, Dajani R, Pearl L, Harwood AJ. Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem Biophys Res Commun. 2002;290:967–72.
Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296:15–19.
Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–50.
De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–64.
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–77.
Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS, et al. Lithium regulates glycogen synthase kinase-3beta in human peripheral blood mononuclear cells: implication in the treatment of bipolar disorder. Biol Psychiatry. 2007;61:216–22.
Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–74.
Haggarty SJ, Singh K, Perlis RH, Karmacharya R. Wnt signaling in mood & psychotic disorders. In: Wnt signaling in development and disease: molecular mechanisms and biological functions. Vol. 29. USA: Wiley; 2014. pp. 379–392.
Haggarty SJ, Singh K, Perlis RH, Karmacharya R. Neuropsychiatric disease-associated genetic variation in the Wnt pathway. In: Wnt signaling in development and disease: molecular mechanisms and biological functions. vol. 30. USA: Wiley; 2014. pp. 393–410.
Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–104.
Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004;29:32–38.
O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–8.
Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30:1223–37.
Gould TD, Einat H, O’Donnell KC, Picchini AM, Schloesser RJ, Manji HK. Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology. 2007;32:2173–83.
Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, et al. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–36.
Pan JQ, Lewis MC, Ketterman JK, Clore EL, Riley M, Richards KR, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36:1397–11.
Harris TJ, Peifer M. Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol. 2005;15:234–7.
Gould TD, Manji HK. The Wnt signaling pathway in bipolar disorder. Neuroscientist. 2002;8:497–511.
Wexler EM, Geschwind DH, Palmer TD. Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008;13:285–92.
Zhao WN, Cheng C, Theriault KM, Sheridan SD, Tsai LH, Haggarty SJ. A high-throughput screen for Wnt/beta-catenin signaling pathway modulators in human iPSC-derived neural progenitors. J Biomol Screen. 2012;17:1252–63.
Maguschak KA, Ressler KJ. Beta-catenin is required for memory consolidation. Nat Neurosci. 2008;11:1319–26.
Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Prog Neurobiol. 2008;86:281–96.
Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. J Neurochem. 2000;75:1729–34.
Choe Y, Pleasure SJ. Wnt signaling regulates intermediate precursor production in the postnatal dentate gyrus by regulating CXCR4 expression. Dev Neurosci. 2012;34:502–14.
Snitow ME, Zanni G, Ciesielski B, Burgess-Jones P, Eisch AJ, O’Brien WT, et al. Adult hippocampal neurogenesis is not necessary for the response to lithium in the forced swim test. Neurosci Lett. 2019;704:67–72.
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–31.
Lipina TV, Kaidanovich-Beilin O, Patel S, Wang M, Clapcote SJ, Liu F, et al. Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse. 2011;65:234–48.
Hennig KM, Fass DM, Zhao WN, Sheridan SD, Fu T, Erdin S, et al. WNT/beta-catenin pathway and epigenetic mechanisms regulate the Pitt-Hopkins syndrome and schizophrenia risk gene TCF4. Mol Neuropsychiatry. 2017;3:53–71.
Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. 2015;134:97–109.
Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105.
Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–73.
O’Brien WT, Huang J, Buccafusca R, Garskof J, Valvezan AJ, Berry GT, et al. Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J Clin Invest. 2011;121:3756–62.
Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. Deletion of GSK3beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA. 2012;109:20732–7.
Latapy C, Rioux V, Guitton MJ, Beaulieu JM. Selective deletion of forebrain glycogen synthase kinase 3beta reveals a central role in serotonin-sensitive anxiety and social behaviour. Philos Trans R Soc Lond B Biol Sci. 2012;367:2460–74.
Kim W, Won SY, Yoon BJ. CRMP2 mediates GSK3beta actions in the striatum on regulating neuronal structure and mania-like behavior. J Affect Disord. 2019;245:1079–88.
Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA. 2008;105:13656–61.
Khlghatyan J, Evstratova A, Chamberland S, Marakhovskaia A, Bahremand A, Toth K, et al. Mental Illnesses-Associated Fxr1 and Its Negative Regulator Gsk3beta Are Modulators of Anxiety and Glutamatergic Neurotransmission. Front Mol Neurosci. 2018;11:119.
Del’Guidice T, Latapy C, Rampino A, Khlghatyan J, Lemasson M, Gelao B, et al. FXR1P is a GSK3beta substrate regulating mood and emotion processing. Proc Natl Acad Sci USA. 2015;112:E4610–9.
Li YC, Panikker P, Xing B, Yang SS, Alexandropoulos C, McEachern EP, et al. Deletion of glycogen synthase kinase-3beta in D2 receptor-positive neurons ameliorates cognitive impairment via nmda receptor-dependent synaptic plasticity. Biol Psychiatry. 2020;87:745–55.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–41.
Fass DM, Shah R, Ghosh B, Hennig K, Norton S, Zhao WN, et al. Effect of inhibiting histone deacetylase with short-chain carboxylic acids and their hydroxamic acid analogs on vertebrate development and neuronal chromatin. ACS Med Chem Lett. 2010;2:39–42.
Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64.
Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J Biol Psychiatry. 2012;13:458–67.
Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, Lopes-Borges J, et al. Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol. 2011;43:207–14.
Kim WY, Kim S, Kim JH. Chronic microinjection of valproic acid into the nucleus accumbens attenuates amphetamine-induced locomotor activity. Neurosci Lett. 2008;432:54–57.
Covington HE III, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–60.
Covington HE III, Vialou VF, LaPlant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett. 2011;493:122–6.
Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang YL, Hennig KM, et al. A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS ONE. 2013;8:e71323.
Covington HE III, Maze I, Vialou V, Nestler EJ. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience. 2015;298:329–35.
Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60.
Wagner FF, Zhang YL, Fass DM, Joseph N, Gale JP, Weiwer M, et al. Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem Sci. 2015;6:804–15.
An WF, Germain AR, Bishop JA, Nag PP, Metkar S, Ketterman J, et al. Discovery of Potent and Highly Selective Inhibitors of GSK3b. Bethesda (MD): Probe Reports from the NIH Molecular Libraries Program; 2010.
Georgievska B, Sandin J, Doherty J, Mortberg A, Neelissen J, Andersson A, et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem. 2013;125:446–56.
Wagner FF, Benajiba L, Campbell AJ, Weiwer M, Sacher JR, Gale JP et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci Transl Med. 2018;10:eaam8460.
Wagner FF, Bishop JA, Gale JP, Shi X, Walk M, Ketterman J, et al. Inhibitors of glycogen synthase kinase 3 with exquisite kinome-wide selectivity and their functional effects. ACS Chem Biol. 2016;11:1952–63.
Griebel G, Stemmelin J, Lopez-Grancha M, Boulay D, Boquet G, Slowinski F, et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci Rep. 2019;9:18045.
Bernard-Gauthier V, Mossine AV, Knight A, Patnaik D, Zhao WN, Cheng C, et al. Structural basis for achieving GSK-3beta inhibition with high potency, selectivity, and brain exposure for positron emission tomography imaging and drug discovery. J Med Chem. 2019;62:9600–17.
Bhat RV, Andersson U, Andersson S, Knerr L, Bauer U, Sundgren-Andersson AK. The conundrum of gsk3 inhibitors: is it the dawn of a new beginning? J Alzheimers Dis. 2018;64:S547–54.
Zhao WN. Discovery of suppressors of CRMP2 phosphorylation reveals compounds that mimic the behavioral effects of lithium on amphetamine-induced hyperlocomotion. Transl Psychiatry. 2020;10:76.
Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, et al. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem. 2010;115:974–83.
Caberlotto L, Carboni L, Zanderigo F, Andreetta F, Andreoli M, Gentile G, et al. Differential effects of glycogen synthase kinase 3 (GSK3) inhibition by lithium or selective inhibitors in the central nervous system. Naunyn Schmiedebergs Arch Pharm. 2013;386:893–903.
Bocchetta A, Bernardi F, Burrai C, Pedditzi M, Del Zompo M. A double-blind study of L-sulpiride versus amitriptyline in lithium-maintained bipolar depressives. Acta Psychiatr Scand. 1993;88:434–9.
Zheng W, Xiang YQ, Li XB, Ungvari GS, Chiu HF, Sun F, et al. Adjunctive huperzine A for cognitive deficits in schizophrenia: a systematic review and meta-analysis. Hum Psychopharmacol. 2016;31:286–95.
Ratia M, Gimenez-Llort L, Camps P, Munoz-Torrero D, Perez B, Clos MV, et al. Huprine X and huperzine A improve cognition and regulate some neurochemical processes related with Alzheimer’s disease in triple transgenic mice (3xTg-AD). Neurodegener Dis. 2013;11:129–40.
Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–9.
Atack JR, Broughton HB, Pollack SJ. Inositol monophosphatase–a putative target for Li+ in the treatment of bipolar disorder. Trends Neurosci. 1995;18:343–9.
Singh N, Halliday AC, Thomas JM, Kuznetsova OV, Baldwin R, Woon EC, et al. A safe lithium mimetic for bipolar disorder. Nat Commun. 2013;4:1332.
Barkus C, Ferland JN, Adams WK, Churchill GC, Cowen PJ, Bannerman DM, et al. The putative lithium-mimetic ebselen reduces impulsivity in rodent models. J Psychopharmacol. 2018;32:1018–26.
Tondo L, Baldessarini RJ. Antisuicidal effects in mood disorders: are they unique to lithium? Pharmacopsychiatry. 2018;51:177–88.
Singh N, Sharpley AL, Emir UE, Masaki C, Herzallah MM, Gluck MA, et al. Effect of the putative lithium mimetic ebselen on brain myo-inositol, sleep, and emotional processing in humans. Neuropsychopharmacology. 2016;41:1768–78.
Masaki C, Sharpley AL, Cooper CM, Godlewska BR, Singh N, Vasudevan SR, et al. Effects of the potential lithium-mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacol (Berl). 2016;233:2655–61.
Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381:1654–62.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address pmhe, Cross-Disorder Group of the Psychiatric Genomics C. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–82. e1411.
Hoffman GE, Bendl J, Voloudakis G, Montgomery KS, Sloofman L, Wang YC, et al. CommonMind Consortium provides transcriptomic and epigenomic data for schizophrenia and bipolar disorder. Sci Data. 2019;6:180.
Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Disco. 2013;12:581–94.
Cipriani A, Saunders K, Attenburrow MJ, Stefaniak J, Panchal P, Stockton S, et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol Psychiatry. 2016;21:1324–32.
Kabir ZD, Martinez-Rivera A, Rajadhyaksha AM. From gene to behavior: L-type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics. 2017;14:588–613.
Brunet G, Cerlich B, Robert P, Dumas S, Souetre E, Darcourt G. Open trial of a calcium antagonist, nimodipine, in acute mania. Clin Neuropharmacol. 1990;13:224–8.
Grunze H, Walden J, Wolf R, Berger M. Combined treatment with lithium and nimodipine in a bipolar I manic syndrome. Prog Neuropsychopharmacol Biol Psychiatry. 1996;20:419–26.
Pazzaglia PJ, Post RM, Ketter TA, George MS, Marangell LB. Preliminary controlled trial of nimodipine in ultra-rapid cycling affective dysregulation. Psychiatry Res. 1993;49:257–72.
Pazzaglia PJ, Post RM, Ketter TA, Callahan AM, Marangell LB, Frye MA, et al. Nimodipine monotherapy and carbamazepine augmentation in patients with refractory recurrent affective illness. J Clin Psychopharmacol. 1998;18:404–13.
Ostacher MJ, Iosifescu DV, Hay A, Blumenthal SR, Sklar P, Perlis RH. Pilot investigation of isradipine in the treatment of bipolar depression motivated by genome-wide association. Bipolar Disord. 2014;16:199–203.
Atkinson LZ, Colbourne L, Smith A, Harmer CH, Nobre AC, Rendell J, et al. The oxford study of calcium channel antagonism, cognition, mood instability and sleep (OxCaMS): study protocol for a randomised controlled, experimental medicine study. Trials. 2019;20:120.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–25.
Renthal W, Maze I, Krishnan V, Covington HE III, Xiao G, Kumar A, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–29.
Wey HY, Gilbert TM, Zurcher NR, She A, Bhanot A, Taillon BD, et al. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci Transl Med. 2016;8:351ra106.
Gilbert TM, Zurcher NR, Wu CJ, Bhanot A, Hightower BG, Kim M, et al. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. J Clin Invest. 2019;129:364–72.
Arjmand S, Behzadi M, Kohlmeier KA, Mazhari S, Sabahi A, Shabani M. Bipolar disorder and the endocannabinoid system. Acta Neuropsychiatr. 2019;31:193–201.
Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29.
McIntyre RS, Soczynska JK, Woldeyohannes HO, Miranda A, Vaccarino A, Macqueen G, et al. A randomized, double-blind, controlled trial evaluating the effect of intranasal insulin on neurocognitive function in euthymic patients with bipolar disorder. Bipolar Disord. 2012;14:697–706.
Loebel A, Xu J, Hsu J, Cucchiaro J, Pikalov A. The development of lurasidone for bipolar depression. Ann N Y Acad Sci. 2015;1358:95–104.
DelBello MP, Goldman R, Phillips D, Deng L, Cucchiaro J, Loebel A. Efficacy and safety of lurasidone in children and adolescents with bipolar I depression: a double-blind, placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 2017;56:1015–25.
Ishiyama T, Tokuda K, Ishibashi T, Ito A, Toma S, Ohno Y. Lurasidone (SM-13496), a novel atypical antipsychotic drug, reverses MK-801-induced impairment of learning and memory in the rat passive-avoidance test. Eur J Pharm. 2007;572:160–70.
Yatham LN, Mackala S, Basivireddy J, Ahn S, Walji N, Hu C, et al. Lurasidone versus treatment as usual for cognitive impairment in euthymic patients with bipolar I disorder: a randomised, open-label, pilot study. Lancet Psychiatry. 2017;4:208–17.
Harvey PD, Ogasa M, Cucchiaro J, Loebel A, Keefe RS. Performance and interview-based assessments of cognitive change in a randomized, double-blind comparison of lurasidone vs. ziprasidone. Schizophr Res. 2011;127:188–94.
Enomoto T, Ishibashi T, Tokuda K, Ishiyama T, Toma S, Ito A. Lurasidone reverses MK-801-induced impairment of learning and memory in the Morris water maze and radial-arm maze tests in rats. Behav Brain Res. 2008;186:197–207.
Horisawa T, Ishibashi T, Nishikawa H, Enomoto T, Toma S, Ishiyama T, et al. The effects of selective antagonists of serotonin 5-HT7 and 5-HT1A receptors on MK-801-induced impairment of learning and memory in the passive avoidance and Morris water maze tests in rats: mechanistic implications for the beneficial effects of the novel atypical antipsychotic lurasidone. Behav Brain Res. 2011;220:83–90.
Van Rheenen TE, Lewandowski KE, Bauer IE, Kapczinski F, Miskowiak K, Burdick KE et al. Current understandings of the trajectory and emerging correlates of cognitive impairment in bipolar disorder: An overview of evidence. Bipolar Disord. 2019;22:13–27.
Cuomo I, Piacentino D, Kotzalidis GD, Lionetto L, De Filippis S. Lacosamide in bipolar disorder: A 30-day comparison to a retrospective control group treated with other antiepileptics. Psychiatry Clin Neurosci. 2018;72:864–75.
Wilson SM, Khanna R. Specific binding of lacosamide to collapsin response mediator protein 2 (CRMP2) and direct impairment of its canonical function: implications for the therapeutic potential of lacosamide. Mol Neurobiol. 2015;51:599–609.
Fava M. The possible antianxiety and mood-stabilizing effects of rufinamide. Psychother Psychosom. 2010;79:194–5.
Mazza M, Marano G, Janiri L. Retigabine-ezogabine: a new treatment option for bipolar disorder? Bipolar Disord. 2019;21:283–4.
Amann B, Sterr A, Vieta E, Stampfer R, Walden J, Grunze H. An exploratory open trial on safety and efficacy of the anticonvulsant retigabine in acute manic patients. J Clin Psychopharmacol. 2006;26:534–6.
Dencker D, Dias R, Pedersen ML, Husum H. Effect of the new antiepileptic drug retigabine in a rodent model of mania. Epilepsy Behav. 2008;12:49–53.
Dencker D, Husum H. Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav Brain Res. 2010;207:78–83.
Kristensen LV, Sandager-Nielsen K, Hansen HH. K(v) 7 (KCNQ) channel openers normalize central 2-deoxyglucose uptake in a mouse model of mania and increase prefrontal cortical and hippocampal serine-9 phosphorylation levels of GSK3beta. J Neurochem. 2012;121:373–82.
Dedic N, Jones PG, Hopkins SC, Lew R, Shao L, Campbell JE, et al. SEP-363856, a novel psychotropic agent with a unique, non-D2 receptor mechanism of action. J Pharm Exp Ther. 2019;371:1–14.
Harmeier A, Obermueller S, Meyer CA, Revel FG, Buchy D, Chaboz S, et al. Trace amine-associated receptor 1 activation silences GSK3beta signaling of TAAR1 and D2R heteromers. Eur Neuropsychopharmacol. 2015;25:2049–61.
Karmacharya R, Lynn SK, Demarco S, Ortiz A, Wang X, Lundy MY, et al. Behavioral effects of clozapine: involvement of trace amine pathways in C. elegans and M. musculus. Brain Res. 2011;1393:91–99.
McNamara RK, Welge JA. Meta-analysis of erythrocyte polyunsaturated fatty acid biostatus in bipolar disorder. Bipolar Disord. 2016;18:300–6.
Saunders EF, Ramsden CE, Sherazy MS, Gelenberg AJ, Davis JM, Rapoport SI. Omega-3 and omega-6 polyunsaturated fatty acids in bipolar disorder: a review of biomarker and treatment studies. J Clin Psychiatry. 2016;77:e1301–8.
Grayson DS, Kroenke CD, Neuringer M, Fair DA. Dietary omega-3 fatty acids modulate large-scale systems organization in the rhesus macaque brain. J Neurosci. 2014;34:2065–74.
Zhao WN, Hylton NK, Wang J, Chindavong PS, Alural B, Kurtser I, et al. Activation of WNT and CREB signaling pathways in human neuronal cells in response to the Omega-3 fatty acid docosahexaenoic acid (DHA). Mol Cell Neurosci. 2019;99:103386.
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Engl J Med. 2019;380:11–22.
Nierenberg AA, Ghaznavi SA, Sande Mathias I, Ellard KK, Janos JA, Sylvia LG. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha as a novel target for bipolar disorder and other neuropsychiatric disorders. Biol Psychiatry. 2018;83:761–9.
Kemp DE, Schinagle M, Gao K, Conroy C, Ganocy SJ, Ismail-Beigi F, et al. PPAR-gamma agonism as a modulator of mood: proof-of-concept for pioglitazone in bipolar depression. CNS Drugs. 2014;28:571–81.
Zeinoddini A, Sorayani M, Hassanzadeh E, Arbabi M, Farokhnia M, Salimi S, et al. Pioglitazone adjunctive therapy for depressive episode of bipolar disorder: a randomized, double-blind, placebo-controlled trial. Depress Anxiety. 2015;32:167–73.
Nutt D. Psychedelic drugs-a new era inpsychiatry? Dialogues Clin Neurosci. 2019;21:139–47.
Carhart-Harris RL, Roseman L, Bolstridge M, Demetriou L, Pannekoek JN, Wall MB, et al. Psilocybin for treatment-resistant depression: fMRI-measured brain mechanisms. Sci Rep. 2017;7:13187.
Carhart-Harris RL, Bolstridge M, Day CMJ, Rucker J, Watts R, Erritzoe DE, et al. Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacol (Berl). 2018;235:399–408.
Madsen MK, Fisher PM, Burmester D, Dyssegaard A, Stenbaek DS, Kristiansen S, et al. Psychedelic effects of psilocybin correlate with serotonin 2A receptor occupancy and plasma psilocin levels. Neuropsychopharmacology. 2019;44:1328–34.
Chouinard G, Young SN, Annable L. A controlled clinical trial of L-tryptophan in acute mania. Biol Psychiatry. 1985;20:546–57.
Lindseth G, Helland B, Caspers J. The effects of dietary tryptophan on affective disorders. Arch Psychiatr Nurs. 2015;29:102–7.
Ritter PS, Marx C, Bauer M, Leopold K, Pfennig A. The role of disturbed sleep in the early recognition of bipolar disorder: a systematic review. Bipolar Disord. 2011;13:227–37.
Plante DT, Winkelman JW. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008;165:830–43.
Seleem MA, Merranko JA, Goldstein TR, Goldstein BI, Axelson DA, Brent DA, et al. The longitudinal course of sleep timing and circadian preferences in adults with bipolar disorder. Bipolar Disord. 2015;17:392–402.
Steinan MK, Scott J, Lagerberg TV, Melle I, Andreassen OA, Vaaler AE, et al. Sleep problems in bipolar disorders: more than just insomnia. Acta Psychiatr Scand. 2016;133:368–77.
Gruber J, Harvey AG, Wang PW, Brooks JO III, Thase ME, Sachs GS, et al. Sleep functioning in relation to mood, function, and quality of life at entry to the systematic treatment enhancement program for bipolar disorder (STEP-BD). J Affect Disord. 2009;114:41–49.
Wirz-Justice A, Benedetti F. Perspectives in affective disorders: Clocks and sleep. Eur J Neurosci. 2019;51:346–65.
Gold AK, Kinrys G. Treating circadian rhythm disruption in bipolar disorder. Curr Psychiatry Rep. 2019;21:14.
Maruani J, Geoffroy PA. Bright light as a personalized precision treatment of mood disorders. Front Psychiatry. 2019;10:85.
Calabrese JR, Guelfi JD, Perdrizet-Chevallier C. Agomelatine bipolar study G. Agomelatine adjunctive therapy for acute bipolar depression: preliminary open data. Bipolar Disord. 2007;9:628–35.
Fornaro M, McCarthy MJ, De Berardis D, De Pasquale C, Tabaton M, Martino M, et al. Adjunctive agomelatine therapy in the treatment of acute bipolar II depression: a preliminary open label study. Neuropsychiatr Dis Treat. 2013;9:243–51.
Yatham LN, Vieta E, Goodwin GM, Bourin M, de Bodinat C, Laredo J, et al. Agomelatine or placebo as adjunctive therapy to a mood stabiliser in bipolar I depression: randomised double-blind placebo-controlled trial. Br J Psychiatry. 2016;208:78–86.
Mahableshwarkar AR, Calabrese JR, Macek TA, Budur K, Adefuye A, Dong X, et al. Efficacy and safety of sublingual ramelteon as an adjunctive therapy in the maintenance treatment of bipolar I disorder in adults: A phase 3, randomized controlled trial. J Affect Disord. 2017;221:275–82.
Zhou TH, Dang WM, Ma YT, Hu CQ, Wang N, Zhang GY, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90–96.
Sit DK, McGowan J, Wiltrout C, Diler RS, Dills JJ, Luther J, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175:131–9.
Phelps J. A powerful non-pharmacologic treatment for mania – virtually. Bipolar Disord. 2016;18:379–82.
Do MTH. Melanopsin and the intrinsically photosensitive retinal ganglion cells: biophysics to behavior. Neuron. 2019;104:205–26.
Kim KY, Rios LC, Le H, Perez AJ, Phan S, Bushong EA, et al. Synaptic specializations of melanopsin-retinal ganglion cells in multiple brain regions revealed by genetic label for light and electron microscopy. Cell Rep. 2019;29:628–44 e626.
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP. Light suppresses melatonin secretion in humans. Science. 1980;210:1267–9.
Jones KA, Hatori M, Mure LS, Bramley JR, Artymyshyn R, Hong SP, et al. Small-molecule antagonists of melanopsin-mediated phototransduction. Nat Chem Biol. 2013;9:630–5.
Henriksen TE, Skrede S, Fasmer OB, Hamre B, Gronli J, Lund A. Blocking blue light during mania - markedly increased regularity of sleep and rapid improvement of symptoms: a case report. Bipolar Disord. 2014;16:894–8.
Henriksen TE, Skrede S, Fasmer OB, Schoeyen H, Leskauskaite I, Bjorke-Bertheussen J, et al. Blue-blocking glasses as additive treatment for mania: a randomized placebo-controlled trial. Bipolar Disord. 2016;18:221–32.
Acknowledgements
We would like to acknowledge helpful discussion and feedback from members of the Haggarty, Perlis, and Karmacharya laboratories, along with members of the Dauten Family Center for Bipolar Treatment Innovation at Massachusetts General Hospital. This work was supported through funding from the Stuart & Suzanne Steele MGH Research Scholars Program, NIH R01MH120227, NIH R01AT009144, NIH R01MH113858. This manuscript is dedicated to the compassionate staff of the Rockyview General Hospital and Foothills Medical Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SJH was or is a member of the scientific advisory board of Rodin Therapeutics, Psy Therapeutics, Frequency Therapeutics, and Souvien Therapeutics, none of whom were involved in the present study. SJH has also received speaking or consulting fees from Amgen, AstraZeneca, Biogen, Merck, Regenacy Pharmaceuticals, Syros Pharmaceuticals, Juvenescence, as well as sponsored research or gift funding from AstraZeneca, JW Pharmaceuticals, and Vesigen unrelated to the content of this manuscript. RHP is a member of the scientific advisory board of Psy Therapeutics, Outermost Therapeutics, and Genomind, and reports consulting fees from Genomind, RID Ventures, Takeda, and Burrage Capital. He holds equity in Psy Therapeutics and Outermost Therapeutics. All of these are unrelated to the content of this manuscript. RK: None.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Haggarty, S.J., Karmacharya, R. & Perlis, R.H. Advances toward precision medicine for bipolar disorder: mechanisms & molecules. Mol Psychiatry 26, 168–185 (2021). https://doi.org/10.1038/s41380-020-0831-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0831-4
- Springer Nature Limited
This article is cited by
-
Progress and Implications from Genetic Studies of Bipolar Disorder
Neuroscience Bulletin (2024)
-
A novel murine model of mania
Molecular Psychiatry (2023)
-
T cells: an emerging cast of roles in bipolar disorder
Translational Psychiatry (2023)
-
Genetic evidence for the “dopamine hypothesis of bipolar disorder”
Molecular Psychiatry (2023)
-
Impulsivity across severe mental disorders: a cross-sectional study of immune markers and psychopharmacotherapy
BMC Psychiatry (2023)