
Abstract
Lithium is the most effective and well established treatment for bipolar disorder, and it has a broad array of effects within cellular pathways. However, the specific processes through which therapeutic effects occur and are maintained in bipolar disorder remain unclear. This paper provides a timely update to an authoritative review of pertinent findings that was published in CNS Drugs in 2013. A literature search was conducted using the Scopus database, and was limited by year (from 2012). There has been a resurgence of interest in lithium therapy mechanisms, perhaps driven by technical advancements in recent years that permit the examination of cellular mechanisms underpinning the effects of lithium—along with the reuptake of lithium in clinical practice. Recent research has further cemented glycogen synthase kinase 3β (GSK3β) inhibition as a key mechanism, and the inter-associations between GSK3β-mediated neuroprotective, anti-oxidative and neurotransmission mechanisms have been further elucidated. In addition to highly illustrative cellular research, studies examining higher-order biological systems, such as circadian rhythms, as well as employing innovative animal and human models, have increased our understanding of how lithium-induced changes at the cellular level possibly translate to changes at behavioural and clinical levels. Neural circuitry research is yet to identify clear mechanisms of change in bipolar disorder in response to treatment with lithium, but important structural findings have demonstrated links to the modulation of cellular mechanisms, and peripheral marker and pharmacogenetic studies are showing promising findings that will likely inform the exploration for predictors of lithium treatment response. With a deeper understanding of lithium’s therapeutic mechanisms—from the cellular to clinical levels of investigation—comes the opportunity to develop predictive models of lithium treatment response and identify novel drug targets, and recent findings have provided important leads towards these goals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Understanding the mechanisms and disentangling the key processes that underpin therapeutic response to lithium in patients with bipolar disorder has proven to be a significant research challenge. |
Recent research has further elucidated lithium’s direct and indirect effects on neuroprotective, anti-oxidative and neurotransmission mechanisms, and glycogen synthase kinase 3β (GSK3β) inhibition is clearly fundamental to its therapeutic mechanisms. |
Uncovering the therapeutic mechanisms of lithium, and in particular the manner in which cellular modulations translate to neural and clinical outcomes, will afford opportunities to identify novel treatment targets and develop more effective and selective medications for patients with bipolar disorder. |
1 Introduction
Bipolar disorder is a common, lifelong mood disorder that poses a considerable burden upon the individual affected and society as a whole. The diagnosis of bipolar disorder gives salience to polarity even though it is recurrence that best defines the illness, and the occurrence of mania that distinguishes it from recurrent depressive disorders.
Lithium is the best and most well established treatment for bipolar disorder. It is particularly effective in maintaining mood stability and preventing mood episodes, and when deemed appropriate is prescribed for life. It is thought to exert its therapeutic, mood stabilising effects by acting on cellular targets and by modulating function within neural pathways (as reviewed previously in Malhi et al. [1]) but its effects are evident at all levels of brain function spanning from clinical phenomenology through functional and structural neural changes to neurochemical and molecular processes (see Fig. 1). However, the specific processes through which its therapeutic effects are effected and maintained remain unclear.

The potential therapeutic effects of lithium can be examined at multiple levels of brain function, from cellular and intracellular changes within neurons and supporting glial cells to neural networks and neurocognition that ultimately results in a spectrum of clinical presentations. In order to understand how neuroprogression at the cellular level results in changes in clinical signs and symptoms, the many intermediate changes at the neurotransmission, neurocircuitry and mood dysregulation and neurocognitive dysfunction levels need to be considered. Based on Malhi and colleagues [1]
In recent years, bipolar disorder has been increasingly conceptualised as an illness that is the result of a step-wise progressive neural deterioration that occurs through a number of cellular pathways. With each episode of illness there is a biological insult that further enhances the likelihood of recurrence. This concept of ‘neuroprogression’ is informative, but it is essentially speculative and remains contentious in the context of bipolar disorder [2–4]. Extrapolating this hypothesis further, it is conceivable that the therapeutic mechanisms of lithium produce ‘neuroprotective’ effects, achieved both directly and indirectly (e.g. [5]). However, elucidating the precise pathophysiology of bipolar disorder and meaningfully dissecting the therapeutic mechanisms of lithium continue to be difficult tasks, for a number of reasons. First, the illness by its very nature is complex because of the many processes involved, which makes the application of diagnostic criteria and segregation of the disorder from its many comorbidities exceedingly difficult—all of which is often further compounded by the use of multiple treatments in clinical practice—added successively as the illness evolves over time (see Fig. 2). Second, translation between levels of understanding—cell to clinical presentation—and between cellular, animal and human models is conceptually and practically difficult, especially when aiming to ascertain clinically meaningful findings. The difficulties are immediately evident when attempting to observe and compare mechanisms at each of these levels of brain function even in pure illness states—namely, mania and depression—and periods of relative mood stability (euthymia). Hence, a more mechanistic as opposed to diagnosis-based approach is needed, perhaps along the lines of the research approach recommended by the NIMH Research Domain Criteria (RDoc) project [6]. Equally, this approach may usefully apply within treatment nomenclature, with treatments being defined by their mechanism rather than on the basis of the ill-defined conditions they are employed to treat [7].

A framework for understanding and disentangling the potential therapeutic actions of lithium from the pathophysiology that determines the course and pattern of illness in bipolar disorder. The schematic represents the theoretical pathophysiology of bipolar disorder as modified by lithium therapy, so as to provide a conceptual template upon which targeted investigations can be conducted. Through experimental manipulation of lithium administration in patients that have been robustly phenotyped and by identifying key mechanisms at each level of understanding (as previously depicted in Fig. 1), within the context of longitudinal research studies—within-subject elucidation of the mechanisms underpinning therapeutic responses to lithium in bipolar disorder may be possible. a Pathophysiology. The pathophysiology of bipolar disorder is presumably in train by the time the illness manifests clinically (transition from purple to white). The precise timing of this with respect to specific pathophysiological mechanisms is unknown but is likely to be at the very latest concurrent. However, the processes that determine a pathophysiological pathway and early maladaptive changes in neural networks are likely to precede full blown clinical presentation. Notably, subsequent to illness onset, any one mechanism is inevitably subject to multiple influences (such as from comorbidities), many of which are likely to compound the pathophysiology of the illness further. Hence, inevitably, the confidence with which the precise nature of the pathophysiology of the illness can be gauged diminishes as it takes hold and progresses. With lithium administration (red), pathophysiological mechanisms change further and characterization of these becomes even more complicated, thus further diminishing the confidence with which the specificity of the changes can be reliably inferred. b Overlapping mechanisms. In order to disentangle the pathophysiological changes that occur in bipolar disorder with lithium treatment, it is necessary to identify component mechanisms according to putative aetiology. These individual mechanisms can be conceptualised as closely bound strands that are intrinsically interrelated but perhaps still separable—with observation over time. But at the same time, differentiating changes in individual component mechanisms over time may enable interrogation of specific therapeutic actions of lithium from those not affected by lithium. However, complicating matters further, the pathophysiology of bipolar disorder is likely not static and evolves as the illness unfolds and treatments are administered. b.1 Mechanisms not modified by lithium. Broadly speaking, some component mechanisms are largely unaltered by lithium administration. These maintain a relatively stable course and collectively represent premorbid functioning (1.1), clinical status and health (1.2) and gradual cumulative illness burden (1.3). The time course of change in these components is relatively slow (years and decades). b.2 Mechanisms modified by lithium. In response to lithium, specific component mechanisms undergo change: improvement, impairment or change in stability. The component mechanisms that improve with lithium response are likely to be a composite of direct effects of the element and those because of overall clinical improvement (2.1). These are likely to occur across overlapping but distinguishable periods. By contrast, changes in otherwise stable components are likely to reflect the impact of adverse effects (2.2) and deterioration in illness. Speculatively, the variability of some component mechanisms may change because of changes in factors that govern mood stability, representing a core therapeutic action of lithium (2.3). The likelihood that many of the changes in component mechanisms are intricately interrelated makes attempts to separate them a tremendous challenge. Adapted with permission from Malhi and colleagues [112]
Beguilingly, lithium is a simple element that has very specific molecular properties, but because of its ubiquity and multiple actions, it produces a diverse range of both positive and negative clinical effects, both short- and long-term, across many different conditions. Due to its diverse effects, research examining the mechanisms of lithium at one specific time point may contradict findings at a separate time point.
Approximately one-third of patients with bipolar I disorder using current criteria can expect to remain clinically well for many years (often decades) on lithium monotherapy. This profound prophylactic effect allows patients to maintain function and good psychological health for long periods of their lives. Referred to as ‘excellent lithium responders’, treating psychiatrists attempt to identify such patients early in the course of illness, and ideally from the outset. Patients who manifest a ‘classic’ pattern of recurrent episodic manic–depressive illness separated by periods of remission, with no comorbid psychiatric disturbances, and have immediate family members who are also ‘excellent’ lithium responders are likely to benefit from lithium therapy and remain well [8]. It therefore follows that in more narrowly defined or ‘classic’ bipolar disorder illness subtypes, a much higher proportion of patients are likely to be lithium responsive, and as such the mechanisms underpinning the effects of lithium are likely to differ when compared with other bipolar disorder and mood disorder subtypes. This is a major problem in lithium research when attempting to classify patients in accordance with the extant classification systems, which in modern times do not adequately capture the ‘classic’ (manic–depressive) subtype [9–12]. However, a continued research focus on the therapeutic mechanisms of lithium will likely facilitate the development of much needed clinically and therapeutically meaningful classification systems, as well as increase investigation of potential differences in pathophysiology between lithium responsive and non-lithium responsive bipolar illness subtypes [13].
Both the tasks of identifying lithium responders and determining therapeutic response are difficult, with no objective biological measures available to assist. Lithium management requires careful monitoring and ongoing adjustment, and clinicians employ trial-and-error and wait-and-see approaches, determining satisfactory therapeutic response retrospectively on the basis of a lack of breakthrough episodes over time. However, given that heritable factors and modification of therapeutic factors appear to play a role in treatment response, research examining both intra- and inter-individual predictive factors may provide beneficial leads towards developing prognostic models concerning lithium therapy. Identifying the pathways through which lithium produces therapeutic effects, from the cell to behaviour, is paramount to understanding lithium therapy and attenuating the difficulty with which it is administered and managed.
The BALANCE (Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder) study in 2010 [14] and some recent naturalistic studies (e.g. [15]), in conjunction with strong recommendations for use in clinical practice guidelines, has led to a resurgence of interest in lithium both in clinical practice and research. Technical advancements and coalescence of basic science strands has meant that research into the cellular mechanisms of lithium response has flourished in recent years (see Fig. 3), prompting this update of our previous CNS Drugs review [1]. The current review focuses on findings concerning the role of brain-derived neurotropic factor (BDNF), glycogen synthase kinase 3 (GSK3β) inhibition and oxidative metabolism as putative targets of lithium mechanisms. In addition, new findings in relation to neurotransmission and cellular signal transduction pathways have been integrated along with research on lithium and circadian rhythms, animal and human models of therapeutic response to lithium and pharmacogenetic findings. With many of these findings, the manner in which heritable factors play a role in therapeutic mechanisms is increasingly coming to light, even though how changes to cellular signal transduction translate to alterations in clinical presentation, particularly in the long-term, remain unknown. Hence, this article aims to provide an updated review of the effects of lithium therapy on neuroprogression across key levels of brain functioning ranging from cellular signal transduction mechanisms to clinical phenomenology (see Fig. 1). Consequently, the article constructs an overarching and updated picture of lithium’s therapeutic mechanisms, and highlights those aspects which may offer accessible leads for the development of predictive models of therapeutic responses to lithium.

The rate at which articles exploring lithium’s mechanism of action have been published in recent years has increased. The line shows the cumulative number of papers identified by the literature search conducted for this review, by year of publication (without limitation on year of publication)
1.1 Method
In order to provide an update, a comprehensive literature search of peer-reviewed publications, written in English and limited by the dates 2012–2016, was conducted using Scopus.Footnote 1 The search syntax used to identify relevant articles was ‘(TITLE (lithium)) AND (TITLE-ABS-KEY (‘neurotransmission’ OR ‘dopamine’ OR ‘glutamate’ OR ‘gamma-aminobutyric acid’ OR ‘NMDA’ OR ‘G-protein’ OR ‘second messenger systems’ OR ‘adenylyl cyclase’ OR ‘phosphoinositide’ OR ‘inositol’ OR ‘protein kinase C’ OR ‘myristoylated alanine-rich C kinase substrate’ OR ‘intracellular calcium’ OR ‘brain derived neurotrophic factor’ OR ‘apoptosis’ OR ‘oxidative stress’ OR ‘mitochondria’ OR ‘bcl-2’ OR ‘glycogen synthase kinase 3’ OR ‘autophagy’ OR ‘gene’)) AND (LIMIT-TO (PUBYEAR, 2016) OR LIMIT-TO (PUBYEAR, 2015) OR LIMIT-TO (PUBYEAR, 2014) OR LIMIT-TO (PUBYEAR, 2013) OR LIMIT-TO (PUBYEAR, 2012))’. Relevant findings were then identified and synthesised in combination with earlier and extant literature (from Malhi et al. [1]).
2 Cellular Mechanisms
Characterising the specific cellular mechanisms that are modulated by lithium is critical to understanding and developing models of clinical response, as well as for advancing future drug discovery. However, linking the cellular mechanisms that underpin putative illness processes and lithium’s therapeutic mechanisms across various cellular, animal and human models, and then relating these back to functions in humans remains a necessarily difficult task (as depicted in Fig. 2). Hence, it is important to consider the many new findings regarding second messenger systems and neuroprotective pathways that have provided much needed evidence and appear to be strengthening existing knowledge. In order to coalesce these recent findings with earlier findings, we have synthesised and updated depictions of the various cellular mechanisms in Fig. 4a–c. A major pathophysiological concept is that of excitotoxicity—the process through which over-excitation of cells results in deleterious by-products causing dysfunction within the cell—which results in significant disruption to the extent that it eventually destroys the cell through processes of programmed cell death, such as apoptosis [3]. Over-excitation of cells may occur by a number of means including through increased excitatory neurotransmission, decreased inhibitory neurotransmission, or both; which is thought to be a major aspect of psychiatric disorder pathophysiology. Lithium is thought to reduce excitotoxicity through diverse direct and indirect pathways.

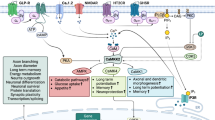
The effects of lithium on cellular mechanisms and neurotransmission. The inhibitory actions of lithium are depicted by red lines and its facilitatory actions are depicted by green arrows; and for illustrative purposes, the links between these mechanisms and higher-order modulatory biological systems (circadian rhythm and HPA axis) are depicted in orange boxes. a GSK3β target and its downstream actions. Lithium is a direct inhibitor of GSK3β, which is potentially one of its primary actions, and which mediates a number of neuroprotective actions. GSK3β inhibition occurs with phosphorylation of GSKβ through a Ca2+channel-mediated pathway, and also with inhibition of Akt phosphorylation. Following this, a cascade of DNA expression then produces neuroprotective proteins though neurotrophic (BDNF, GDNF) and growth factor pathways (Bcl-2, IGF) and cellular structural changes including remyelination though B-catenin and astrocyte protection, along with anti-oxidative molecules (glutathione), thus preventing lipid perioxidisation, and inhibition of pro-inflammatory factors (including IL-6) and direct (mPTP) and indirect (AY1R-p53, NRF2) anti-apoptotic actions, some of which occur through improved mitochondrial function. GSK3β phosphorylation modulates expression of clock genes, providing a link to the circadian rhythm system. Note, GSK3β inhibition alone does not produce the full therapeutic effect, other direct and indirect targets are involved. b The phosphoinositide cycle. Lithium inhibits the phosphoinositide cycle by upregulating ImPase 1, which ameliorates mitochondrial dysfunction and also mediates GSK3β inhibition through a DAG-PKC-MARCKS pathway. b.i The PI cycle is activated following stimulation of the cell surface receptor by a neurotransmitter. PLC mediates the hydrolysis of PIP2 to the secondary messengers DAG and IP3. These then activate downstream signalling pathways. ImPase and IPPase facilitate recycling of IP3 back into mI, which then allows the PI cycle to continue. Lithium inhibits cellular mI by (1) blocking the reuptake of inositol via inhibition of the SMIT and (2) via direct inhibition of IPPase and ImPase. Overall, this results in the inhibition of transmembrane signalling, improved mitochondrial function and prevention of apoptosis. b.ii In particular, apoptosis is reduced by decreased intracellular Ca2+ release from the mitochondria by reduced IP3. The mTOR protein is activated by lithium and is a negative regulator of autophagy and therefore inhibits this process. Lithium-induced depletion of IP3 also induces autophagy; however, its inhibitory effects through the mTOR protein are more potent. Changes to PKC mediates expression in the adrenal glands, thus providing a link to the HPA axis. c The AC/cAMP system. Lithium modulates this system in several ways: initially, basal levels of AC and cAMP are increased. Consequently, when a cell is stimulated, large fluctuations of AC and cAMP that would normally occur are minimised, therefore stabilizing the system. The CREB transcription factor is an important downstream target of the AC system and is activated by lithium though inhibition of cAMP and facilitation of CREB co-activators, which then facilitates expression of neuroprotective and anti-oxidative molecules. d Neurotransmission. Lithium modulates pre-synaptic and post-synaptic neurotransmission. Lithium inhibits glutamate neurotransmission and decreases phosphorylation of subunits of NMDA receptors. In terms of dopaminergic neurotransmission, lithium prevents dysregulated dopamine release and thus decreases metabolism resulting in reactive oxygen species (ROS). Decreased G-protein coupled dopamine stimulation manifests GSK3β inhibition through β-arrestin 2. Excitation due to G-protein-coupled reception is regulated by lithium, through regulation of G-protein-gated potassium channels. Along with inhibited excitatory neurotransmission, lithium’s effects on inhibitory GABAergic transmission are apparent but not as potent. Lithium’s modulation of Acet and Gly neurotransmission are also potential therapeutic mechanisms. AC adenyl cyclase, Acet acetylcholine, Akt phos. protein kinase B phosphorylation, Astro. astrocytes, AT1R angiotensin II type 1 receptor, Bcl-2 B-cell lymphoma 2, BDNF brain-derived neurotrophic factor, Ca 2+ Chan calcium ion channel, cAMP cyclic adenosine monophosphate, Clock Circadian clock genes and circadian rhythm modulation, CREB cAMP response element binding, DA dopamine, DAG diaglycerol, DNA deoxyribonucleic acid, GDNF glial cell line-derived neurotrophic factor, GSK3B glycogen synthase kinase-3β, Glu glutamine, Gly glycine, HPA hypothalamic–pituitary–adrenal axis, IGF insulin growth factor, IL-6 interleukin 6, ImPase inositol monophosphate 1-phosphatase, Ins inositol, IP inositol phosphate, IPPase inositol phosphate 1-phosphatase, IP2 inositol bisphosphate, IP3 inositol triphosphate, K + Chan potassium ion channel, Li + lithium ion, Lipid periox. lipid perioxidisation, MARCKS myristoylated alanine-rich c kinase substrate, mI myoinositol, mPTP mitochondrial permeability transition pore, mTOR mammalian target of rapamycin, NRF2 nuclear factor E2-related factor 2, p53 tumour protein p53, PI phosphoinositide, PIP2 phosphoinositol 4-5-biphosphate, PKC protein kinase C, PLC phospholipase C, Proinflam. proinflammation, ROS reactive oxygen species, SMIT sodium myo-inositol transporter
2.1 Neuroprotective Pathways Mediated by Direct Inhibition of Glycogen Synthase Kinase 3β (GSK3β)
Lithium has a suite of effects on a number of neuroprotective pathways through its direct inhibition of GSK3β (see Fig. 4a). GSK3β is involved in gene transcription and thus has many effects on neuroprotective cellular mechanisms. Recent studies show that GSK3β inhibition modifies neural systems (e.g. [16]) and is critical to clinical responses to lithium [17–20]. However, a recent study [21] investigating the actions of two selective GSK3β inhibitors in rats revealed that GSK3β inhibition alone does not produce lithium-like behavioural and cellular effects, but instead produces normalisation of cellular and behavioural effects of excitotoxicity. Additionally, GSK3β inhibition alone did not increase BDNF levels, whereas lithium administration did [21]. This suggests that more than GSK3β inhibition alone likely mediates therapeutic responses to lithium and that at least this one action results in neuroprotective effects. Recent studies have examined the manner in which GSK3β inhibition is modulated by lithium treatment. These studies show that lithium clinical response is predicted by GSK3β gene expression [17] and phosphorylation [18, 19]. In a recent study of platelets collected from bipolar disorder patients, lithium increased phosphorylated GSK3β (GSK3β proteins that have been ‘turned off’, perhaps through Akt activation) and this increase was found to correlate with clinical improvement of depression symptoms [19]. A study with gene knockout mice has shown that phosphorylation of GSK3β by lithium occurs via the activation of a Ca2+ permeable cation channel-mediated pathway (see Fig. 4a) and behavioural results suggest this action may underpin lithium’s anti-manic effects [18]. In the suprachiasmatic nucleus of mice, GSK3β inhibition with lithium restores circadian rhythm gene expression (see Fig. 4a; [22]). But lithium’s actions on GSK3β are modulatory and to achieve GSK3β inhibition, a recent study on rat hippocampal slices has shown that lithium also has bimodal indirect actions: one of inhibition of RAC-alpha serine/threonine-protein kinase (Akt) phosphorylation and the other via β-catenin [23], perhaps through cyclic adenosine monophosphate (cAMP) response element binding (CREB) effectors (see Fig. 4a). Further, through wider multi-protein complexes, lithium increases Akt activity, which then phosphorylates GSK3β to inhibit its activity. This action of lithium has been shown in the mouse striatum, through modulation of dopaminergic cell function [24]. Similarly, in order to illustrate neuroprotective effects along this Akt-GSK3β pathway, a recent cell culture study demonstrated that lithium attenuated dephosphorylation of the Akt-GSK3β-mammalian target of rapamycin (mTOR) pathway after methamphetamine insult [25].
2.1.1 Brain-Derived Neurotropic Factor (BDNF)
BDNF is an essential protein that governs therapeutic changes to neural structures. In a recent cell culture study, neurons exposed to chronic but not acute lithium treatment had increased BDNF; providing a possible explanation for the 6- to 10-day delay in therapeutic response observed clinically [26]. Independently, glial cell line-derived neurotrophic factor (GDNF) increases with lithium in astrocyte cultures. Therefore, the neuroprotective effects of lithium are perhaps not limited to neurons and extend to astrocytes [26]. However, in a study of patients with bipolar disorder, GDNF levels were negatively correlated with lithium levels in euthymia, contrary to BDNF levels which were positively correlated [27]. Interestingly, this did not hold during mania, in which GDNF and lithium levels were positively correlated. This suggests that lithium perhaps has differential effects on neurotropic factors depending on the degree of excitotoxicity reflected by mood. A recent finding that lithium normalises acute amphetamine-induced mania in rats, but does so without altering BDNF levels [28], partially supports this suggestion. In a model of hippocampal degeneration, lithium increased BDNF and protected cells as well as ameliorated depressive behavioural deficits [29]. Similarly, lithium improved BDNF levels in a model of cognitive deficit in rats [30]. This shows that lithium acts to prevent cellular degeneration that may underpin dysfunction in mood disorder, through upregulation of BDNF. This may be explained in part by protection against acute excitotoxic insult, whereby increased synthesis and mRNA expression of BDNF abolishes the potential toxic effects of any future insult [31]. Conversely, antidepressants have been shown to exert neuroprotective effects by increasing BDNF levels in patients, and lithium augments this process [32]. The complexity of lithium response and its effects on BDNF is perhaps explained in part by variants on the BDNF gene’s promoter IV region, which promotes the expression of BDNF [33]. Clearly, this modulatory role needs to be better understood given that in hippocampal neurons, this action protects against excitatory glutamate toxicity [34].
2.1.2 Oxidative Metabolism
Recent work has shown that lithium reverses and repairs oxidative stress exacted by excitotoxicity in rats [35]. The excitotoxic effects of excessive dopamine transmission are a consequence of dopamine metabolism, which produces reactive oxygen species. In a recent study, lithium reversed and prevented oxidative stress by increasing the antioxidant glutathione in the striatum and the prefrontal cortex [36]. Specifically, lithium reverses dysfunction in DNA methylation—a process which typically represses gene transcription—caused by oxidative stress by-products [37]. A recent study with excellent lithium responders and family members suggests that increases in glutathione may be involved in this process [38], perhaps via the regulation of glutathione perioxidase precursor gene expression [39].
New findings using cell lines show that chronic lithium treatment reduces the expression of stress proteins, explaining its long-term neuroprotective effects [40]. Additionally, inhibition of GSK3β with lithium reduces proinflammatory molecules that result from acute neurotoxin exposure [41]. Further, lithium attenuates immune responses to stress by regulating interleukin 6 and methane metabolism pathways [42]. Lithium inhibits oxidative stress in healthy controls, suggesting that lithium has specific anti-oxidative effects regardless of condition [43]. In cell lines, a recent study has shown that lithium increased cell viability under oxidative stress conditions and that, when combined with haloperidol, lithium did not have this protective effect [44]. In the haloperidol-only arm of the study, cells were also not protected. This suggests that anti-oxidative actions are specific to lithium and may not necessarily occur when used in combination with antipsychotics.
Replicating earlier findings [45], a study in bipolar disorder patients showed that lithium decreases lipid peroxidation levels, and that this effect is associated with clinical response [46]. Decreases in lipid peroxidation attenuate disruption to protein function, and in a recent study in rats, lithium prevented disruption to the vesicular monoamine transporter 2 protein (VMAT2) in frontal cortex neurons, which is critical to forming vesicles for neurotransmission [47]—a finding that has implications for understanding behavioural changes with lithium treatment.
A recent study has demonstrated that lithium also ameliorates mitochondrial dysfunction in patients with bipolar disorder, thus reversing the effects of oxidative stress [48]. Novel findings show that nuclear factor E2-related factor 2 (NRF2) mediates the expression of genes involved in oxidative cytoprotection [49]. A recent study has elucidated a novel lithium neuroprotective mechanism whereby NRF2 is activated and microRNA-34a (miR-34a) is suppressed, which in turn protects against apoptosis [50].
Similarly, recent findings have shown that activation of the brain angiotension II type 1 receptor (AT1R), which leads to oxidative and inflammatory activity, is likely involved in deleterious effects of mania [51]. Promisingly, the AT1R antagonist candesartan has lithium-like neuroprotective effects, raising the possibility of new mood-stabilising agents targeting this receptor [51].
2.1.3 Apoptosis
Apoptosis is the process of cell death caused by signal transduction. Recent findings demonstrate that lithium prevents apoptosis in the hippocampus, through neuroprotective and anti-inflammatory pathways [52]. It does so by modulating GSK3β, which regulates the mitochondrial permeability transition pore (mPTP), which can trigger cell death [53] and tumour protein p53 (p53), which acts to signal apoptosis following DNA damage [54], such as that brought about by oxidative stress.
B-cell lymphoma 2 (Bcl-2) is another key regulator of apoptosis. In addition to increasing Bcl-2 in neurons, chronic lithium administration increases Bcl-2 in astrocytes, which are also critical to neuronal survival [55]. Bcl-2 gene (BCL2L1) expression appears to differentiate lithium responders from non-responders [56], and therefore lithium response may be defined by survival of neurons and their supporting cell structures. Interestingly, valproate also has anti-apoptotic properties, suggesting that mood stabilisation may occur with decreases in apoptosis [57]. When cells are exposed to toxic insult, lithium preserves mitochondrial function and prevents the accumulation of reactive oxygen species, and thereby stalls cell apoptosis [58].
2.1.4 Other Protective Factors
As identified previously [1], the effect of lithium on other neurotrophic factors has been studied to a lesser extent, but some recent studies have investigated this potential mechanism. For example, insulin-like growth factor (IGF) appears to play a role in overall lithium responsiveness, with cell lines from lithium non-responders showing increased lithium sensitivity when IGF is added [59]. Furthermore, IGF-1 is over-expressed in lithium responders relative to non-responders [60], and it is known to be a cell survival factor that prevents apoptosis.
2.1.5 Cell Structure and Glia
In recent years, there has been an increase in research interest in lithium’s effects on neuronal structures and glia, which serve to maintain brain tissue integrity [61]. A recent study in mice treated with lithium demonstrated increased numbers of neurons and glial cells and increased astrocyte density in the hippocampus, compared with control mice [62]. However, it is uncertain whether this would translate to effects in humans. Additionally, the fact that research employing different cell imaging methods has shown null results for the generation of neurons is problematic [63]. The neuroprotective effect of lithium on astrocytes specifically is likely to be an independent effect, with recent findings showing GDNF increasing with lithium in astrocyte cultures [26]. Separately, myelination is an important aspect of neuroprotection and neuroplasticity, and lithium works to stimulate remyelination through β-catenin and CREB effectors, and thus can act on reparation of demyelinated pathways [64]. In addition to myelination stimulation, lithium reduces axon length; increases axonal spreading; and increases neural growth, size, and branching through a β-catenin pathway [65]; as well as actin remodelling of dendrites [66]. Recent findings suggest that the inhibition of GSK3β by lithium plays a significant role in reparation of axon and myelin integrity in white matter tracts in those with bipolar disorder [16], and that lithium also increases expression of proteins involved in dopaminergic and glutamatergic pathways [67]. Interestingly, lithium also reduces neuroinflammation by inhibiting microglial pro-inflammatory cytokines [68], and thus regulates microglia interactions with neurons [69].
2.2 Second Messenger Systems
Although the cascade effects of GSK3β inhibition are extensive, GSK3β inhibition alone does not account for the therapeutic effects of lithium; there are, in addition, a number of important second messenger systems involved (see Fig. 4b, c).
2.2.1 The Phosphoinositide Cycle, Protein Kinase C (PKC) and Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) and Intracellular Ca2+
2.2.1.1 The Phosphoinositide Cycle
Inositol depletion produces mitochondrial dysfunction and is thought to be involved, at least in part, in the pathophysiology of bipolar disorder. Lithium is thought to inhibit inositol monophosphatase 1 (ImPase 1; see Fig. 4b), which in turn ameliorates inositol depletion-related mitochondrial dysfunction by reregulating the phosphoinositide cycle and reducing inositol triphosphate (IP3) levels. This is important as IP3 has previously been shown to be a direct effector of autophagy [70]. Examining the potential mechanisms underpinning therapeutic effects using an animal model, upregulation of ImPase 1 [71] in addition to lithium treatment after inositol depletion [72] results in gene expression and behavioural changes, suggesting a potential role of the phosphoinositide cycle in the therapeutic effects of lithium administration [71]. In a recent study of gene knockout mice, the links between inositol metabolism and the effects of lithium on behavioural assays were demonstrated [73]. Translating to humans, in a recent magnetic resonance spectroscopy study on bipolar disorder patients, remitters had increased myo-inositol levels after lithium treatment in comparison with non-remitters [74]. Following these leads, ImPase is being examined as a potential therapeutic drug target [75], and recent research examining the ImPase inhibitor ebselen is further elucidating the role of this target in therapeutic responses [76, 77]. The SESTD1 gene, which is involved in the regulation of phospholipids—including components within the phosphoinositide cycle—has been found to be associated with lithium response in a clinical study [78]. Although the inositol depletion hypothesis of bipolar disorder pathophysiology still needs to overcome a number of conceptual challenges (e.g. inositol dietary availability), more specified accounts of the roles of ImPase and IP3 are providing important, clinically relevant insights.
2.2.1.2 PKC and MARCKS
Protein kinase C (PKC) inhibition underpins reparative neuronal plasticity after dopaminergic hyperexcitation and is thought to be one of lithium’s essential mechanisms. New findings show that lithium acts to inhibit PKC through an intricate myristoylated alanine-rich C kinase substrate (MARCKS) pathway, initiated by its inhibition of GSK3β ([79]; see Fig. 4b.i). Hence, PKC is being investigated as a potential therapeutic drug target [80]. Interestingly, because of PKC inhibition, lithium has also been considered to be a regulator of expression of corticotrophins in the adrenal glands [81]. However, previous studies in patients with affective disorders stabilised on lithium have shown limited effect on attenuation of intermittent hypothalamic-pituitary-adrenal (HPA) axis dysregulation, using the dexamethasone suppression test paradigm [82]. Hence, future prospective studies should investigate the role of lithium in attenuating chronic HPA axis dysregulation.
2.2.1.3 Intracellular Ca2+
Intracellular Ca2+ homeostasis is central to the maintenance of cellular functioning during excitation (e.g. dopaminergic signalling). In bipolar disorder, Ca2+ homeostasis is impaired and results in apoptosis. Recent findings show that lithium downregulates the transient receptor potential channel 3 (TRPC3), thus modulating Ca2+ disturbances ([83] see Fig. 4b.ii). Recent research is linking the temporal dynamics of Ca2+ with lithium’s effects on circadian rhythm [84]. Although lithium restores calcium signalling, it is of note that variants of the L-type calcium channel (LTCC) genes modulate lithium’s amplification of circadian rhythm, potentially explaining variability in the therapy’s effect on circadian rhythm [84].
2.2.2 The Adenyl Cyclase (AC) and Cyclic Adenosine Monophosphate (cAMP) System
Adenyl cyclase (AC) and cyclic adenosine monophosphate (cAMP) are activated with excitatory neurotransmission, but in bipolar disorder the sensitivity of the system is reduced because of hyperexcitation. A recent study [85] has shown that lithium enhances cAMP-induced CREB-dependent gene transcription (see Fig. 4c). Additionally, lithium but not aripiprazole decreases phosphorylation of CREB in the prefrontal cortex in rats, potentially counteracting neural plasticity deficits due to stress [86]. Specifically, lithium supports CREB co-activators, which enhances cAMP-induced CREB-dependent gene transcription (see Fig. 4c). Finally, recent findings in cell lines from patients, and both affected and unaffected relatives show that abnormalities in phosphorylated CREB signalling may be related to bipolar disorder and lithium responsiveness, suggesting a role for heritable factors [87].
3 Neurotransmission
Lithium is known to modulate neurotransmission, pre-synaptically and post-synaptically (see Fig. 4d). Dopamine, glutamate and GABA were highlighted previously. New findings are further examining the manner in which lithium achieves this, as well as indicating possible effects lithium has on cholinergic and glycinergic function. Along with studies that tease apart mechanisms that change with lithium administration, it is also important to find mechanisms that do not change in order to determine specificity. For example, a recent study has demonstrated that functions within presynaptic vesicles, at least in hippocampal neurons, are not directly affected by acute or chronic lithium treatment [88]. However, the involvement of presynaptic effects in the mechanisms of lithium cannot be ruled out entirely given that synapsins, which regulate vesicles and make them available for neurotransmitter release, are implicated in lithium response [89]. Therefore, in sum, the manner in which lithium-induced neurotransmission changes lead to specific clinical effects remains unknown (as depicted in Fig. 2).
3.1 Glutamate and NMDA Receptors
Glutamate is an excitatory neurotransmitter. In patched rat hippocampal neurons, recent research has shown that lithium acts preferentially on presynaptic glutamate terminals (see Fig. 4d), to inhibit excitatory postsynaptic currents [90]. In cell lines, lithium increases the active regulatory elements such as the enhancers and promoters of genes involved in glutamate neurotransmission [91]. Translating these findings to bipolar disorder patients, lithium decreases glutamate levels depending on lithium plasma concentration [92]. Post-synaptically, lithium decreases phosphorylation of subunits of NMDA receptors in the prefrontal cortex and ventral striatum ([86]; see Fig. 4d) and decreases NMDA-induced cytoskeletal deterioration [66]. This is important because it has been previously found that the effects of lithium on NMDA receptor activation also have indirect long-term consequences for enhancing serotonergic and reducing dopaminergic neurotransmission, which in turn inactivates the NMDA receptor [93]. Interestingly, in a study of bipolar disorder patients on lithium, glutamate decarboxylase-like protein (GADL1) gene variants predicted good response [94]; however, these findings have not been replicated by other groups [78, 95] and some have suggested that this gene is involved more so in kidney function (which is important for maintaining lithium therapeutic levels) than neuroprotective or therapeutic effects per se [96].
3.2 Dopamine and G-Protein Coupled Receptors
Lithium prevents excessive dopamine release in the prefrontal cortex in mice, underpinning its behavioural effects [97]. However, a recent study with mice treated with a dopamine transporter inhibitor demonstrated attenuation, but not complete reversal, of manic-like behaviour with lithium [98]. In terms of anhedonic behaviour, however, lithium reverses impaired dopaminergic activity underpinning this behaviour in rats [99]. These findings suggest regulation of dopaminergic function is an important therapeutic mechanism of lithium (see Fig. 4d), and further recent research has demonstrated the manner in which lithium achieves this. Another pathway through which lithium can inhibit GSK3β and produce antimanic and antidepressant behavioural changes is the dopamine 2 receptor (D2) and its associated G-protein signalling pathway through β-arrestin 2 ([100]; see Fig. 4c). Furthermore, a recent study has reported that the glucagon-like peptide-1 receptor (GLP1R), which is a G-protein coupled receptor, is involved in lithium-mediated regulation of dopamine release [101]. This is important because excitation due to G-protein coupled stimulation is regulated by lithium, through regulation of G protein-gated potassium channels ([102]; see Fig. 4d).
3.3 GABA and GABA Receptors
GABA is an inhibitory neurotransmitter. In the Wakita et al. [90] study, lithium’s effects on GABAergic transmission were apparent but not as potent as its effects on inhibition of excitatory glutamate neurotransmission (see Fig. 4d). This suggests that lithium’s net inhibition of neural excitation perhaps occurs more so via regulation of excitatory glutamatergic neurotransmission as opposed to inhibitory GABAergic neurotransmission.
3.4 Acetylcholine and Glycine
In addition to further addressing dopaminergic, glutamatergic, and GABAergic systems, recent findings indicate that lithium has other potentially therapeutic effects on acetylcholine and glycine neurotransmission. For example, a recent study showed that, in addition to attenuation of manic-like behaviour in mice through dopaminergic pathways, lithium also attenuates depression-like behaviour through cholinergic pathways [98]. In terms of glycine neurotransmission, it is interesting that glycine transporters on the neural cell surface are expressed differentially with lithium administration, through a mechanism of GSK3β inhibition [103].
4 Higher-Order Modulatory Biological Systems
In recent years, there has been an increase in findings linking lithium’s basic cellular changes with consequential changes to higher-order biological systems involved in the pathophysiology of bipolar disorder, such as circadian rhythms and the HPA axis. However, the specificity of these treatment-related modulations to bipolar disorder illness pathophysiology itself, as opposed to other mood disorder subtypes, is still unknown (as depicted in Fig. 1).
4.1 Circadian Rhythm
Recent studies investigating the link between lithium administration and amelioration of circadian rhythm disturbances have found that lithium enhances the resynchronisation of rhythms [104] through the modulation of clock gene expression [105]. In fact, recent studies have shown that circadian components are essential for typical lithium response. In knockout mice, circadian components were important in mediating lithium’s effects on mood behaviours [106]. Additionally, chronic lithium modulates expression of certain clock genes [107], and gene variants are associated with lithium response [108]. Lithium stimulates transcription of clock genes through stimulation of expression promoters [109] and, notably, GSK3β phosphorylation (see Fig. 4a) in the suprachiasmatic nucleus [22].
4.2 Hypothalamic–Pituitary–Adrenal (HPA) Axis
Recent findings show lithium’s cellular effects may have consequences for activation of the HPA axis. Specifically, lithium’s effect on PKC regulates the expression of corticotrophins in the human adrenal glands ([81]; see Fig. 4b), and lithium mediates stress reactivity via ankyrin 3 (Ank3) regulation of corticosterone. This is important as corticosteroids are thought to play an important role in the pathogenesis of bipolar disorder [110].
5 Neurocircuitry and Neurocognition
Bipolar disorder is associated with dysfunctional neural mechanisms that underpin and drive neurocognitive processes [111], and it is in this context the effects of lithium on cellular and neurotransmission mechanisms need to be considered (see Fig. 5). A recent review examined prominent models and outlined the key neural networks and neuropsychological processes putatively involved in mood disorders [111]. As such, a major consideration in this type of research is the involvement of illness burden factors, such as medical and psychiatric comorbidities. Given the limited data thus far (as reviewed elsewhere [111, 112]), longitudinal studies on bipolar disorder and illness burden factors are required in order to better delineate the effects of lithium on neurocircuitry and neurocognition. One particular aspect that is of particular importance is the normalisation of the cognitive and emotional neural networks implicated in bipolar disorder by the effects of lithium [1, 113]. Investigating lithium’s effects on functional networks is necessarily difficult and therefore advancement of our understanding is slow and remains limited as the question continues to be understudied. Nevertheless, it is an important field and predicting treatment response using fMRI along with other clinical and biological parameters is likely to be of use prognostically [113]. This is partly supported by promising findings from recent structural imaging studies that continue to show the normalising effects of lithium treatment on grey matter abnormalities in bipolar disorder [114–116], and the possibility of identifying structural imaging-based response markers [117]. Furthermore, these effects of lithium are likely to be specific and appear to be unique to the element. For example, a recent study showed that lithium administration in rats specifically increases cortical grey matter, in contrast to the effects of the antipsychotic haloperidol which decreases it [118]. Intriguingly and importantly, when treatment was withdrawn, haloperidol-induced reductions were normalised, while the lithium-induced increases were maintained. This suggests that the effects of lithium are longer term, pointing to its neuroprotective actions; however, this needs to be robustly verified and also tested in different clinical populations in order to determine the specificity of lithium’s therapeutic effects but also the specificity for bipolar disorder illness. Neurocircuitry studies are also now linking cellular neuroprotective mechanisms to macro-level changes in the brain. Specifically, through aforementioned GSK3β-mediated neurotrophic pathways, lithium plays a major role in repairing white matter tracts [16] and preserving grey matter [119] in patients with bipolar disorder. Each of these effects may contribute to amelioration of the functional integrity of neural networks [5, 120, 121]. Additionally, recent work has shown decreases in myo-inositol in the hippocampus [122] and anterior cingulate cortex [74] of patients undergoing lithium treatment, suggesting that lithium has putative neuroprotective effects in key cognitive and emotional regions. Further, recent structural MRI studies of hippocampal volumes have shown protective effects of lithium in bipolar disorder patients [5, 121], building on earlier population studies of reduced dementia risk in psychiatric patients treated with lithium [123, 124].

Translating understanding from cellular and neurotransmission levels to neurocircuitry and neurocognition. Through lithium’s actions on cellular and neurotransmission mechanisms (blue circle), amelioration of neurocircuitry (yellow circle) and neurocognitive dysfunction underpinning mood dysregulation (red circle) and clinical presentation arises (purple circle). In particular, the structure and function of neurocircuitry and regions involved in cognition and emotion will change with lithium therapy, consistent with improved clinical presentation. Lithium’s neuroprotective and anti-apoptotic actions and modulation of neurotransmission improves the structural and functional integrity of frontal, limbic and striatal regions. With the use of novel human cellular models (black circle; e.g. drawing inferences from iPSC [induced pluripotent stem-cell]), researchers may be able to investigate therapeutic responses to lithium through each level of understanding—from cell to clinical presentation—simultaneously, and over time. DA dopamine, Glu glutamine, Li + lithium ion
Taken together, these recent advances have mapped important links between key cellular mechanisms and neurocircuitry and neurocognition; however, further research that identifies the functional and clinical implications of these is needed—using, for example, functional imaging studies on patients that have been robustly phenotyped. In order to continue to disentangle the mechanisms that underpin therapeutic responses to lithium from the pathophysiology of bipolar disorder, research will need to also examine intermediary aspects such as neurotransmission together with the aforementioned higher order systemic mechanisms.
6 Summarising Lithium’s Effects on Neuroprotection and Neurotransmission
In sum, there have been many new research findings that have contributed to a greater understanding of the potential therapeutic mechanisms of lithium treatment. Lithium’s action of GSK3β inhibition and its suite of consequences are being mapped with ever-increasing detail, and findings regarding its neuroprotective effects seem to be particularly promising for the development of therapeutic response markers in bipolar disorder. However, disentangling the effects of lithium from those of the underlying illness remains challenging (as depicted in Fig. 2), and this limits our ability to translate findings across levels of understanding (as depicted in Fig. 1). At the same time, a stronger appreciation of these mechanisms affords the opportunity to determine predictive models of lithium treatment response and identify novel drug targets, and recent findings have certainly provided important new leads towards achieving this goal.
7 Towards Predictive Models of Treatment Response
In parallel to the new research on neuroprotection, neurotransmission, and neurocircuitry mechanisms underpinning the effects of lithium, there has been a surge of studies developing and testing animal and human models. These models have essentially shown that behaviours and mental state changes associated with bipolar disorder can be reversed with lithium administration, possibly through the modulation of cellular mechanisms.
7.1 Animal Models
A mixed-state model in mice based on circadian rhythm knock out has been developed, and may be useful in exploring lithium non-response [106]. Already in animal models of lithium responders, heterozygous GSK3B gene knock-out mice have been shown to have differential behavioural and molecular responses to chronic lithium administration [125].
Some of the more specific behavioural models of mania in animals are derived from the effects of amphetamine. In these, lithium reverses manic-like locomotor activity, through partial reversal of neurodegenerative [28, 80] and anti-oxidative [35, 36] activity, largely in the prefrontal cortex. Additionally, manic-like reward stimulation is attenuated by lithium through changes to CREB and glutamatergic activation [86].
Similarly, a neurotoxic trimethyltin model of depression has been recently developed through degeneration of hippocampal neurons; this may also serve as a model of progressive neurodegeneration [29]. In this model, in rats, lithium protects hippocampal neurons from degeneration through up-regulation of BDNF and down-regulation of tumour necrosis factor, and decreases depression-related locomotor immobility in the forced swim and tail suspension tests [29]. In addition, animal models of chronic stress-induced anhedonia show restoration of dopaminergic reward pathways with lithium administration [99]. Related to this, in a genetically prone model of depression in rats, lithium has restorative effects on hippocampal DNA expression processes [126].
Bipolar disorder is associated with a range of neurocognitive deficits, some of which may be protected from further deterioration, worsened, or reversed with lithium treatment. A recent review suggests that lithium potentially acts on psychomotor and processing speed, and verbal learning, memory, and fluency, though the specific effects of the illness are difficult to disentangle [112]. Working along these lines, researchers have recently created an animal model of induced cognitive deficit [30]. In this model, lithium normalises learning and memory impairment, attenuates oxidative stress, and up-regulates BDNF levels [30]. These findings now need to be replicated in bipolar disorder patients.
7.2 Human Models
7.2.1 Cellular Models
Given that the cellular pathways involved in the actions of lithium are being increasingly characterised, activation of these pathways is now being studied in humans in vivo. In a study of cell lines from bipolar disorder patients, activation of pathways are apparent from the first dose [127], and abnormalities in phosphorylated CREB signalling may be related to lithium response [87]. Additionally, recent work has shown that levels of phosphorylated GSK3β in platelets was associated with clinical improvement in depression symptoms [19]. Interestingly, recent work has shown that offspring of excellent lithium responders undergoing lithium treatment have greater BDNF levels and lower IL-6 levels than offspring of partial or non-responders [128]. Such findings offer promising leads for detecting and predicting lithium responses from an early stage in treatment.
The emergence of induced pluripotent stem-cell (iPSC) technology is also providing novel and exciting avenues for biological human models of bipolar disorder [129] and lithium therapy [130]. Specifically, Mertens and colleagues [130] induced hippocampal-like neuron cell lines from stem cells collected from patients with bipolar disorder. These cells were hyperexcitable relative to healthy controls, putatively providing a working biological model of bipolar disorder. Astonishingly, when the cells were treated with lithium, only those derived from lithium responders showed a reversal of hyperexcitability. Future investigation along these lines may afford opportunities to further examine the mechanisms that underlie clinical lithium treatment responses as well as the discovery of novel drug targets and methods to predict response. Essentially, these techniques could be applied prospectively in different patient groups, including excellent lithium responders, and across different mood disorder subtypes, while treatments are co-administered clinically. This will enable researchers to examine therapeutic response to lithium through each level of understanding, simultaneously, and over time (as depicted in Fig. 5). These developments are likely to enable the necessary investigations for further understanding and disentangling of the potential therapeutic effects of lithium from those caused by the illness course.
In terms of novel treatments arising from human model research, some researchers are suggesting that stem cells themselves may have therapeutic application in this area [61]. Another emerging line of human model research is that of lithium mimetics; for example, with ebselen [131], which may allow researchers to disentangle the therapeutic mechanisms of lithium and to explore targets with greater specificity.
7.2.2 Pharmacogenetic Models
Since the demonstration that excellent lithium response may be inherited [132], researchers have been examining pharmacogenetic models of lithium response. More recently, it has been shown that lithium responsiveness in parents may be a heritable trait related to the developmental trajectory of certain mental disorders in their offspring [133]. Aside from this, and now extrapolating from the aforementioned cellular research, it is becoming evident that gene variability of many of the targets along the neuroprotective pathways is implicated in lithium therapeutic outcomes. These include variants of the GSK3β gene [16, 17, 20] and GSK3β-modulating genes [18], BDNF [33] and Bcl-2 [56] genes, but also the serotonin transporter-linked polymorphic region (5-HTTLPR) in bipolar depression [134] and prophylaxis [135]. Such findings provide important leads for gene–brain–behaviour models, and form the necessary foundation for constructing links from cellular changes to neurocircuitry functioning and ultimately clinical manifestations. In this vein, recent studies have shown that GSK3B gene variants are associated with lithium-induced changes to white matter tracts [16] and grey matter volumes [136]. In order to capture many of the interactive cellular and systemic changes that occur with lithium, a recent study has examined lithium response explained by candidate gene variants across GSK3β, phosphoinositol, HPA axis, and glutamatergic pathways [137]. The study in bipolar disorder found that genetic variants related to GSK3β (GSK3B), phosphoinositol (INPP1; IMPA2), and glutamatergic (GRIK2) pathways were associated with lithium response, while the candidate HPA axis-related genes were not associated with responsivity (not FKBP5, CRHR2 nor CRHR1). However, these have not been well replicated [78, 95]. In a large genome-wide association study, lithium response was associated with two long non-coding RNA genes, which are thought to be important regulators of gene expression in the central nervous system [95]. Together with clinical parameters and understanding of functional neurocircuitry, pharmacogenetic analysis can provide important leads towards more complete models of bipolar disorder and its treatment with lithium, through characterisation of illness and treatment processes at each level of investigation.
8 Future Directions
Here we have reviewed recent literature pertaining to the potential therapeutic mechanisms of lithium treatment on cellular mechanisms, neurotransmission, and the higher-order modulatory systems, which has provided new insights and has further refined understanding. Further research should focus on examining the consequences of change within cellular mechanisms on neurotransmission, in order to ascertain the manner in which pre-synaptic and post-synaptic neurotransmission is modified through second messenger and neuroprotective pathways. A surge in animal and human model research, along with the development of human iPSC models (e.g. [130]), is allowing the development of predictive models of cellular modification with putative links to behavioural modification. For example, this kind of research has promise for providing links to illness subtypes and responsiveness, the effects of lithium withdrawal on different affective disorder clinical subtypes, and therapeutic effects in other presentations such as psychosis within which lithium exhibits some benefit. In order to understand the intermediary processes involved in these therapeutic changes, research efforts need to be directed towards understanding functional neurocircuitry and neurocognition (see Fig. 5). These aspects have been relatively understudied, even though structural studies have shown promising findings. Thus, it is important to examine the impact of structural changes on function.
With an increase in clinical applicability of more sophisticated models and the opportunity to map them longitudinally, researchers will ideally be able to identify correlates of clinical markers, including neuroimaging, and peripheral and genetic markers. Future studies should aim to track modifications over time through each level of understanding, from first dose, titration and initial response, to longer-term treatment outcomes, looking for predictors of response that indicate changes supporting therapeutic processes at each time point—especially now that there are promising avenues to begin doing so with iPSC. Insights from such research will enable deeper understanding and afford opportunities to better intervene. In addition to the continued exploration of treatment targets and their specificity, reverse engineering of therapeutic mechanisms may permit novel management strategies, new drug target discovery, and adjunctive treatment methodologies.
9 Conclusions
Since our previous CNS Drugs review [1], there has been a surge in interest in the potential cellular mechanisms underpinning therapeutic responses to lithium treatment. In particular, through an increase in animal and human model research, there is a greater appreciation of the association between these cellular mechanisms and behavioural outcomes. Recent research has unearthed greater complexity within the overall picture showing an increased number of mediators and inter-relations; however, at the same time, the specificity of lithium’s mechanisms is becoming clearer. Recent findings have shown that GSK3β inhibition with lithium mediates many of the previously determined cellular and now higher-order biological mechanisms, and the sophistication with which excitatory neurotransmission is dampened to a greater extent than inhibitory neurotransmission is an indication of the subtlety of effects delivered by this deceptively simple element. While there are opportunities to further understand the underpinning intricate cellular mechanisms, research into the intermediate effects on neural circuitry and clinical neurocognitive function will allow for further development of predictive human models for lithium response. This will facilitate the clinical translation of findings and ultimately improve the targeted use of lithium in the management of mood disorders.
Notes
Literature search was conducted on March 7, 2016.
References
Malhi GS, Tanious M, Das P, Coulston CM, Berk M. Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs. 2013;27:135–53.
Berk M. Neuroprogression: pathways to progressive brain changes in bipolar disorder. Int J Neuropsychopharmacol. 2009;12:441–5.
Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. 2011;35:804–17.
Frank E, Nimgaonkar VL, Phillips ML, Kupfer DJ. All the world’s a (clinical) stage: rethinking bipolar disorder from a longitudinal perspective. Mol Psychiatry. 2015;20:23–31.
Hajek T, Bauer M, Simhandl C, Rybakowski J, O’Donovan C, Pfennig A, et al. Neuroprotective effect of lithium on hippocampal volumes in bipolar disorder independent of long-term treatment response. Psychol Med. 2014;44:507–17.
Insel TR. The NIMH Research Domain Criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry. 2014;171:395–7.
Zohar J, Stahl S, Möller H-J, Blier P, Kupfer D, Yamawaki S, et al. A review of the current nomenclature for psychotropic agents and an introduction to the neuroscience-based nomenclature. Eur Neuropsychopharmacol. 2015;25:2318–25.
Gershon S, Chengappa KR, Malhi GS. Lithium specificity in bipolar illness: a classic agent for the classic disorder. Bipolar Disord. 2009;11:34–44.
Malhi GS, Porter RJ. Are ‘buy-polar’ forces and “try-polar” thinking expanding bipolarity? Aust N Z J Psychiatry. 2014;48:697–700.
Malhi GS, Berk M. Diagnosing bipolar disorder: defining thresholds and setting boundaries. Aust N Z J Psychiatry. 2014;48:500–4.
Malhi GS, Das P, Gessler D, Outhred T, Fritz K. The mixed features of DSM-5. Aust N Z J Psychiatry. 2015;49:842–3.
Sara GE, Malhi GS. Trends in diagnosis of bipolar disorder: have the boundaries changed? Aust N Z J Psychiatry. 2015;49:1021–8.
Malhi GS, Geddes JR. Carving bipolarity using a lithium sword. Br J Psychiatry. 2014;205:337–9.
Geddes JR, Goodwin GM, Rendell J, Azorin J-M, Cipriani A, Ostacher MJ, et al. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375:385–95.
Simhandl C, König B, Amann BL. A prospective 4-year naturalistic follow-up of treatment and outcome of 300 bipolar I and II patients. J Clin Psychiatry. 2014;75:254–62 (quiz 263).
Benedetti F, Bollettini I, Barberi I, Radaelli D, Poletti S, Locatelli C, et al. Lithium and GSK3-β promoter gene variants influence white matter microstructure in bipolar disorder. Neuropsychopharmacology. 2013;38:313–27.
Iwahashi K, Nishizawa D, Narita S, Numajiri M, Murayama O, Yoshihara E, et al. Haplotype analysis of GSK-3β gene polymorphisms in bipolar disorder lithium responders and nonresponders. Clin Neuropharmacol. 2014;37:108–10.
Jang Y, Lee SH, Lee B, Jung S, Khalid A, Uchida K, et al. TRPM2, a susceptibility gene for bipolar disorder, regulates glycogen synthase kinase-3 activity in the brain. J Neurosci. 2015;35:11811–23.
de Sousa RT, Zanetti MV, Talib LL, Serpa MH, Chaim TM, Carvalho AF, et al. Lithium increases platelet serine-9 phosphorylated GSK-3β levels in drug-free bipolar disorder during depressive episodes. J Psychiatr Res. 2015;62:78–83.
Lin Y-F, Huang M-C, Liu H-C. Glycogen synthase kinase 3β gene polymorphisms may be associated with bipolar I disorder and the therapeutic response to lithium. J Affect Disord. 2013;147:401–6.
Caberlotto L, Carboni L, Zanderigo F, Andreetta F, Andreoli M, Gentile G, et al. Differential effects of glycogen synthase kinase 3 (GSK3) inhibition by lithium or selective inhibitors in the central nervous system. Naunyn Schmiedeberg’s Arch Pharmacol. 2013;386:893–903.
Kinoshita C, Miyazaki K, Ishida N. Chronic stress affects PERIOD2 expression through glycogen synthase kinase-3β phosphorylation in the central clock. NeuroReport. 2012;23:98–102.
Tian N, Kanno T, Jin Y, Nishizaki T. Lithium potentiates GSK-3β activity by inhibiting phosphoinositide 3-kinase-mediated Akt phosphorylation. Biochem Biophys Res Commun. 2014;450:746–9.
Harrison LM, Muller SH, Spano D. Effects of the Ras homolog Rhes on Akt/protein kinase B and glycogen synthase kinase 3 phosphorylation in striatum. Neuroscience. 2013;236:21–30.
Wu J, Zhu D, Zhang J, Li G, Liu Z, Sun J. Lithium protects against methamphetamine-induced neurotoxicity in PC12 cells via Akt/GSK3β/mTOR pathway. Biochem Biophys Res Commun. 2015;465:368–73.
Emamghoreishi M, Keshavarz M, Nekooeian AA. Acute and chronic effects of lithium on BDNF and GDNF mRNA and protein levels in rat primary neuronal, astroglial and neuroastroglia cultures. Iran J Basic Med Sci. 2015;18:240–6.
Tunca Z, Ozerdem A, Ceylan D, Yalçın Y, Can G, Resmi H, et al. Alterations in BDNF (brain derived neurotrophic factor) and GDNF (glial cell line-derived neurotrophic factor) serum levels in bipolar disorder: the role of lithium. J Affect Disord. 2014;166:193–200.
Stertz L, Fries GR, Aguiar BW de, Pfaffenseller B, Valvassori SS, Gubert C, et al. Histone deacetylase activity and brain-derived neurotrophic factor (BDNF) levels in a pharmacological model of mania. Rev Bras Psiquiatr. 2014;36:39–46.
Moghadas M, Edalatmanesh MA. Protective effect of lithium chloride against trimethyltin-induced hippocampal degeneration and comorbid depression in rats. Comp Clin Pathol. 2014;24:1165–75.
Sharma S, Taliyan R. Synergistic effects of GSK-3β and HDAC inhibitors in intracerebroventricular streptozotocin-induced cognitive deficits in rats. Naunyn Schmiedeberg’s Arch Pharmacol. 2014;388:337–49.
Croce N, Mathé AA, Gelfo F, Caltagirone C, Bernardini S, Angelucci F. Effects of lithium and valproic acid on BDNF protein and gene expression in an in vitro human neuron-like model of degeneration. J Psychopharmacol. 2014;28:964–72.
Ricken R, Adli M, Lange C, Krusche E, Stamm TJ, Gaus S, et al. Brain-derived neurotrophic factor serum concentrations in acute depressive patients increase during lithium augmentation of antidepressants. J Clin Psychopharmacol. 2013;33:806–9.
Hing B, Davidson S, Lear M, Breen G, Quinn J, McGuffin P, et al. A polymorphism associated with depressive disorders differentially regulates brain derived neurotrophic factor promoter IV activity. Biol Psychiatry. 2012;71:618–26.
Dwivedi T, Zhang H. Lithium-induced neuroprotection is associated with epigenetic modification of specific BDNF gene promoter and altered expression of apoptotic-regulatory proteins. Front Neurosci. 2015;8:525.
da-Rosa DD, Valvassori SS, Steckert AV, Ornell F, Ferreira CL, Lopes-Borges J, et al. Effects of lithium and valproate on oxidative stress and behavioral changes induced by administration of m-AMPH. Psychiatry Res. 2012;198:521–6.
Macêdo DS, de Lucena DF, Queiroz AIG, Cordeiro RC, Araújo MM, Sousa FC, et al. Effects of lithium on oxidative stress and behavioral alterations induced by lisdexamfetamine dimesylate: relevance as an animal model of mania. Prog Neuropsychopharmacol Biol Psychiatry. 2013;43:230–7.
Toledano E, Ogryzko V, Danchin A, Ladant D, Mechold U. 3′-5′ Phosphoadenosine phosphate is an inhibitor of PARP-1 and a potential mediator of the lithium-dependent inhibition of PARP-1 in vivo. Biochem J. 2012;443:485–90.
Huzayyin AA, Andreazza AC, Turecki G, Cruceanu C, Rouleau GA, Alda M, et al. Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychopharmacol. 2014;17:561–9.
Farah R, Khamisy-Farah R, Amit T, Youdim MBH, Arraf Z. Lithium’s gene expression profile, relevance to neuroprotection A cDNA microarray study. Cell Mol Neurobiol. 2013;33:411–20.
Nciri R, Desmoulin F, Allagui MS, Murat J-C, Feki AE, Vincent C, et al. Neuroprotective effects of chronic exposure of SH-SY5Y to low lithium concentration involve glycolysis stimulation, extracellular pyruvate accumulation and resistance to oxidative stress. Int J Neuropsychopharmacol. 2013;16:365–76.
Wang H-M, Zhang T, Li Q, Huang J-K, Chen R-F, Sun X-J. Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem Int. 2013;63:345–53.
Watanabe S, Iga J, Nishi A, Numata S, Kinoshita M, Kikuchi K, et al. Microarray analysis of global gene expression in leukocytes following lithium treatment. Hum Psychopharmacol. 2014;29:190–8.
Khairova R, Pawar R, Salvadore G, Juruena MF, de Sousa RT, Soeiro-de-Souza MG, et al. Effects of lithium on oxidative stress parameters in healthy subjects. Mol Med Rep. 2012;5:680–2.
Gawlik-Kotelnicka O, Mielicki W, Rabe-Jabłońska J, Lazarek J, Strzelecki D. Impact of lithium alone or in combination with haloperidol on oxidative stress parameters and cell viability in SH-SY5Y cell culture. Acta Neuropsychiatrica. 2016;28:38–44.
Banerjee U, Dasgupta A, Rout JK, Singh OP. Effects of lithium therapy on Na+-K+-ATPase activity and lipid peroxidation in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2012;37:56–61.
de Sousa RT, Zarate CA, Zanetti MV, Costa AC, Talib LL, Gattaz WF, et al. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J Psychiatr Res. 2014;50:36–41.
Tan H, Young LT, Shao L, Che Y, Honer WG, Wang J-F. Mood stabilizer lithium inhibits amphetamine-increased 4-hydroxynonenal-protein adducts in rat frontal cortex. Int J Neuropsychopharmacol. 2012;15:1275–85.
de Sousa RT, Streck EL, Zanetti MV, Ferreira GK, Diniz BS, Brunoni AR, et al. Lithium increases leukocyte mitochondrial complex I activity in bipolar disorder during depressive episodes. Psychopharmacology. 2015;232:245–50.
Rizak J, Tan H, Zhu H, Wang JF. Chronic treatment with the mood-stabilizing drug lithium up-regulates nuclear factor E2-related factor 2 in rat pheochromocytoma PC12 cells in vitro. Neuroscience. 2014;256:223–9.
Alural B, Ozerdem A, Allmer J, Genc K, Genc S. Lithium protects against paraquat neurotoxicity by NRF2 activation and miR-34a inhibition in SH-SY5Y cells. Front Cell Neurosci. 2015;9:209.
de Souza Gomes JA, de Souza GC, Berk M, Cavalcante LM, de Sousa FCF, Budni J, et al. Antimanic-like activity of candesartan in mice: possible involvement of antioxidant, anti-inflammatory and neurotrophic mechanisms. Eur Neuropsychopharmacol. 2015;25:2086–97.
Liechti FD, Stüdle N, Theurillat R, Grandgirard D, Thormann W, Leib SL. The mood-stabilizer lithium prevents hippocampal apoptosis and improves spatial memory in experimental meningitis. PLoS One. 2014;9:e113607.
Tanno M, Kuno A, Ishikawa S, Miki T, Kouzu H, Yano T, et al. Translocation of glycogen synthase kinase-3β (GSK-3β), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J Biol Chem. 2014;289:29285–96.
Ngok-Ngam P, Watcharasit P, Thiantanawat A, Satayavivad J. Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells. Cell Mol Biol Lett. 2013;18:58–74.
Keshavarz M, Emamghoreishi M, Nekooeian AA, Warsh J, Zare HR. Increased bcl-2 protein levels in rat primary astrocyte culture following chronic lithium treatment. Iran J Med Sci. 2013;38:255–62.
Beech RD, Leffert JJ, Lin A, Sylvia LG, Umlauf S, Mane S, et al. Gene-expression differences in peripheral blood between lithium responders and non-responders in the Lithium Treatment-Moderate dose Use Study (LiTMUS). Pharmacogenomics J. 2013;14:182–91.
Gupta A, Schulze TG, Nagarajan V, Akula N, Corona W, Jiang X-Y, et al. Interaction networks of lithium and valproate molecular targets reveal a striking enrichment of apoptosis functional clusters and neurotrophin signaling. Pharmacogenomics J. 2012;12:328–41.
Hou L, Xiong N, Liu L, Huang J, Han C, Zhang G, et al. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015;16:82.
Milanesi E, Hadar A, Maffioletti E, Werner H, Shomron N, Gennarelli M, et al. Insulin-like growth factor 1 differentially affects lithium sensitivity of lymphoblastoid cell lines from lithium responder and non-responder bipolar disorder patients. J Mol Neurosci. 2015;56:681–7.
Squassina A, Costa M, Congiu D, Manchia M, Angius A, Deiana V, et al. Insulin-like growth factor 1 (IGF-1) expression is up-regulated in lymphoblastoid cell lines of lithium responsive bipolar disorder patients. Pharmacol Res. 2013;73:1–7.
Ferensztajn-Rochowiak E, Rybakowski JK. The effect of lithium on hematopoietic, mesenchymal and neural stem cells. Pharmacol Rep. 2016;68:224–30.
Rajkowska G, Clarke G, Mahajan G, Licht CM, van de Werd HJ, Yuan P, et al. Differential effect of lithium on cell number in the hippocampus and prefrontal cortex in adult mice: a stereological study. Bipolar Disord. 2016;18:41–51.
Kara N, Narayanan S, Belmaker RH, Einat H, Vaidya VA, Agam G. Chronic lithium treatment enhances the number of quiescent neural progenitors but not the number of DCX-positive immature neurons. Int J Neuropsychopharmacol. 2015;18:pyv003–3.
Meffre D, Massaad C, Grenier J. Lithium chloride stimulates PLP and MBP expression in oligodendrocytes via Wnt/β-catenin and Akt/CREB pathways. Neuroscience. 2015;284:962–71.
Nciri R, Boujbiha MA, Jbahi S, Allagui MS, Elfeki A, Vincent C, et al. Cytoskeleton involvement in lithium-induced SH-SY5Y neuritogenesis and the role of glycogen synthase kinase 3β. Aging Clin Exp Res. 2015;27:255–63.
Calabrese B, Halpain S. Lithium prevents aberrant NMDA-induced F-actin reorganization in neurons. NeuroReport. 2014;25:1331–7.
de Bartolomeis A, Tomasetti C, Cicale M, Yuan P-X, Manji HK. Chronic treatment with lithium or valproate modulates the expression of Homer1b/c and its related genes Shank and Inositol 1,4,5-trisphosphate receptor. Eur Neuropsychopharmacol. 2012;22:527–35.
Dong H, Zhang X, Dai X, Lu S, Gui B, Jin W, et al. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J Neuroinflamm. 2014;11:140.
Yu Z, Ono C, Aiba S, Kikuchi Y, Sora I, Matsuoka H, et al. Therapeutic concentration of lithium stimulates complement C3 production in dendritic cells and microglia via GSK-3 inhibition. Glia. 2015;63:257–70.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11.
Damri O, Sade Y, Toker L, Bersudsky Y, Belmaker RH, Agam G, et al. Molecular effects of lithium are partially mimicked by inositol-monophosphatase (IMPA)1 knockout mice in a brain region-dependent manner. Eur Neuropsychopharmacol. 2015;25:425–34.
Shtein L, Agam G, Belmaker RH, Bersudsky Y. Inositol-deficient food augments a behavioral effect of long-term lithium treatment mediated by inositol monophosphatase inhibition: an animal model with relevance for bipolar disorder. J Clin Psychopharmacol. 2015;35:175–7.
Toker L, Bersudsky Y, Plaschkes I, Chalifa-Caspi V, Berry GT, Buccafusca R, et al. Inositol-related gene knockouts mimic lithium’s effect on mitochondrial function. Neuropsychopharmacology. 2014;39:319–28.
Machado-Vieira R, Gattaz WF, Zanetti MV, de Sousa RT, Carvalho AF, Soeiro-de-Souza MG, et al. A longitudinal (6-week) 3T (1)H-MRS study on the effects of lithium treatment on anterior cingulate cortex metabolites in bipolar depression. Eur Neuropsychopharmacol. 2015;25:2311–7.
Agam G, Almog O. Calbindin D28k and S100B have a similar interaction site with the lithium-inhibitable enzyme inositol monophosphatase-1: a new drug target site. J Med Chem. 2015;58:2042–4.
Singh N, Halliday AC, Thomas JM, Kuznetsova OV, Baldwin R, Woon ECY, et al. A safe lithium mimetic for bipolar disorder. Nat Commun. 2013;4:1332.
Singh N, Sharpley AL, Emir UE, Masaki C, Herzallah MM, Gluck MA, et al. Effect of the putative lithium mimetic ebselen on brain myo-inositol, sleep, and emotional processing in humans. Neuropsychopharmacology. 2016;41:1768–78.
Song J, Bergen SE, Di Florio A, Karlsson R, Charney A, Ruderfer DM, et al. Genome-wide association study identifies SESTD1 as a novel risk gene for lithium-responsive bipolar disorder. Mol Psychiatry. 2015:1–8.
Tsui MMK, Tai WCS, Wong WY, Hsiao WLW. Selective G2/M arrest in a p53Val135-transformed cell line induced by lithium is mediated through an intricate network of MAPK and β-catenin signaling pathways. Life Sci. 2012;91:312–21.
Cechinel-Recco K, Valvassori SS, Varela RB, Resende WR, Arent CO, Vitto MF, et al. Lithium and tamoxifen modulate cellular plasticity cascades in animal model of mania. J Psychopharmacol. 2012;26:1594–604.
Kovzun EI, Lukashenya OS, Pushkarev VM, Mikosha AS, Tron’ko ND. Effect of ions of potassium and lithium on NO synthase expression in the human adrenal cortex. Bull Exp Biol Med. 2014;156:332–4.
Deshauer D, Grof E, Alda M, Grof P. Patterns of DST positivity in remitted affective disorders. Biol Psychiatry. 1999;45:1023–9.
Zaeri S, Farjadian S, Emamghoreishi M. Decreased levels of canonical transient receptor potential channel 3 protein in the rat cerebral cortex after chronic treatment with lithium or valproate. Res Pharm Sci. 2015;10:397–406.
McCarthy MJ, Le Roux MJ, Wei H, Beesley S, Kelsoe JR, Welsh DK. Calcium channel genes associated with bipolar disorder modulate lithium’s amplification of circadian rhythms. Neuropharmacology. 2015;101:439–48.
Heinrich A, von der Heyde AS, Böer U, Phu DT, Tzvetkov M, Oetjen E. Lithium enhances CRTC oligomer formation and the interaction between the CREB coactivators CRTC and CBP–implications for CREB-dependent gene transcription. Cell Signal. 2013;25:113–25.
Mavrikaki M, Schintu N, Kastellakis A, Nomikos GG, Svenningsson P, Panagis G. Effects of lithium and aripiprazole on brain stimulation reward and neuroplasticity markers in the limbic forebrain. Eur Neuropsychopharmacol. 2014;24:630–8.
Alda M, Shao L, Wang J-F, Lopez de Lara C, Jaitovich-Groisman I, Lebel V, et al. Alterations in phosphorylated cAMP response element-binding protein (pCREB) signaling: an endophenotype of lithium-responsive bipolar disorder? Bipolar Disord. 2013;15:824–31.
Lueke K, Kaiser T, Svetlitchny A, Welzel O, Wenzel EM, Tyagarajan S, et al. Basic presynaptic functions in hippocampal neurons are not affected by acute or chronic lithium treatment. J Neural Transm. 2014;121:211–9.
Cruceanu C, Alda M, Grof P, Rouleau GA, Turecki G. Synapsin II is involved in the molecular pathway of lithium treatment in bipolar disorder. PLoS One. 2012;7:e32680.
Wakita M, Nagami H, Takase Y, Nakanishi R, Kotani N, Akaike N. Modifications of excitatory and inhibitory transmission in rat hippocampal pyramidal neurons by acute lithium treatment. Brain Res Bull. 2015;117:39–44.
Higgins GA, Allyn-Feuer A, Barbour E, Athey BD. A glutamatergic network mediates lithium response in bipolar disorder as defined by epigenome pathway analysis. Pharmacogenomics. 2015;16:1547–63.
Zanetti MV, Otaduy MC, de Sousa RT, Gattaz WF, Busatto GF, Leite CC, et al. Bimodal effect of lithium plasma levels on hippocampal glutamate concentrations in bipolar II depression: a pilot study. Int J Neuropsychopharmacol. 2015;18:pyu058–8.
Ghasemi M, Dehpour AR. The NMDA receptor/nitric oxide pathway: a target for the therapeutic and toxic effects of lithium. Trends Pharmacol Sci. 2011;32:420–34.
Chen C-H, Lee C-S, Lee M-TM, Ouyang W-C, Chen C-C, Chong M-Y, et al. Variant GADL1 and response to lithium therapy in bipolar I disorder. N Engl J Med. 2014;370:119–28.
Hou L, Heilbronner U, Degenhardt F, Adli M, Akiyama K, Akula N, et al. Genetic variants associated with response to lithium treatment in bipolar disorder: a genome-wide association study. Lancet. 2016;387:1085–93.
Birnbaum R, Shin JH. Variant GADL1 and response to lithium in bipolar I disorder. N Engl J Med. 2014;370:1855–60.
Ago Y, Tanaka T, Kita Y, Tokumoto H, Takuma K, Matsuda T. Lithium attenuates methamphetamine-induced hyperlocomotion and behavioral sensitization via modulation of prefrontal monoamine release. Neuropharmacology. 2012;62:1634–9.
van Enkhuizen J, Milienne-Petiot M, Geyer MA, Young JW. Modeling bipolar disorder in mice by increasing acetylcholine or dopamine: chronic lithium treats most, but not all features. Psychopharmacology. 2015;232:3455–67.
Marchese G, Scheggi S, Secci ME, De Montis MG, Gambarana C. Anti-anhedonic activity of long-term lithium treatment in rats exposed to repeated unavoidable stress. Int J Neuropsychopharmacol. 2013;16:1611–21.
Del’ Guidice T, Beaulieu J-M. Selective disruption of dopamine D2-receptors/beta-arrestin2 signaling by mood stabilizers. J Recept Signal Transduct Res. 2015;35:224–32.
Fortin SM, Chartoff EH, Roitman MF. The aversive agent lithium chloride suppresses phasic dopamine release through central GLP-1 receptors. Neuropsychopharmacology. 2016;41:906–15.
Farhy Tselnicker I, Tsemakhovich V, Rishal I, Kahanovitch U, Dessauer CW, Dascal N. Dual regulation of G proteins and the G-protein-activated K+ channels by lithium. Proc Natl Acad Sci. 2014;111:5018–23.
Jiménez E, Núñez E, Ibáñez I, Zafra F, Aragón C, Giménez C. Glycine transporters GlyT1 and GlyT2 are differentially modulated by glycogen synthase kinase 3β. Neuropharmacology. 2015;89:245–54.
McCarthy MJ, Wei H, Marnoy Z, Darvish RM, McPhie DL, Cohen BM, et al. Genetic and clinical factors predict lithium’s effects on PER2 gene expression rhythms in cells from bipolar disorder patients. Transl Psychiatry. 2013;3:e318.
McCarthy MJ, Nievergelt CM, Kelsoe JR, Welsh DK. A survey of genomic studies supports association of circadian clock genes with bipolar disorder spectrum illnesses and lithium response. PLoS One. 2012;7:e32091.
Schnell A, Sandrelli F, Ranc V, Ripperger JA, Brai E, Alberi L, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32:1075–89.
Kittel-Schneider S, Schreck S, Ziegler C, Weißflog L, Hilscher M, Schwarz R, et al. Lithium-induced clock gene expression in lymphoblastoid cells of bipolar affective patients. Pharmacopsychiatry. 2015;48:145–9.
Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, Hauser J. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16:151–8.
Kim SH, Yu HS, Park HG, Ahn YM, Kim YS, Lee YH, et al. Egr1 regulates lithium-induced transcription of the Period 2 (PER2) gene. Biochimica et Biophysica Acta (BBA) Mol Basis Dis. 2013;1832:1969–79.
Leussis MP, Berry-Scott EM, Saito M, Jhuang H, de Haan G, Alkan O, et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol Psychiatry. 2013;73:683–90.
Malhi GS, Byrow Y, Fritz K, Das P, Baune BT, Porter RJ, et al. Mood disorders: neurocognitive models. Bipolar Disord. 2015;17(Suppl 2):3–20.
Malhi GS, McAulay C, Gershon S, Gessler D, Fritz K, Das P, et al. The Lithium Battery: assessing the neurocognitive profile of lithium in bipolar disorder. Bipolar Disord. 2016;18:102–15.
Outhred T, Kemp AH, Malhi GS. Physiological correlates of bipolar spectrum disorders and their treatment. In: Kumari V, Boutros N, Bob P, editors. Current topics in behavioral neurosciences. Berlin, Heidelberg: Springer; 2014. p. 47–102.
Giakoumatos CI, Nanda P, Mathew IT, Tandon N, Shah J, Bishop JR, et al. Effects of lithium on cortical thickness and hippocampal subfield volumes in psychotic bipolar disorder. J Psychiatr Res. 2015;61:180–7.
Hartberg CB, Jørgensen KN, Haukvik UK, Westlye LT, Melle I, Andreassen OA, et al. Lithium treatment and hippocampal subfields and amygdala volumes in bipolar disorder. Bipolar Disord. 2015;17:496–506.
Baykara B, Inal-Emiroglu N, Karabay N, Çakmakçı H, Cevher N, Şentürk Pilan B, et al. Increased hippocampal volumes in lithium treated adolescents with bipolar disorders: a structural MRI study. J Affect Disord. 2012;138:433–9.
Selek S, Nicoletti M, Zunta-Soares GB, Hatch JP, Nery FG, Matsuo K, et al. A longitudinal study of fronto-limbic brain structures in patients with bipolar I disorder during lithium treatment. J Affect Disord. 2013;150:629–33.
Vernon AC, Natesan S, Crum WR, Cooper JD, Modo M, Williams SCR, et al. Contrasting effects of haloperidol and lithium on rodent brain structure: a magnetic resonance imaging study with postmortem confirmation. Biol Psychiatry. 2012;71:855–63.
Benedetti F, Poletti S, Radaelli D, Locatelli C, Pirovano A, Lorenzi C, et al. Lithium and GSK-3β promoter gene variants influence cortical gray matter volumes in bipolar disorder. Psychopharmacology. 2015;232:1325–36.
Pfennig A, Alda M, Young T, MacQueen G, Rybakowski J, Suwalska A, et al. Prophylactic lithium treatment and cognitive performance in patients with a long history of bipolar illness: no simple answers in complex disease-treatment interplay. Int J Bipolar Disord. 2014;2:1.
Hajek T, Cullis J, Novak T, Kopecek M, Hoschl C, Blagdon R, et al. Hippocampal volumes in bipolar disorders: opposing effects of illness burden and lithium treatment. Bipolar Disord. 2012;14:261–70.
Berger GE, Wood SJ, Ross M, Hamer CA, Wellard RM, Pell G, et al. Neuroprotective effects of low-dose lithium in individuals at ultra-high risk for psychosis. A longitudinal MRI/MRS study. Curr Pharm Des. 2012;18:570–5.
Kessing LV, Forman JL, Andersen PK. Does lithium protect against dementia? Bipolar Disord. 2010;12:87–94.
Kessing LV, Sondergard L, Forman JL, Andersen PK. Lithium treatment and risk of dementia. Arch Gen Psychiatry. 2008;65:1331–5.
O’Brien WT, Klein PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009;37:1133–8.
Wei YB, Backlund L, Wegener G, Mathé AA, Lavebratt C. Telomerase dysregulation in the hippocampus of a rat model of depression: normalization by lithium. Int J Neuropsychopharmacol. 2015;18:pyv002.
Hayashi A, Le Gal K, Södersten K, Vizlin-Hodzic D, Ågren H, Funa K. Calcium-dependent intracellular signal pathways in primary cultured adipocytes and ANK3 gene variation in patients with bipolar disorder and healthy controls. Mol Psychiatry. 2015;20:931–40.
Ferensztajn E, Skibinska M, Kaczmarek M, Losy J, Rybakowski JK. Neurobiology and temperament in the offspring of excellent lithium responders. World J Biol Psychiatry. 2015;16:272–7.
Madison JM, Zhou F, Nigam A, Hussain A, Barker DD, Nehme R, et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry. 2015;20:703–17.
Mertens J, Wang Q-W, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–9.
Masaki C, Sharpley AL, Godlewska BR, Berrington A, Hashimoto T, Singh N, et al. Effects of the potential lithium-mimetic, ebselen, on brain neurochemistry: a magnetic resonance spectroscopy study at 7 tesla. Psychopharmacology. 2016;233:1–8.
Grof P, Duffy A, Cavazzoni P, Grof E, Garnham J, MacDougall M, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63:942–7.
Duffy A, Horrocks J, Doucette S, Keown-Stoneman C, McCloskey S, Grof P. The developmental trajectory of bipolar disorder. Br J Psychiatry. 2014;204:122–8.
Benedetti F, Dallaspezia S, Lorenzi C, Pirovano A, Radaelli D, Locatelli C, et al. Gene-gene interaction of glycogen synthase kinase 3-β and serotonin transporter on human antidepressant response to sleep deprivation. J Affect Disord. 2012;136:514–9.
Tharoor H, Kotambail A, Jain S, Sharma PSVN, Satyamoorthy K. Study of the association of serotonin transporter triallelic 5-HTTLPR and STin2 VNTR polymorphisms with lithium prophylaxis response in bipolar disorder. Psychiatr Genet. 2013;23:77–81.
Benedetti F, Bollettini I, Poletti S, Locatelli C, Lorenzi C, Pirovano A, et al. White matter microstructure in bipolar disorder is influenced by the serotonin transporter gene polymorphism 5-HTTLPR. Genes Brain Behav. 2015;14:238–50.
Mitjans M, Arias B, Jiménez E, Goikolea JM, Sáiz PA, García-Portilla MP, et al. Exploring genetic variability at PI, GSK3, HPA, and glutamatergic pathways in lithium response: association with IMPA2, INPP1, and GSK3B genes. J Clin Psychopharmacol. 2015;35:600–4.
Acknowledgments
Ms Danielle Gessler provided assistance with the literature search.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research is supported by an NHMRC Program Grant (APP1073041), the Sydney Medical School Foundation, SPARK Sydney, the Ramsay Health Research and Teaching Fund and Grant PRG-0-090-14 awarded to GSM from the American Foundation for Suicide Prevention. The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Foundation for Suicide Prevention.
Conflict of interest
GSM has received grant or research support from NHMRC, NSW Health, AstraZeneca, Eli Lilly & Co., Organon, Pfizer, Servier and Wyeth; has delivered talks at Abbot, AstraZeneca, Eli Lilly & Co., Janssen Cilag, Lundbeck, Pfizer, Ranbaxy, Servier and Wyeth sponsored meetings; and has served on advisory boards for AstraZeneca, Eli Lilly & Co., Janssen Cilag, Lundbeck and Servier. TO declares no conflict of interest.
Rights and permissions
About this article

Cite this article
Malhi, G.S., Outhred, T. Therapeutic Mechanisms of Lithium in Bipolar Disorder: Recent Advances and Current Understanding. CNS Drugs 30, 931–949 (2016). https://doi.org/10.1007/s40263-016-0380-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-016-0380-1