
Abstract
Glycogen synthase kinase (GSK3) is a constitutively active serine-threonine kinase associated to neurological and psychiatric disorders. GSK3 inhibition is considered a mediator of the efficacy of the mood-stabiliser lithium. This study aimed at comparing the central nervous system effect of lithium with the selective GSK3 inhibitors AZ1080 and compound A in biochemical, cellular, and behavioural tests. Collapsin response mediator protein 2 is a neuron-specific GSK3 substrate. Lithium, AZ1080, and compound A inhibited its phosphorylation in rat primary neurons with different pIC50. After systemic treatments with lithium or GSK3 inhibitors to assess specific functional responses, phosphorylation was unchanged in adult rat brain, while it was strongly inhibited by GSK3 inhibitors in pups, differently from lithium. Lithium may exert neurotrophic effect by increasing brain-derived neurotrophic factor (BDNF) levels: in the present experimental conditions, lithium exerted opposite effects on plasma BDNF levels compared to GSK3 inhibitors, suggesting this effect might not be necessarily mediated by GSK3 inhibition alone. While plasma thyroid-stimulating hormone and luteinising hormone were not affected by lithium, they were decreased by selective inhibitors. GH and prolactin displayed similar responses towards reduction. Follicle-stimulating hormone levels were not altered by treatments, whereas melatonin was specifically increased by AZ1080. Lithium impaired mouse spontaneous locomotion and decreased amphetamine-induced hyper-locomotion. AZ1080 had no effects on locomotion, while compound A reduced spontaneous locomotor activity without effects on amphetamine-induced hyper-locomotion. The present results indicate that a broad correlation between the effects of lithium and selective GSK3 inhibitors could not be devised, suggesting alternative mechanisms, whereas overlapping results could be obtained in specific assays.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The serine/threonine kinase glycogen synthase kinase 3 (GSK3) was originally identified as a regulator of glycogen metabolism (Embi et al. 1980). During the past decade, the interest for GSK3 has appreciably grown, since accumulating evidence supports its involvement in a plethora of cellular and physiological events, including Wnt and Hedgehog signalling, transcription, insulin action, cell division cycle, response to DNA damage, cell death and survival, development, neuronal functions, circadian rhythms, and others (reviewed in Rayasam et al. 2009).
Within the brain, GSK3 is widely distributed and constitutively active, maintaining many of its cellular targets in an inactive state through inhibitory phosphorylation (Grimes and Jope 2001; Jope and Johnson 2004). Given the widespread brain distribution of GSK3 and its central biological role in multiple cellular pathways, it is not surprising that GSK3 is considered a potential target for pharmacological intervention in human neuropathological and psychiatric conditions (Martinez and Perez 2008; Catapano and Manji 2008; Duman and Voleti 2012). Interestingly, several mood stabilisers directly or indirectly inhibit this kinase (Phiel and Klein 2001; Zhang et al. 2003) and similarities exist between GSK3 inactivation and treatment with mood stabilisers, such as lithium or valproic acid (Chen et al. 1999; Leng et al. 2008). Specifically, lithium, used as a mood stabiliser in bipolar disorder patients for nearly 50 years was shown to act as a direct (Klein and Melton 1996) as well as an indirect (Beaulieu et al. 2004; Chalecka-Franaszek and Chuang 1999; De Sarno and Jope 2002) GSK3 inhibitor. Nevertheless, it remains to be fully elucidated if lithium exerts its therapeutic effects mainly through GSK3 inhibition. Indeed, lithium is not a selective GSK3 inhibitor and its therapeutic effects could be due to its actions on multiple targets, so far only partially identified (O’Brien and Klein 2009).
Understanding which among the effects exerted by lithium are mediated by GSK3 inhibition is critical to figure out the molecular mechanisms of its therapeutic benefits. Moreover, this knowledge could assist in the challenging task of developing new medicines that target this kinase. On the other hand, investigations aimed at comparing the effects of lithium treatments with GSK3 inhibitors face the challenge of potential lack of specificity of these very inhibitors.
The present study was aimed at comparing the effect of lithium targeting the selective inhibition of GSK3 with the selective compounds AZ1080 and compound A in biochemical, cellular, and behavioural tests centred on the nervous system.
Methods
Animals
Adult (200–250 g) and 16-day-old Sprague–Dawley rats (Charles River, Calco, Italy), and adult (18–20 g) C57Bl/6 J mice (Charles River Labs) were used as experimental subjects and were kept under standard lighting conditions (12:12, lights on at 6:00 am), at a constant room temperature (21 ± 2 °C) and with food and water available ad libitum. All experimental procedures were carried out in accordance with Italian Law (Legislative Decree No. 116, 27 January 1992), which acknowledges the European Directive 86/609/EEC, and were fully compliant with GlaxoSmithKline policy on the care and use of laboratory animals and codes of practice. All efforts were made to minimise the number of experimental animals used and their suffering.
Drugs
Lithium and d-amphetamine were purchased from Sigma-Aldrich (St. Louis, MO, USA). AZ1080 was synthesised in GlaxoSmithKline from WO2003082853 A1 (Berg et al. 2003). 1H-thieno[2,3-c]pyrazol-3-yl derivative (compound A) was synthesised in GlaxoSmithKline based on patent WO2004007504 A1 (Tonani et al. 2004). GSK3 inhibition activity was tested in a GSK3 β biochemical assay and selectivity was assessed in a panel of 40 kinases. Lithium was dissolved in saline and injected i.p. (10 mL/kg) at 50, 100, or 200 mg/kg. AZ1080 was prepared in 1 % Methocell (Sigma-Aldrich) and administered p.o. (10 mL/kg) at 3, 10, or 30 mg/kg. Compound A was suspended in 12.5 % Captisol® (CyDex Inc., Lenexa, KS, USA) citrate buffer pH 5.5 solution and injected i.p. (10 mL/kg) at 10, 30, or 100 mg/kg. d-amphetamine was dissolved in saline and administered i.p. at 2 mg/kg (10 mL/kg).
Primary cortical neurons
Primary cortical neurons were obtained from embryonic days 18/19 Sprague–Dawley rats. Rat brains were dissected out and cortices were quickly isolated at 4 °C in HBSS pH 7.3 buffer containing 10 mM HEPES, 100 U/mL penicillin and 100 μg/mL streptomycin (HBSSH buffer, cell culture reagents from Gibco, Life Technologies Italia, Monza, Italy). Cells were treated with HBSSH buffer added with Trypsin 0.1 % at 37 °C for 10 min. In the last 5 min of incubation, cells were treated also with 166 μg/mL of DNAse I. After a single wash with HBSSH buffer containing 10 % FBS (PAA, GE Healthcare Life Sciences, Little Chalfont, UK) and two additional washes with HBSSH buffer, cells were mechanically dissociated by triturating with Pasteur pipettes. Cortical neurons were then placed in poly-l-lysine coated 12- or 96-well plates at the density of 800 cells/cm2 in serum-free neurobasal medium supplemented with B27 supplement, 500 μM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. In all assays, neuronal primary cultures were used after 1 week in culture and drug treatment was performed during 24 h.
Pharmacological treatments
In separate experiments, adult and pup rats were dosed with either vehicle or compounds at the tested doses (n = 7–8/group). Based on pharmacokinetic data, animals were sacrificed either at 1.5 (lithium) or 4 h (AZ1080 and compound A) after treatment.
Western blotting
The effect of GSK3 inhibition was assessed by measuring collapsin response mediator protein 2 (CRMP2) phosphorylation using an antibody specific for phosphorylation on Thr 509 and Thr 514 (Cole et al. 2006). For in vitro experiments, primary cortical neurons were obtained and treated as described above. For ex vivo experiments, brain regions were obtained from young as well as adult rats. Animals were sacrificed, brains were removed, and selected regions (cerebral cortex, hippocampus, and cerebellum) were rapidly dissected out, frozen in dry ice, and stored at −80 °C. Total proteins were extracted in RIPA buffer supplemented with protease (Complete Mini, Roche Applied Science, Monza, Italy) and phosphatase inhibitors (Sigma-Aldrich; Di Daniel et al. 2005). Cytoplasmic proteins were extracted and their levels were assessed by Western blotting as previously described (Culbert et al. 2001). Preliminary experiments were carried out in extracts prepared from cortical primary cultures, SH-SY5Y cells, and rat brain regions (cortex, hippocampus, and cerebellum), to ensure that treatment with inhibitors did not affect total CRMP2 levels. The following primary antibodies were used: anti-CRMP2 Phospho Tht 509 + Thr 514 (Dundee Consortium, http://www.lifesci.dundee.ac.uk/research/dstt/) S374B, 1:5,000; polyclonal anti-CRMP2 (Sigma-Aldrich) 1:20,000; anti-GAPDH purified in mouse (Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1:5,000; anti- β-Catenin (Abcam, Cambridge, UK) 1:1,000. Protein levels were normalised to GADPH and quantification was performed using Odyssey or MCID software.
Peripheral hormone levels
Rats were sacrificed by quick decapitation and trunk blood was collected in Microtainer BD K2EDTA tubes (Becton Dickinson Italia, Milano, Italy) with a protease inhibitor cocktail (Sigma-Aldrich) and a DPPIV protease inhibitor (Millipore, Billerica, MA, USA). After 10-min centrifugation at 1,800×g, 4 °C, plasma was collected, split into aliquots, and stored at −80 °C. Analytes were measured by Luminex technology on a Bio-Plex instrument (Bio-Rad, Hercules, CA, USA). Prolactin, luteinising hormone (LH), follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH), growth hormone (GH), and brain-derived neurotrophic factor (BDNF) levels were measured with Rat Pituitary Panel Milliplex kits (Millipore); melatonin levels were determined with Rat Stress Hormone Milliplex kits (Millipore) following manufacturer’s instructions.
Measurement of locomotor activity
Behavioural data were collected using 12 VersaMax Animal Activity Monitors (AccuScan Model RXYZXCM-16, Columbus, OH, USA). Each chamber (40 × 40 × 30.5 cm) was made of clear Plexiglas and covered with a perforated Plexiglas lid. Infrared monitoring sensors were located every 2.53 cm along the perimeter (16 infrared beams along each side) and 2.5 cm above the floor. Two additional sets of 16 sensors were located 8.0 cm above the floor on opposite sides. Data were collected and analysed by a VersaMax Analyzer (AccuScan Model CDA-8, Columbus, OH, USA) which sent information to a computer for analysis. Locomotion was evaluated under illuminated conditions in the light phase of the light/dark cycle. On test day, mice (n = 12/group) were individually habituated to the activity chambers for 2 h. Mice were then administered with either drug or vehicle and returned to the activity chamber for additional 30 min recordings (habituation). At the end of the habituation, mice were challenged with either d-amphetamine or saline and tested for further 90 min (challenge). The activity chambers were thoroughly cleansed after each animal. Locomotor activity was measured in terms of the horizontal activity (number of beam breaks in the lower infrared sensors). Total activity counts were obtained during either the habituation or the challenge phase.
Delayed gastric emptying
Animals were weighed and sacrificed promptly after the end of the locomotor activity recordings. To assess the occurrence of gastroparesis (i.e., a potential side effect of GSK3 inhibition), stomachs were dissected out excluding the intra-abdominal part of the oesophagus and the mesentery from the final weight. Stomach weight was then analysed as percentage of body weight.
Data analysis
The inhibition of CRMP2 phosphorylation in primary cortical neurons was analysed with nonlinear regression analysis using GraphPad Prism 5.0 (GraphPad Software, CA, USA). IC50 values were determined by fitting data to a four-parameter logistic equation (sigmoidal dose–response variable slope). Data were then expressed as pIC50 values (negative logarithm of IC50) and reported as mean pIC50 ± SEM. Protein and hormone levels, total horizontal activity counts, and stomach weight values were analysed by one-way ANOVAs with a cutoff value of p < 0.05. Data were log-transformed before analysis when necessary to fulfil homoscedaticity requirements. If ANOVAs suggested statistically significant differences, Dunnett’s tests versus values obtained in vehicle-treated controls were performed as post hoc tests. Statistical analyses were conducted using Statistica V8 (Statsoft, Inc. Tulsa, OK, USA).
Results
The first step to compare the effects of lithium and other GSK3 inhibitors in the central nervous system was based on an in vitro model. The pharmacological parameters of GSK3 inhibition were assessed in rat primary cortical neurons by measuring phosphorylation levels of CRMP2, a regulating component of neurite outgrowth and direct neuronal GSK3 substrate (Cole et al. 2004; Yoshimura et al. 2005; Cole et al. 2007). CRMP2 is phosphorylated by GSK3 at Ser518, Thr514, and Thr509, while previous priming is required by Cdk5 phosphorylation at Ser522 (Yoshimura et al. 2005). The anti-phosphoCRMP2 antibody used in these experiments recognised Thr 509 and Thr 514 (Cole et al. 2006), thus ensuring that GSK3 activity was specifically detected. Results showed that 24-h treatments with each GSK3 inhibitor reduced CRMP2 phosphorylation following a dose–response curve (Fig. 1). As expected, pIC50 values differed for each pharmacological treatment (lithium, 2.98 ± 0.12; AZ1080, 6.69 ± 0.11; and compound A, 8.63 ± 0.23).

a Concentration response curves of phospho-CRMP2 protein following 24-h treatment with GSK3 inhibitors AZ1080, compound A, and lithium in primary cortical neurons. Each data point represents the mean of three independent experiments. b Representative immunoblotting images of primary cortical neurons treated with GSK3 inhibitors or lithium
Subsequently, we aimed to compare the effects on CRMP2 phosphorylation in rat brain as a consequence of systemic treatment with either lithium or GSK3 inhibitors. Phosphorylated CRMP2 (pCRMP2) levels were measured in cortex, hippocampus, and cerebellum of adult rats after pharmacological treatment. The end-points were established by performing preliminary pharmacokinetic measures to identify peak exposures in brain. Surprisingly, no inhibition could be induced by any treatment, although brain exposures in the micromolar range were achieved. Since CRMP2 is enriched in the developing nervous system (Yoshimura et al. 2005), additional experiments were performed in young animals. In contrast to results obtained in adults, in young rats CRMP2 phosphorylation was strongly inhibited by AZ1080 [F(1,12) = 23.3744, p = 0.0004; Fig. 2a] as well as by compound A [F(1,9) = 10.8283, p = 0.0094; Fig. 2c]. Lithium treatment did not affect CRMP2 phosphorylation in young rats [F(1,12) = 0.0014, ns] as well as in adults (not shown).

Phospo-CRMP2 protein in 16-day-old pups and adult rat hippocampus 4 h following 100 mg/kg i.p. treatment with the GSK3 inhibitors AZ1080 (a and b) and compound A (c and d) mean ± SEM (n = 6–7/group). ***p < 0.001; ∼p < 0.09. Values are expressed in integrated intensities (II, the sum of the intensity values for all pixels enclosed by a feature, multiplied by the area of the feature) measured by Odyssey. e–f Representative images of immunoblot experiments measuring hippocampal phospho-CRMP2 levels after treatment with AZ1080 in 16-day-old pups (e) or adult rats (f)
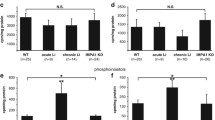
As a further step, we addressed the issue of whether treatments with lithium or GKS3 inhibitors differently affected brain-mediated modulation of peripheral hormones. The objective was to investigate their potential roles in pharmacokinetic/pharmacodinamic assays for drug development or as peripheral biomarkers of central GKS3 inhibition. Therefore, after systemic administrations of lithium or GSK3 inhibitors to rats, plasma levels of BDNF, pituitary hormones, and melatonin were measured and compared between treatments. A number of hormones appeared to respond differently to lithium or GSK3 inhibitors treatments. For instance, a trend towards increased levels of BDNF was observed in lithium-treated rats [F(1,14) = 4.15, p = 0.06, Fig. 3a]. In contrast, lower BDNF levels were observed in animals treated with either AZ1080 or compound A [AZ1080: F(1,14) = 5.69, p = 0.03; compound A: F(1,11) = 4.71, p = 0.053, Fig. 3a]. Whereas GH levels were not significantly altered by lithium treatment [F(1,12) = 0.39, p = 0.5, Fig. 3b], lower GH levels were detected in rats treated with either GSK3 inhibitor [AZ1080: F(1,14) = 4.75, p = 0.047; compound A: F(1,12) = 4.08, p = 0.066, Fig. 3b]. Likewise, TSH levels were not affected by lithium treatment [F(1,14) = 0.14, p = 0.7, Fig. 3c], while lower levels were revealed in rats treated with either AZ1080 or compound A [AZ1080: F(1,13) = 6.40, p = 0.025; compound A: F(1,12) = 5.97, p = 0.031, Fig. 3c]. Again showing different patterns, lithium treatment had no influence on LH levels [F(1,14) = 0.75, p = 0.4 Fig. 3d], while significantly lower LH levels were detected after treatment with either GSK3 inhibitor [AZ1080: F(1,13) = 104.6, p < 0.0001; compound A: F(1,11) = 7.31, p = 0.02, Fig. 3d].

Blood hormone levels in rats after treatment with 200 mg/kg lithium, 100 mg/kg AZ1080, or 100 mg/kg compound A. Separate experiments were performed for each compound and statistical analyses were carried out by comparing Li+ or GSK3 inhibitors with respective vehicle-treated controls (n = 7–8/group). In this figure, a summary of results is displayed, by expressing hormone changes as percentage of levels measured in the respective control animals. a BDNF [controls, 398.4 ± 51.29 (average ± SEM) pg/ml]; b GH (controls, 3.77 ± 1.11 ng/ml); c TSH (controls, 2.33 ± 0.38 ng/ml); d LH (controls, 801.2 ± 115.7 pg/ml); e FSH (controls, 23.14 ± 1.25 ng/ml); f prolactin (controls, 2.12 ± 0.39 ng/ml), g melatonin (controls, 59.41 ± 13.0 ng/ml). ***p < 0.001; **p < 0.01; *p < 0.05; ˜0.053 < p < 0.066
In contrast, other hormones displayed more similar responses. FSH levels were not altered by any treatment [lithium: F(1,14) = 2.60, ns; AZ1080: F(1,14) = 0.02, ns; compound A: F(1,12) = 2.08, n.s.; Fig. 3e]. Prolactin levels were significantly lower in rats treated either with lithium or AZ1080 [lithium: F(1,12) = 6.57, p = 0.025; AZ1080: F(1,14) = 21.30, p = 0.0004, Fig. 3f], while the trend towards reduction by compound A was not significant [compound A: F(1,12) = 2.29, ns, Fig. 3f]. Melatonin levels were not influenced by lithium [F(1,13) = 0.26, ns, Fig. 3g] or compound A treatment [F(1,12) = 0.13, ns, Fig. 3g], whereas a statistically significant rise was found after treatment with AZ1080 [F(1,14) = 12.27, p = 0.003, Fig. 3g], possibly a specific effect of the compound. Finally, we used a model of amphetamine-induced hyperactivity in mice as a well-established behavioural test to investigate in vivo efficacy of the compounds. In this assay, we also assessed the potential for gastroparesis, a potential liability of pharmacological inhibition of GSK3.
During the habituation phase, lithium significantly impaired mouse spontaneous locomotor activity [F(4,43) = 13.44, p < 0.01], at 100 mg/kg (p < 0.05) and 200 mg/kg (p < 0.001), but not at 50 mg/kg. During the challenge phase, lithium significantly decreased the d-amphetamine-induced hyper-locomotion [F(1,43) = 21.57, p < 0.001] at all doses tested (50 mg/kg, p < 0.001; 100 mg/kg, p < 0.001; 200 mg/kg, p < 0.001; Fig. 4a–b). Delayed gastric emptying (DGE) was not evaluated in these subjects, since lithium has been shown to induce gastroparesis in rodents at doses above 60 mg/kg (McCann et al. 1989; Table 1). AZ1080 had no significant effects at any of the tested doses during the habituation phase [F(4,43) = 1.21, ns] as well as during the d-amphetamine-induced hyperactivity [F(1,43) = 66.85, p < 0.001] (Fig. 4c–d). A trend for DGE was observed for AZ1080 [F(1, 34) = 2.41, p = 0.084] that reached significant levels at the highest dose tested (30 mg/kg, p < 0.05; Table 1). Compound A had a significant effect during the habituation phase [F(1,43) = 7.71, p < 0.001] at all tested doses as it reduced mouse basal locomotor activity (10 mg/kg, p = 0.053; 30 mg/kg, p < 0.01; 100 mg/kg, p < 0.01). On the other hand, compound A did not have any effect on the hyper-locomotion induced by d-amphetamine [F(1,43) = 26.06, p < 0.001] (Fig. 4e–f) and, in the present conditions, it had no DGE effects [F(1, 39) = 0.23, ns] (Table 1).

Locomotor activity time course (a, c, e) and total values (b, d, f) measured in C57BL/6 J mice and recorded as horizontal activity counts. Mice (n = 12/group) received d-amphetamine challenge followed by either vehicle administration or a GSK3 inhibitor (lithium (a–b), AZ1080 (c–d), compound A (e–f)). GSK3 inhibitors were i.p. administered at three doses 30 min prior to d-amphetamine i.p. injection. ∼p = 0.053; *p < 0.05; **p < 0.01; ***p < 0.001 versus vehicle group during baseline; ### p < 0.001 vs. d-amphetamine
Discussion
GSK3 inhibition is considered as a relevant mechanism of action of the antimanic activity exerted by lithium (Jope 2003). In the present study, different approaches were adopted to compare the effects of selective GSK3 inhibitors with lithium in molecular, biochemical, and behavioural assays performed in the nervous system.
Initially, the consequences of the inhibition of GSK3 were compared with lithium treatments in primary cortical neuronal cultures, where the levels of phosphorylation of the GSK3 substrate CRMP2 were analysed. CRMP2 is a protein enriched in the central nervous system that binds to microtubules and regulates axon outgrowth in neurons. Lithium dose-dependently inhibited Thr-514-CRMP2 phosphorylation in the present study in agreement with previous findings (Lim et al. 2010). Available data demonstrated direct and indirect actions of lithium on GSK3 (Jope 2003), the former exerted by competing for a magnesium binding site (Ryves et al. 2002). Nevertheless, lithium impinges on multiple intracellular targets that extend well beyond GSK3 (Quiroz et al. 2004). Therefore, the observed efficacy of both selective GSK3 inhibitors, AZ1080 and compound A, further confirms the pharmacological inhibition of CRMP2 phosphorylation as being GSK3 mediated. Moreover, these findings support the use of CRMP2 phosphorylation assays in primary neurons as a tool for investigating the pharmacological activity of potential new GSK3 inhibitors for neurological or psychiatric indications.
To compare the inhibitory activity of lithium and GSK3 inhibitors in an in vivo system, adult rats were treated with therapeutically relevant doses of lithium or with GSK3 inhibitors. The objective of the designed experiments was to obtain adequate compound exposures in brain to induce GSK3 inhibition. Since no data were available about the pharmacokinetic properties of GSK3 inhibitors, we performed preliminary experiments to establish the best administration routes and end-point timings for both compounds by analysing blood and brain concentrations in different conditions. As a result, AZ1080 oral administration and i.p. administration of compound A achieved micromolar concentrations in the brain 4 h after dosing; we therefore carried out subsequent experiments following this design. Notwithstanding the different administration route, the results obtained can be compared across compounds, since we ascertained that comparable compound concentrations were reached in the central nervous system. pCRMP2 levels were evaluated in different brain regions such as cortex, hippocampus, and cerebellum. Nevertheless, in these conditions, none of the treatments could effectively inhibit CRMP2 phosphorylation. However, since CRMP2 exerts a major role in brain development, it is possible that the lack of efficacy on pCRMP2 in adult rats depend on a developmental resistance to de-phosphorylation. Indeed, in 16-day-old rats, AZ1080 and compound A induced considerable inhibition of CRMP2 phosphorylation. This age-dependent susceptibility to de-phosporylation could represent an adaptation to prevent undesired effects in adults, since phosphorylation of CRMP2 by GSK3 reflects a deactivating post-transcriptional modification. In contrast, lithium treatment did not affect pCRMP levels at both rat ages, showing a different activity in comparison to more selective compounds in these experimental conditions. On the other hand, previous findings suggest that repeated treatments are often required to induce noticeable in vivo effects due to GSK3 inhibition by lithium treatment, such as increased β-catenin levels (Gould et al. 2004), decreased Tau aggregation (Pérez et al. 2003), and decreased amyloid β (Su et al. 2004). Therefore, further studies are warranted to assess phosphorylation levels of GSK3 substrates such as CRMP2 or β-catenin following chronic treatments. Interestingly, β-catenin has been implicated in brain development, cognitive activity, and dendritic growth (Coyle-Rink et al. 2002; Yu and Malenka 2004) and its accumulation is considered a potential marker of in vivo inhibition of GSK3. Indeed, in preliminary experiments, AZ1080 treatment induced cytoplasmic β-catenin accumulation (data not shown), thus suggesting its potential value as a read out of GSK3 inhibition in adult animals.
In an effort to establish the effects of GSK3 functional inhibition based upon whole animal responses, we compared the effects of lithium or AZ1080 and compound A in peripheral hormone levels. Available data suggested that lithium administration affects blood levels of multiple hormones (Banerji et al. 1983; Seggie et al. 1985; Smythe et al. 1979). Interestingly, this approach highlighted a divergent panel of hormonal alterations due to either lithium or more selective GSK3 inhibitor treatments. Specifically, lithium induced a trend towards increased BDNF, whereas both GSK3 inhibitors significantly reduced its plasma levels. Since BDNF can cross the blood–brain barrier through a transport system (Pan et al. 1998), it is likely that equilibrium is reached between peripheral and central levels. In vitro studies in cultured neurons demonstrated that lithium treatment increased BDNF levels (Hashimoto et al. 2002) by transcriptional regulation mediated through GSK3 inhibition (Yasuda et al. 2009). Nevertheless, data obtained in vivo suggest that a more complicated pattern is observed when the system maintains its natural complexity, since a general trend toward increased BDNF levels is specifically displayed only in discrete brain regions and with different patterns in distinct disease models (Fukumoto et al. 2001; Angelucci et al. 2003; Jacobsen and Mørk 2004; Frey et al. 2006; Omata et al. 2008; Hammonds and Shim 2009). A contribution to this complexity is also provided by the finding that lithium may exert contrasting regulation in glial cells as compared to neurons (Nishino et al. 2012). In line with our findings in rats, the assessment of blood BDNF levels after lithium treatment in patients showed that an increase is revealed in some instances (Leyhe et al. 2009; de Sousa et al. 2011), although divergent results are reported as well (Yoshimura et al. 2007; Suwalska et al. 2010).
A large body of evidence supports the notion that peripheral BDNF levels are altered in psychiatric and neurological diseases (Fernandes et al. 2011; Diniz and Teixeira 2011) and lithium neurotrophic activity may play a major role in mediating its beneficial effects (Quiroz et al. 2010). Therefore, it is important to highlight that our findings suggest that lithium neurotrophic effects on BDNF levels may not be shared by GSK3 inhibition exerted by more selective compounds.
Similarly to BDNF, TSH, and LH levels showed a different response to lithium treatment as compared to selective GSK3 inhibitors. While both AZ1080 and compound A treatments reduced plasma TSH and LH levels, lithium administration did not affect them.
In lithium-treated patients, hypothyroidism may be frequently induced by direct inhibition of thyroid hormones production, with increased TSH as a response to thyroid reduced functionality (Bou Khalil and Richa 2011). It is possible that this effect cannot be detected after a single lithium administration. Nevertheless, the opposite regulation in the direction of reduced TSH levels after treatment with selective GSK3 inhibitors suggest that the multiplicity of effects promoted by lithium do not overlap with exclusive GSK3 inhibition.
Moreover, the treatment with selective GSK3 inhibitors induced an unexpected pattern of gonadotropic hormone levels. Indeed, the respective releasing hormone GnRH has been shown to avail of a signal transduction pathway that involves GSK3 inhibition and β-catenin increase in gonadotrophs (Gardner et al. 2007). Therefore, the observed reduction of LH levels and stable FSH imply that additional layers of control occur in vivo.
A second group of peripheral hormones showed more similar patterns after treatment with lithium or selective GSK3 inhibitors, such as GH, prolactin, and FSH. Previous data reported that lithium treatment significantly inhibited both prolactin and GH secretion in the rat (Smythe et al. 1979), in agreement with our findings.
GH release is controlled by GHRH, somatostatin, and ghrelin (Anderson and Scanes 2012). Somatostatin-induced inhibition of GH release is mediated through GSK3 signal transduction (Khattak et al. 2010), thus suggesting that an interference with this pathway may be responsible of the observed reduction of GH levels.
Prolactin levels are also reduced by both lithium and selective GSK3 inhibitors, implying that this action is indeed mediated by GSK3 inhibition. Nevertheless, it is surprising that dopamine-induced inhibition of prolactin release is potentiated by GSK3 inhibition, which is known to antagonise dopamine-dependent behaviours (Beaulieu et al. 2004).
Finally, several pieces of evidence suggest that a number of behavioural effects of lithium in rodents could be related to GSK3 inhibition. Among them, lithium treatment is able to restore normal activity in the psychostimulant-induced hyperactivity test. Previous findings in a transgenic mouse model demonstrated that this action is mediated by GSK3 at least in part (Ahnaou and Drinkenburg 2011). Other studies based on pharmacological inhibition of GSK3 provided further support to this hypothesis (Miller et al. 2009; Enman and Unterwald 2012; Kozikowski et al. 2011; Xu et al. 2011; Kalinichev and Dawson 2011). Thus, mice were tested in amphetamine-induced hyperlocomotor activity. Lithium at clinically relevant doses confirmed its effects in this model, in line with previous studies (Cox et al. 1971; Lerer et al. 1984). On the other hand, the GSK3 inhibitors AZ1080 and compound A were not effective, although compound A induced a decrease of mouse spontaneous activity. The present results did not confirm previous publications showing efficacy of GSK3 inhibitors in this model. Procedural differences, such as mouse strain and/or psychostimulant substance used, knowing to differentially affect the outcome of stimulant-induced hyperlocomotion, could explain the observed discrepancy (Gould et al. 2007; Miller et al. 2009). On the other hand, since lithium demonstrated activity in our experimental conditions, it is possible that additional components of the multiple molecular targets of lithium may be involved in the mechanism of reducing amphetamine-induced hyperlocomotion.
Since historical data implicate GSK3 in metabolic function and disorders (Rayasam et al. 2009) and recent indications imply a specific role in gastric secretion (Rotte et al. 2010), stomach weight was assessed in mice treated with selective GSK3 inhibitors. DGE was observed following AZ1080 but not compound A treatment. These findings cast doubts that a simple correlation could be revealed between the effects on locomotor impairment and gastroparesis, since these processes were differently affected by AZ1080 and compound A.
In conclusion, the present study compares the effects of lithium and selective GSK3 inhibitors in molecular, biochemical, and behavioural assays performed in the nervous system. Overall, the present results suggest that a general correlation between lithium activities and the effects of selective GSK3 inhibitors could not be outlined, although overlapping results could be obtained in specific assays. Therefore, our findings suggest that the development of new medicines targeting GSK3 for neurological or psychiatric indications will face the challenging task of identifying suitable methods to assess their action.
References
Ahnaou A, Drinkenburg WH (2011) Disruption of glycogen synthase kinase-3-beta activity leads to abnormalities in physiological measures in mice. Behav Brain Res 221:246–52
Anderson LL, Scanes CG (2012) Nanobiology and physiology of growth hormone secretion. Exp Biol Med 237:126–42
Angelucci F, Aloe L, Jiménez-Vasquez P, Mathé AA (2003) Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int J Neuropsychopharmacol 6:225–31
Banerji TK, Parkening TA, Collins TJ, Rassoli A (1983) Lithium-induced changes in the plasma and pituitary levels of luteinizing hormone, follicle stimulating hormone and prolactin in rats. Life Sci 33:1621–7
Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A 101:5099–50104
Berg S, Hellberg S, Nylöf M, Xue Y (2003) WO2003082853 A1
Bou Khalil R, Richa S (2011) Thyroid adverse effects of psychotropic drugs: a review. Clin Neuropharmacol 34:248–55
Catapano LA, Manji HK (2008) Kinases as drug targets in the treatment of bipolar disorder. Drug Discov Today 13:295–302
Chalecka-Franaszek E, Chuang DM (1999) Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA 96:8745–50
Chen G, Huang LD, Jiang YM, Manji HK (1999) The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem 72:1327–30
Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C (2004) GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem 279:50176–80
Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernández F, Eickholt BJ, Nikolic M, Sutherland C (2006) Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J Biol Chem 281:16591–8
Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, Taylor M, LaFrancois J, Gunn-Moore F, Verkhratsky A, Oddo S, LaFerla F, Giese KP, Dineley KT, Duff K, Richardson JC, Yan SD, Hanger DP, Allan SM, Sutherland C (2007) Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem 103:1132–44
Cox C, Harrison-Read PE, Steinberg H, Tomkiewicz M (1971) Lithium attenuates drug-induced hyperactivity in rats. Nature 232:336–8
Coyle-Rink J, Del Valle L, Sweet T, Khalili K, Amini S (2002) Developmental expression of Wnt signaling factors in mouse brain. Cancer Biol Ther 1:640–5
Culbert AA, Brown MJ, Frame S, Hagen T, Cross DA, Bax B, Reith AD (2001) GSK-3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and beta-catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett 507:288–94
De Sarno P, Li X, Jope RS (2002) Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43:1158–64
de Sousa RT, van de Bilt MT, Diniz BS, Ladeira RB, Portela LV, Souza DO, Forlenza OV, Gattaz WF, Machado-Vieira R (2011) Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett 494:54–6
Di Daniel E, Mudge AW, Maycox PR (2005) Comparative analysis of the effects of four mood stabilizers in SH-SY5Y cells and in primary neurons. Bipolar Disord 7:33–741
Diniz BS, Teixeira AL (2011) Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromolecular Med 13:217–22
Duman RS, Voleti B (2012) Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci 35:47–56
Embi N, Rylatt DB, Cohen P (1980) Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 107:519–27
Enman NM, Unterwald EM (2012) Inhibition of GSK3 attenuates amphetamine-induced hyperactivity and sensitization in the mouse. Behav Brain Res 231:217–25
Fernandes BS, Gama CS, Ceresér KM, Yatham LN, Fries GR, Colpo G, de Lucena D, Kunz M, Gomes FA, Kapczinski F (2011) Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res 45:995–1004
Frey BN, Andreazza AC, Ceresér KM, Martins MR, Valvassori SS, Réus GZ, Quevedo J, Kapczinski F (2006) Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci 79:281–6
Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S (2001) Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology 158:100–6
Gardner S, Maudsley S, Millar RP, Pawson AJ (2007) Nuclear stabilization of beta-catenin and inactivation of glycogen synthase kinase-3beta by gonadotropin-releasing hormone: targeting Wnt signaling in the pituitary gonadotrope. Mol Endocrinol 21:3028–38
Gould TD, Chen G, Manji HK (2004) In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29:32–8
Gould D, Einat H, O'Donnell KC, Picchini AM, Schloesser RJ, Manji HK (2007) Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology 32:2173–83
Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65:391–426
Hammonds MD, Shim SS (2009) Effects of 4-week treatment with lithium and olanzapine on levels of brain-derived neurotrophic factor, B-cell CLL/lymphoma 2 and phosphorylated cyclic adenosine monophosphate response element-binding protein in the sub-regions of the hippocampus. Basic Clin Pharmacol Toxicol 105:113–9
Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM (2002) Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43:1173–9
Jacobsen JP, Mørk A (2004) The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res 1024:183–92
Jope RS (2003) Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 24:441–3
Jope RS, Johnson GV (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29:95–102
Kalinichev M, Dawson LA (2011) Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int J Neuropsychopharmacol 14:1051–67
Khattak MN, Buchfelder M, Kleindienst A, Schöfl C, Kremenevskaja N (2010) CRH and SRIF have opposite effects on the Wnt/β-catenin signalling pathway through PKA/GSK-3β in corticotroph pituitary cells. Cancer Invest 28:797–805
Klein PS, Melton DA (1996) A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93:8455–9
Kozikowski AP, Gunosewoyo H, Guo S, Gaisina IN, Walter RL, Ketcherside A, McClung CA, Mesecar AD, Caldarone B (2011) Identification of a glycogen synthase kinase-3β inhibitor that attenuates hyperactivity in CLOCK mutant mice. ChemMedChem 6:1593–602
Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM (2008) Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci 28:2576–88
Lerer B, Globus M, Brik E, Hamburger R, Belmaker RH (1984) Effect of treatment and withdrawal from chronic lithium in rats on stimulant-induced responses. Neuropsychobiology 11:28–32
Leyhe T, Eschweiler GW, Stransky E, Gasser T, Annas P, Basun H, Laske C (2009) Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J Alzheimers Dis 16:649–56
Lim YW, Yoon SY, Choi JE, Kim SM, Lee HS, Choe H, Lee SC, Kim DH (2010) Maintained activity of glycogen synthase kinase-3beta despite of its phosphorylation at serine-9 in okadaic acid-induced neurodegenerative model. Biochem Biophys Res Commun 395:207–12
Martinez A, Perez DI (2008) GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer’s disease? J Alzheimers Dis 15:181–91
McCann MJ, Verbalis JG, Stricker EM (1989) LiCl and CCK inhibit gastric emptying and feeding and stimulate OT secretion in rats. Am J Physiol 256:R463–8
Miller JS, Tallarida RJ, Unterwald EM (2009) Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacology 56:1116–23
Nishino S, Ohtomo K, Numata Y, Sato T, Nakahata N, Kurita M (2012) Divergent effects of lithium and sodium valproate on brain-derived neurotrophic factor (BDNF) production in human astrocytoma cells at therapeutic concentrations. Prog Neuropsychopharmacol Biol Psychiatry 39:17–22
O'Brien WT, Klein PS (2009) Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans 37:1133–8
Omata N, Murata T, Takamatsu S, Maruoka N, Mitsuya H, Yonekura Y, Fujibayashi Y, Wada Y (2008) Neuroprotective effect of chronic lithium treatment against hypoxia in specific brain regions with upregulation of cAMP response element binding protein and brain-derived neurotrophic factor but not nerve growth factor: comparison with acute lithium treatment. Bipolar Disord 10:360–8
Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J (2003) Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis 5:301–8
Phiel CJ, Klein PS (2001) Molecular targets of lithium action. Annu Rev Pharmacol Toxicol 41:789–813
Quiroz JA, Gould TD, Manji HK (2004) Molecular effects of lithium. Mol Interv 4:259–72
Quiroz JA, Machado-Vieira R, Zarate CA Jr, Manji HK (2010) Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology 62:50–60
Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A (2009) Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol 156:885–98
Rotte A, Pasham V, Eichenmüller M, Yang W, Qadri SM, Bhandaru M, Lang F (2010) Regulation of basal gastric acid secretion by the glycogen synthase kinase GSK3. J Gastroenterol 45:1022–32
Ryves WJ, Dajani R, Pearl L, Harwood AJ (2002) Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem Biophys Res Commun 290:967–72
Seggie J, Werstiuk ES, Joshi M (1985) Lithium and twenty-four hour rhythms of serum corticosterone, prolactin and growth hormone in pigmented eye rats. Prog Neuropsychopharmacol Biol Psychiatry 9:755–8
Smythe GA, Brandstater JF, Lazarus L (1979) Acute effects of lithium on central dopamine and serotonin activity reflected by inhibition of prolactin and growth hormone secretion in the rat. Aust J Biol Sci 32:329–34
Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B (2004) Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 43:6899–908
Suwalska A, Sobieska M, Rybakowski JK (2010) Serum brain-derived neurotrophic factor in euthymic bipolar patients on prophylactic lithium therapy. Neuropsychobiology 62:229–34
Tonani R, Bindi S, Fancelli D, Pittalà V, D’ Anello M (2004) WO2004007504 A1
Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ (1998) Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology 37:1553–1561
Xu CM, Wang J, Wu P, Xue YX, Zhu WL, Li QQ, Zhai HF, Shi J, Lu L (2011) Glycogen synthase kinase 3β in the nucleus accumbens core is critical for methamphetamine-induced behavioral sensitization. J Neurochem 118:126–39
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM (2009) The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry 14:51–9
Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120:137–49
Yoshimura R, Tsuji K, Ueda N, Nakamura J (2007) Increase of plasma brain-derived neurotrophic factor levels in two psychotic depressed patients responding to lithium addition to paroxetine treatment. Neuropsychiatr Dis Treat 3:683–6
Yu X, Malenka RC (2004) Multiple functions for the cadherin/catenin complex during neuronal development. Neuropharmacology 47:779–86
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS (2003) Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem 278:33067–77
Acknowledgments
The authors wish to thank Claudio Righetti for technical support and GlaxoSmithKline DMPK scientists for measuring blood and brain levels of GSK3 inhibitors after systemic administrations. The authors were GlaxoSmithKline full-time employees when this investigation was performed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caberlotto, L., Carboni, L., Zanderigo, F. et al. Differential effects of glycogen synthase kinase 3 (GSK3) inhibition by lithium or selective inhibitors in the central nervous system. Naunyn-Schmiedeberg's Arch Pharmacol 386, 893–903 (2013). https://doi.org/10.1007/s00210-013-0893-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-013-0893-9