
Abstract
Purpose of Review
Modern social life often demands aberrant light exposures (i.e., jet lag, shift work, or nocturnal life style), which results in desynchronization and misalignment of circadian rhythms. Experimental and epidemiological data suggest that circadian disruption, caused by genetic manipulations or forced light/dark conditions, promotes cancerogenesis. Human genetic studies highlight the contribution of individual clock genes to this process, though the exact function is somewhat controversial.
Recent Findings
Multiple reports demonstrate an association of genetic variations within clock genes with risk of tumor development. Mutations or deregulated expression of clock genes are frequently detected in different tumors and often show correlation with cancer progression and patient prognosis. Cellular studies report contradictory results that clock genes can inhibit as well as support tumor growth and proliferation in different cells.
Summary
Clock genes appear to have multifaceted functions during cancer development and can act both as tumor suppressors or promote cancerogenesis depending on the particular type of tumor. However, the exact conditions and factors which determine such behavior remain elusive and must be investigated in future studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In essentially all living organisms, from cyanobacteria to humans, physiological and behavior responses are manifested in 24-h cycles, thereby, improving metabolic fitness and survival under daily changes of food availability, temperature, and light. The circadian system in mammals has a complex hierarchical structure with central clock and peripheral oscillators. The central clock resides in the suprachiasmatic nuclei (SCN) of the brain and perceives light information directly from the retina via optical nerves [1]. In contrast, other organs contain peripheral clocks, which encompass single-cell oscillators found in virtually all cells of the body or cultured cell lines [2]. Entrainment of peripheral oscillators by SCN via humoral and neuronal pathways, or temperature, is crucial to maintain synchrony of central and peripheral body clocks between each other and the external environment [2]. Alternative entraining cues, such as food, can also reset peripheral clocks independent of SCN and shift the balance in the internal synchrony [3].
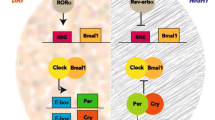
The molecular apparatus of the circadian system consists of several clock genes interlocked in the cell autonomous network of transcriptional-translational feedback loops (TTLs). Transcription factors CLOCK and BMAL1 heterodimerize to induce expression of negative regulators Periods (PER1-3) and Cryptochromes (CRY1-2). Translated PERs and CRYs gradually accumulate in the cytoplasm and form macromolecular complexes up to 1 MDa, that later translocate into the nucleus to repress CLOCK and BMAL1 [4]. Consequent degradation of PERs and CRYs allows CLOCK and BMAL1 to launch a new cycle of transcriptional activity, resulting in 24 h oscillations of transcription [5]. Additionally, a family of nuclear receptors REV-ERBs (α and β) and RORs (α, β, and γ) controls rhythmic expression of BMAL1 gene and other targets through competitive binding to cognate response elements in the promoter region [6]. The molecular clockwork impacts cellular and tissue physiology via the large network of clock-controlled genes (CCGs), whose rhythmic expression is regulated by different clock factors on transcriptional and posttranscriptional levels [7, 8].
Tumor-Protective Function of the Circadian Clock
Extensive experimental evidence, obtained on laboratory animals, demonstrates that disruption of circadian rhythms often leads to malfunction of diverse physiological processes [9]. Thus, the question whether circadian disturbances affect human health in similar manner became a subject of intensive investigations. Indeed, multiple epidemiological studies showed that impaired function of the circadian clock promotes development of different disorders, such as metabolic syndrome, cardiovascular disease and cancer [10]. Light exposure at night, shorter sleep duration, and irregular food intake, along with high caloric diets are inevitable attributes of the modern postindustrial society, and cause desynchronization of body clocks. In turn, circadian disruption impacts multiple aspects of human health potentially leading to impaired metabolism and cancer [11, 12]. Indeed, prolonged shift work and chronic jet lag were associated with development of cancer [13,14,15]. Accumulating evidence in this field prompted the World Health Organization (WHO) to include shift work in the officially recognized list of cancerogenic factors as “probably carcinogenic to humans” [12, 16].
Animal models of circadian disruption, such as chronic jet lag or constant light exposure, clearly demonstrate that the presence of an intact circadian clock system exerts a tumor suppressive function [17,18,19, 20•]. Furthermore, genetic ablation of clock genes in mice resulted in higher frequency of spontaneous and induced tumors [21, 22••]. Consistently, expression of certain clock genes, such as Per2, also suppressed proliferation in murine cancer cells [23]. Together, these findings suggest that synchrony of the circadian system and intact molecular clocks are required to maintain both healthy homeostasis in cell division and to prevent cancer.
Influence of Human Clock Genes on Cancer
Genomic alterations involving clock genes, such as point mutations or copy number variations (CNV), are frequently found in different human cancers (Table 1). As a consequence, tumors often show deregulated expression of clock genes (Table 1). Additionally, human geneticists identified in circadian genes polymorphic regions and small genetic variations, such as single nucleotide polymorphisms (SNPs), which may profoundly affect the function of the gene [25]. In turn, certain polymorphic alleles showed higher frequency of occurrence in different cancer samples, correlating with their malignancy and clinical outcome.
BMAL1
In mice, genetic ablation of single clock gene Bmal1, the partner of Clock, yielded arrhythmic locomotor activity accompanied by multiple pathological conditions, including accelerated aging and cancer [21, 26, 27, 28•]. Tissue-specific deletion of Bmal1 in skin resulted in disrupted proliferation of keratinocytes, resembling a pre-cancerous state [29]. In line with these observations, several studies reported reduced expression of BMAL1 in different types of human malignancies [30,31,32,33]. Moreover, certain genetic variants of BMAL1 gene (rs2278749) negatively correlated with risk of breast cancer [34]. Finally, ectopic expression of BMAL1 efficiently inhibited proliferation of cancer cells, highlighting the tumor suppressor function of this gene [35, 36].
Concurrently, other groups reported results contrasting with aforementioned previous findings. For instance, expression of BMAL1 in malignant pleural mesothelioma appeared to be higher than in normal tissues [37••]. Furthermore, BMAL1 gene was identified as a survival factor for several tumors, since it was required to prevent differentiation of cancer cells and facilitated their mitosis [37••, 38••], suggesting that BMAL1 can also promote tumor growth under certain conditions.
CLOCK
Monoallelic and biallelic mutations of human CLOCK, resulting in reduced expression of this gene, were reported in many cases of colorectal cancer [39]. In particular, the truncating mutation CLOCK T8, was observed in 47 of 53 cases of colorectal cancer, and led to production of aberrant protein. CLOCK T8 lacked a transactivation domain and acted in a dominant-negative manner, perhaps, similar to Clock∆19 mutation in mice [39, 40]. SNP in the 3’UTR of the CLOCK gene (rs1801260), which was previously associated with diurnal preference [41], also correlated with development of the colorectal cancer [42]. CLOCK SNPs rs1801260 and rs3749474 associated with survival in patients with the colorectal cancer [43]. Interestingly, the latter SNP (rs3749474) and another CLOCK SNP (rs3805151) also significantly correlated with risk of breast cancer [44,44,45], suggesting that CLOCK gene polymorphisms may serve as prognostic markers for cancer patients.
It is worthwhile to mention that certain types of breast and colorectal cancers showed higher expression of CLOCK gene compared to normal tissues [46, 47, 48••]. Additionally, CLOCK gene variants (SNPs rs7698022 and rs11932595) significantly associated with more aggressive ER/PR-negative tumors, which did not respond to hormone therapy and had poorer prognosis [46]. Moreover, similar to BMAL1, CLOCK appeared to be required for proliferation of certain tumors [38••, 49], arguing against its role as a tumor suppressor gene.
NPAS2
NPAS2 is considered as a functional homolog of CLOCK with partially overlapping role in the TTLs mechanism [50]. Polymorphisms in the promoter and coding regions (rs2305160, Ala394Thr) of NPAS2 gene were significantly associated with increased risk of certain tumors, such as breast cancer, non-Hodgkin’s lymphomas, hepatocellular carcinomas, and melanomas [51,52,53,54]. Colorectal cancer showed lower expression of NPAS2 compared to healthy tissues, negatively correlating with tumor size, stage and metastasis. Subsequent depletion of NPAS2 with siRNA resulted in increased proliferation, migration and invasiveness of tumor cells [55].
In contrast, NPAS2 expression in hepatocellular carcinoma cells was upregulated, compared to peri-tumoral tissues, and positively correlated with both tumor progression and poorer prognosis. Furthermore, knockdown of NPAS2 in these cells yielded decreased tumor growth and proliferation, whereas NPAS2 overexpression produced an opposite effect [56], asserting that NPAS2 is necessary for development of certain tumors.
PERs
PERs were the first clock genes linked to the cancer development in humans and mice [57,58,59]. The initial evidence hinting to the tumor suppressor function of mammalian PERs originated from experiments on Per2m/m mice, which developed tumors after γ-irradiation [57]. Indeed, PER2 gene, transfected in sarcoma cells, suppressed proliferation and tumor growth in a dose-dependent manner [60]. Moreover, antiproliferative properties were also attributed to PER1, which suppressed cell growth in several tumor cell lines, and showed reduced expression in lung cancer [61]. Consistently, other cancers also exhibited decreased activity of PER genes [59, 62,63,64,65,66]. Moreover, lower levels of PERs often correlated with higher malignancy and poorer survival [30, 63]. Remarkably, a conserved residue in human PER2, Serine 662, mutated to Glycine in case of Familial advanced sleep-phase syndrome (FASPS) [67], played a prominent role in tumorigenesis. Transfection of PER2 S662G and S662D variants markedly increased oncogenic transformation and decreased apoptosis in Per2−/− mouse embryonic fibroblasts (MEFs) [68].
A variable number tandem repeat (VNTR) sequence within PER3 gene (rs57875989), containing 4 or 5 copies of a 54-bp sequence (18 amino acids), was previously linked with diurnal preference (PER3 4/4 evening and 5/5 morning phenotype) and sleep homeostasis [69]. The same VNTR region showed significant association with higher risk of breast cancer for PER3 5/5 carriers [58]. Moreover, individuals carrying five alleles in PER3 were also more susceptible to formation of adenomas [70]. Besides this observation, several other polymorphic regions in PER3 gene were shown to modify risk of cancer. For instance, SNP (rs228729) significantly correlated with risk of lung cancer [64], whereas SNP (rs2640908) associated with overall survival in patients with hepatocellular carcinoma [71].
Nevertheless, expression of PERs is not generally reduced during cancerogenesis. Significantly higher levels of PER2 mRNA and protein were detected in samples from gastric cancer [72]. Similarly, expression of PER1 and PER2 positively correlated with tumor size in colorectal carcinomas [73]. Therefore, although PERs exhibit mostly antioncogenic properties, it might not be the case in some specific tumor types.
CRYs
The link between cancer and CRY genes is particularly interesting, since mammalian CRYs have high sequence homology to photolyases, enzymes involved in repair of the light-induced DNA damage [74]. Indeed, lower expression levels of CRY1 and CRY2 were reported in different cancers [75]. Polymorphisms in CRY1 (rs1056560) and CRY2 (rs1401417) genes also significantly modified risk of breast cancer [45]. SNP (rs1056560) in 3′UTR region of CRY1 gene correlated with overall survival in gastric cancer and modulated patients’ response to platinum-based adjuvant chemotherapy [76]. Several genetic variants of CRY2 showed significant association with susceptibility to non-Hodgkin’s lymphomas [77].
Interestingly, loss of Cry proteins in murine models significantly reduced the risk of cancer [78]. In addition, higher expression of CRY1 correlated with poor prognosis in patients with colorectal cancer [79], suggesting tumor promoting functions for CRY1.
REV-ERBs and RORs
Activation of both REV-ERBα and β by a synthetic agonist (SR9011) suppressed the viability of breast cancer cells, hinting at the tumor suppressor potential of these transcriptional regulators [80]. Nevertheless, certain breast cancer tumors showed amplification of the genomic region containing REV-ERBα gene [81, 82]. Proliferation of highly aggressive ERBB2-positive breast cancer cells was dependent on REV-ERBα [83]. Moreover, pharmacological inhibition of REV-ERBs in cancer cells led to enhanced cytotoxicity and improved sensitivity to anti-cancer drugs [84].
A family of RORs include three members (RORα, RORβ, and RORγ), which antagonize REV-ERBs and activate transcription of target genes [85]. The function of RORs, however, spans beyond the regulation of circadian rhythms, since RORs are also implicated in differentiation and maturation of various cell types [86]. Mounting evidence suggests that RORs may play a tumor suppressor role in cancer related pathways [87]. Indeed, ligand-induced activation or transfection of exogenous RORα inhibited proliferation and tumor growth in different cancer lines [88,89,90]. Mice with targeted deletion of Rorγ were highly susceptible to development of aggressive highly metastatic T-cell lymphomas [91]. Finally, different tumors and cancer cell lines showed reduced expression levels of RORs, often correlating with higher malignancy and poor prognosis [92,93,94,95,96,97].
Accordingly, multiple genetic variations within RORs genes were associated with risk of cancer, such as SNPs in RORα (rs7164773, rs10519097, rs1482057, and rs12914272 for breast cancer) and RORβ (rs3903529, rs3750420, and rs7867494 for breast cancer) [34, 44, 98].
Notably, a particular isoform of RORα, RORα2, promoted cell motility and migration in breast cancer cells and showed elevated protein abundance in breast cancer tumors [99]. Additionally, expression of RORβ was found to be increased in the highly metastatic leiomyosarcoma of uterus [100]. In turn, prostate cancer showed higher expression of RORγ, correlating with malignancy and metastasis. Subsequent siRNA-depletion of RORγ or treatment with its chemical antagonists impaired viability of prostate cancer cells and hampered tumor growth [101••].
TIMELESS
Although, TIMELESS is a mammalian homolog of the bona fide component of the Drosophila clock, it does not have clearly assigned function within mammalian TTLs [102]. Nevertheless, TIMELESS was suggested to mediate DNA damage induced resetting of the circadian clock and facilitate coupling of the circadian oscillator with the cell cycle [103, 104]. Furthermore, TIMELESS had a critical role in DNA damage-mediated activation of both check-point kinases CHK1 and CHK2, thus, influencing proliferation and sensitivity to anti-cancer drugs in tumor cells [103, 105,106,107]. Consistent with this, deregulated expression of TIMELESS was documented in several tumors, such as lung and breast cancers, negatively correlating with patient survival [106, 107]. Genetic variants of TIMELESS gene (rs2291738 and rs7302060) were also significantly associated with development of breast cancer [108].
Conclusions
Experimental studies, in combination with epidemiological and genetic data, favor the concept that human circadian clock and, in particular, clock genes play an important role during tumorigenesis. According to the prevailing hypothesis, clock genes manifest predominantly antioncogenic properties and inhibit uncontrolled proliferation via homeostatic regulation of cell division. Altered expression or mutations of clock genes per se are rather unlikely to be a primary driver of cancer, though these aspects may definitely contribute to growth and progression of tumors via disrupted temporal control of physiology at the local and systemic levels. On the other hand, clock genes may also support tumor growth under certain conditions, since many cancers express high levels of clock genes and require them for survival. Perhaps, these effects depend on the type of cancer and the tissue of tumor origin. Indeed, low or high expression levels of several clock genes in tumors from different tissues may have opposite prognostic values (Fig. 1). For instance, high expression of PER1 in stomach cancer had a negative prognosis on survival of patients, whereas in the case of liver and cervical cancers—higher expression of PER1 was linked to a more favorable prognosis [109,110,111]. Similar phenomena can be also observed for CRY2 and REV-ERBα (Fig. 1), suggesting that individual mutational signatures occurring in different cancers may define the overall influence of clock genes on tumor physiology. Thus, future research, focused on the exact molecular mechanisms determining such tumor-specific behavior, will help us to improve our comprehension of mutually driven interactions between the circadian clock and cancer.

Prognostic value of clock gene expression in different cancers. Kaplan–Meier survival graphs of patient cohorts, grouped upon expression levels of PER1, CRY2, and REV-ERBα. Images and data available from v17.proteinatlas.org
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol. 2010;72(1):551–77. https://doi.org/10.1146/annurev-physiol-021909-135919.
Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35(1):445–62. https://doi.org/10.1146/annurev-neuro-060909-153128.
Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14(23):2950–61. https://doi.org/10.1101/gad.183500.
Aryal RP, Kwak PB, Tamayo AG, Gebert M, Chiu PL, Walz T, et al. Macromolecular assemblies of the mammalian circadian clock. Mol Cell. 2017;67(5):770–82 e6. https://doi.org/10.1016/j.molcel.2017.07.017.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18(3):164–79. https://doi.org/10.1038/nrg.2016.150.
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110(2):251–60. https://doi.org/10.1016/S0092-8674(02)00825-5.
Eckel-Mahan K, Sassone-Corsi P. Metabolism and the circadian clock converge. Physiol Rev. 2013;93(1):107–35. https://doi.org/10.1152/physrev.00016.2012.
Robles MS, Humphrey SJ, Mann M. Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 2017;25(1):118–27. https://doi.org/10.1016/j.cmet.2016.10.004.
Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9(10):764–75. https://doi.org/10.1038/nrg2430.
Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26(10):R432–43. https://doi.org/10.1016/j.cub.2016.04.011.
Stevens RG, Brainard GC, Blask DE, Lockley SW, Motta ME. Breast cancer and circadian disruption from electric lighting in the modern world. CA Cancer J Clin. 2014;64(3):207–18. https://doi.org/10.3322/caac.21218.
Wang F, Zhang L, Zhang Y, Zhang B, He Y, Xie S, et al. Meta-analysis on night shift work and risk of metabolic syndrome. Obes Rev. 2014;15(9):709–20. https://doi.org/10.1111/obr.12194.
Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Night work and breast cancer risk: a systematic review and meta-analysis. Eur J Cancer. 2005;41(13):2023–32. https://doi.org/10.1016/j.ejca.2005.05.010.
Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. Night work and risk of breast cancer. Epidemiology. 2006;17(1):108–11. https://doi.org/10.1097/01.ede.0000190539.03500.c1.
Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, et al. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J Natl Cancer Inst. 2001;93(20):1563–8. https://doi.org/10.1093/jnci/93.20.1563.
Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, et al. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8(12):1065–6. https://doi.org/10.1016/S1470-2045(07)70373-X.
Filipski E, Delaunay F, King VM, Wu MW, Claustrat B, Grechez-Cassiau A, et al. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 2004;64(21):7879–85. https://doi.org/10.1158/0008-5472.CAN-04-0674.
Popovich IG, Zabezhinski MA, Panchenko AV, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Exposure to light at night accelerates aging and spontaneous uterine carcinogenesis in female 129/Sv mice. Cell Cycle. 2013;12(11):1785–90. https://doi.org/10.4161/cc.24879.
Yasuniwa Y, Izumi H, Wang KY, Shimajiri S, Sasaguri Y, Kawai K, et al. Circadian disruption accelerates tumor growth and angio/stromagenesis through a Wnt signaling pathway. PLoS One. 2010;5(12):e15330. https://doi.org/10.1371/journal.pone.0015330.
• Van Dycke KC, Rodenburg W, van Oostrom CT, van Kerkhof LW, Pennings JL, Roenneberg T, et al. Chronically alternating light cycles increase breast cancer risk in mice. Curr Biol. 2015;25(14):1932–7. https://doi.org/10.1016/j.cub.2015.06.012. This study demonstrated that internal desynchronization and sleep disturbance, induced by shift work, lead to cancer development and obesity.
Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One. 2010;5(6):e10995. https://doi.org/10.1371/journal.pone.0010995.
•• Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell. 2016;30(6):909–24. https://doi.org/10.1016/j.ccell.2016.10.007. This study showed that chronic circadian disruption induces spontaneous hepatocarcinogenesis via global gene deregulation and metabolic disruption.
Hua H, Wang Y, Wan C, Liu Y, Zhu B, Yang C, et al. Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci. 2006;97(7):589–96. https://doi.org/10.1111/j.1349-7006.2006.00225.x.
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D83. https://doi.org/10.1093/nar/gkw1121.
Shastry BS. SNPs: impact on gene function and phenotype. Methods Mol Biol. 2009;578:3–22. https://doi.org/10.1007/978-1-60327-411-1_1.
Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103(7):1009–17. https://doi.org/10.1016/S0092-8674(00)00205-1.
Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20(14):1868–73. https://doi.org/10.1101/gad.1432206.
• Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, et al. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016;24(2):324–31. https://doi.org/10.1016/j.cmet.2016.07.001. This study showed that loss of circadian genes or chronic jet lag promotes lung tumorigenesis.
Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, et al. Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc Natl Acad Sci USA. 2012;109(29):11758–63. https://doi.org/10.1073/pnas.1209592109.
Hsu CM, Lin SF, Lu CT, Lin PM, Yang MY. Altered expression of circadian clock genes in head and neck squamous cell carcinoma. Tumour Biol. 2012;33(1):149–55. https://doi.org/10.1007/s13277-011-0258-2.
Taniguchi H, Fernandez AF, Setien F, Ropero S, Ballestar E, Villanueva A, et al. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009;69(21):8447–54. https://doi.org/10.1158/0008-5472.CAN-09-0551.
Yang MY, Chang JG, Lin PM, Tang KP, Chen YH, Lin HY, et al. Downregulation of circadian clock genes in chronic myeloid leukemia: alternative methylation pattern of hPER3. Cancer Sci. 2006;97(12):1298–307. https://doi.org/10.1111/j.1349-7006.2006.00331.x.
Fu L, Kettner NM. The circadian clock in cancer development and therapy. Prog Mol Biol Transl Sci. 2013;119:221–82. https://doi.org/10.1016/B978-0-12-396971-2.00009-9.
Zienolddiny S, Haugen A, Lie JA, Kjuus H, Anmarkrud KH, Kjaerheim K. Analysis of polymorphisms in the circadian-related genes and breast cancer risk in Norwegian nurses working night shifts. Breast Cancer Res. 2013;15(4):R53. https://doi.org/10.1186/bcr3445.
Tang Q, Cheng B, Xie M, Chen Y, Zhao J, Zhou X, et al. Circadian clock gene Bmal1 inhibits tumorigenesis and increases paclitaxel sensitivity in tongue squamous cell carcinoma. Cancer Res. 2017;77(2):532–44. https://doi.org/10.1158/0008-5472.CAN-16-1322.
Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, Walton ZE, et al. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab. 2015;22(6):1009–19. https://doi.org/10.1016/j.cmet.2015.09.003.
•• Elshazley M, Sato M, Hase T, Yamashita R, Yoshida K, Toyokuni S, et al. The circadian clock gene BMAL1 is a novel therapeutic target for malignant pleural mesothelioma. Int J Cancer. 2012;131(12):2820–31. https://doi.org/10.1002/ijc.27598. In this study, the authors demonstrated that BMAL1 was overexpressed in cancer cells and supported their proliferation.
•• Puram RV, Kowalczyk MS, de Boer CG, Schneider RK, Miller PG, McConkey M, et al. Core circadian clock genes regulate leukemia stem cells in AML. Cell. 2016;165(2):303–16. https://doi.org/10.1016/j.cell.2016.03.015. This work showed that Bmal1 and Clock were required for growth of leukemia cells and loss Bmal1 gene selectively inhibited leukemia progression.
Alhopuro P, Bjorklund M, Sammalkorpi H, Turunen M, Tuupanen S, Bistrom M, et al. Mutations in the circadian gene CLOCK in colorectal cancer. Mol Cancer Res. 2010;8(7):952–60. https://doi.org/10.1158/1541-7786.MCR-10-0086.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280(5369):1564–9. https://doi.org/10.1126/science.280.5369.1564.
Katzenberg D, Young T, Finn L, Lin L, King DP, Takahashi JS, et al. A CLOCK polymorphism associated with human diurnal preference. Sleep. 1998;21(6):569–76. https://doi.org/10.1093/sleep/21.6.569.
Karantanos T, Theodoropoulos G, Gazouli M, Vaiopoulou A, Karantanou C, Stravopodis DJ, et al. Association of the clock genes polymorphisms with colorectal cancer susceptibility. J Surg Oncol. 2013;108(8):563–7. https://doi.org/10.1002/jso.23434.
Zhou F, He X, Liu H, Zhu Y, Jin T, Chen C, et al. Functional polymorphisms of circadian positive feedback regulation genes and clinical outcome of Chinese patients with resected colorectal cancer. Cancer. 2012;118(4):937–46. https://doi.org/10.1002/cncr.26348.
Benna C, Helfrich-Forster C, Rajendran S, Monticelli H, Pilati P, Nitti D, et al. Genetic variation of clock genes and cancer risk: a field synopsis and meta-analysis. Oncotarget. 2017;8(14):23978–95. https://doi.org/10.18632/oncotarget.15074.
Dai H, Zhang L, Cao M, Song F, Zheng H, Zhu X, et al. The role of polymorphisms in circadian pathway genes in breast tumorigenesis. Breast Cancer Res Treat. 2011;127(2):531–40. https://doi.org/10.1007/s10549-010-1231-2.
Hoffman AE, Yi CH, Zheng T, Stevens RG, Leaderer D, Zhang Y, et al. CLOCK in breast tumorigenesis: genetic, epigenetic, and transcriptional profiling analyses. Cancer Res. 2010;70(4):1459–68. https://doi.org/10.1158/0008-5472.CAN-09-3798.
Wang L, Chen B, Wang Y, Sun N, Lu C, Qian R, et al. hClock gene expression in human colorectal carcinoma. Mol Med Rep. 2013;8(4):1017–22. https://doi.org/10.3892/mmr.2013.1643.
•• Yu JZ, Sun N, Bei YB, Li XB, Lu C, Hua LC. Circadian gene hCLOCK contributes to progression of colorectal carcinoma and is directly regulated by tumorsuppressive microRNA124. Mol Med Rep. 2017;16(6):7923–30. https://doi.org/10.3892/mmr.2017.7596.
Li A, Lin X, Tan X, Yin B, Han W, Zhao J, et al. Circadian gene clock contributes to cell proliferation and migration of glioma and is directly regulated by tumor-suppressive miR-124. FEBS Lett. 2013;587(15):2455–60. https://doi.org/10.1016/j.febslet.2013.06.018.
DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007;10(5):543–5. https://doi.org/10.1038/nn1884.
Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, et al. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008;107(3):421–5. https://doi.org/10.1007/s10549-007-9565-0.
Zhu Y, Leaderer D, Guss C, Brown HN, Zhang Y, Boyle P, et al. Ala394Thr polymorphism in the clock gene NPAS2: a circadian modifier for the risk of non-Hodgkin's lymphoma. Int J Cancer. 2007;120(2):432–5. https://doi.org/10.1002/ijc.22321.
Yuan P, Wang S, Zhou F, Wan S, Yang Y, Huang X, et al. Functional polymorphisms in the NPAS2 gene are associated with overall survival in transcatheter arterial chemoembolization-treated hepatocellular carcinoma patients. Cancer Sci. 2014;105(7):825–32. https://doi.org/10.1111/cas.12428.
Franzoni A, Markova-Car E, Devic-Pavlic S, Jurisic D, Puppin C, Mio C, et al. A polymorphic GGC repeat in the NPAS2 gene and its association with melanoma. Exp Biol Med. 2017;242(15):1553–8. https://doi.org/10.1177/1535370217724093.
Xue X, Liu F, Han Y, Li P, Yuan B, Wang X, et al. Silencing NPAS2 promotes cell growth and invasion in DLD-1 cells and correlated with poor prognosis of colorectal cancer. Biochem Biophys Res Commun. 2014;450(2):1058–62. https://doi.org/10.1016/j.bbrc.2014.06.104.
Yuan P, Li J, Zhou F, Huang Q, Zhang J, Guo X, et al. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. 2017;8(3):e2704. https://doi.org/10.1038/cddis.2017.131.
Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111(1):41–50. https://doi.org/10.1016/S0092-8674(02)00961-3.
Zhu Y, Brown HN, Zhang Y, Stevens RG, Zheng T. Period3 structural variation: a circadian biomarker associated with breast cancer in young women. Cancer Epidemiol Biomarkers Prev. 2005;14(1):268–70.
Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis. 2005;26(7):1241–6. https://doi.org/10.1093/carcin/bgi075.
Miyazaki K, Wakabayashi M, Hara Y, Ishida N. Tumor growth suppression in vivo by overexpression of the circadian component, PER2. Genes Cells. 2010;15(4):351–8. https://doi.org/10.1111/j.1365-2443.2010.01384.x.
Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006;22(3):375–82. https://doi.org/10.1016/j.molcel.2006.03.038.
Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu J, et al. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009;69(19):7619–25. https://doi.org/10.1158/0008-5472.CAN-08-4199.
Liu B, Xu K, Jiang Y, Li X. Aberrant expression of Per1, Per2 and Per3 and their prognostic relevance in non-small cell lung cancer. Int J Clin Exp Pathol. 2014;7(11):7863–71.
Couto P, Miranda D, Vieira R, Vilhena A, De Marco L, Bastos-Rodrigues L. Association between CLOCK, PER3 and CCRN4L with nonsmall cell lung cancer in Brazilian patients. Mol Med Rep. 2014;10(1):435–40. https://doi.org/10.3892/mmr.2014.2224.
Yeh KT, Yang MY, Liu TC, Chen JC, Chan WL, Lin SF, et al. Abnormal expression of period 1 (PER1) in endometrial carcinoma. J Pathol. 2005;206(1):111–20. https://doi.org/10.1002/path.1756.
Lin YM, Chang JH, Yeh KT, Yang MY, Liu TC, Lin SF, et al. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol Carcinog. 2008;47(12):925–33. https://doi.org/10.1002/mc.20446.
Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291(5506):1040–3. https://doi.org/10.1126/science.1057499.
Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19(3):397–405. https://doi.org/10.1038/cdd.2011.103.
Dijk DJ, Archer SN. PERIOD3, circadian phenotypes, and sleep homeostasis. Sleep Med Rev. 2010;14(3):151–60. https://doi.org/10.1016/j.smrv.2009.07.002.
Alexander M, Burch JB, Steck SE, Chen CF, Hurley TG, Cavicchia P, et al. Case-control study of the PERIOD3 clock gene length polymorphism and colorectal adenoma formation. Oncol Rep. 2015;33(2):935–41. https://doi.org/10.3892/or.2014.3667.
Zhao B, Lu J, Yin J, Liu H, Guo X, Yang Y, et al. A functional polymorphism in PER3 gene is associated with prognosis in hepatocellular carcinoma. Liver Int. 2012;32(9):1451–9. https://doi.org/10.1111/j.1478-3231.2012.02849.x.
Hu ML, Yeh KT, Lin PM, Hsu CM, Hsiao HH, Liu YC, et al. Deregulated expression of circadian clock genes in gastric cancer. BMC Gastroenterol. 2014;14(1):67. https://doi.org/10.1186/1471-230X-14-67.
Momma T, Okayama H, Saitou M, Sugeno H, Yoshimoto N, Takebayashi Y, et al. Expression of circadian clock genes in human colorectal adenoma and carcinoma. Oncol Lett. 2017;14(5):5319–25. https://doi.org/10.3892/ol.2017.6876.
Thompson CL, Sancar A. Photolyase/cryptochrome blue-light photoreceptors use photon energy to repair DNA and reset the circadian clock. Oncogene. 2002;21(58):9043–56. https://doi.org/10.1038/sj.onc.1205958.
Mazzoccoli G, Colangelo T, Panza A, Rubino R, De Cata A, Tiberio C, et al. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol Cancer. 2016;15(1):6. https://doi.org/10.1186/s12943-016-0492-8.
Qu F, Qiao Q, Wang N, Ji G, Zhao H, He L, et al. Genetic polymorphisms in circadian negative feedback regulation genes predict overall survival and response to chemotherapy in gastric cancer patients. Sci Rep. 2016;6(1):22424. https://doi.org/10.1038/srep22424.
Hoffman AE, Zheng T, Stevens RG, Ba Y, Zhang Y, Leaderer D, et al. Clock-cancer connection in non-Hodgkin’s lymphoma: a genetic association study and pathway analysis of the circadian gene cryptochrome 2. Cancer Res. 2009;69(8):3605–13. https://doi.org/10.1158/0008-5472.CAN-08-4572.
Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106(8):2841–6. https://doi.org/10.1073/pnas.0813028106.
Yu H, Meng X, Wu J, Pan C, Ying X, Zhou Y, et al. Cryptochrome 1 overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS One. 2013;8(4):e61679. https://doi.org/10.1371/journal.pone.0061679.
Wang Y, Kojetin D, Burris TP. Anti-proliferative actions of a synthetic REV-ERBalpha/beta agonist in breast cancer cells. Biochem Pharmacol. 2015;96(4):315–22. https://doi.org/10.1016/j.bcp.2015.06.010.
Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10(6):529–41. https://doi.org/10.1016/j.ccr.2006.10.009.
Davis LM, Harris C, Tang L, Doherty P, Hraber P, Sakai Y, et al. Amplification patterns of three genomic regions predict distant recurrence in breast carcinoma. J Mol Diagn. 2007;9(3):327–36. https://doi.org/10.2353/jmoldx.2007.060079.
Kourtidis A, Jain R, Carkner RD, Eifert C, Brosnan MJ, Conklin DS. An RNA interference screen identifies metabolic regulators NR1D1 and PBP as novel survival factors for breast cancer cells with the ERBB2 signature. Cancer Res. 2010;70(5):1783–92. https://doi.org/10.1158/0008-5472.CAN-09-1550.
De Mei C, Ercolani L, Parodi C, Veronesi M, Lo Vecchio C, Bottegoni G, et al. Dual inhibition of REV-ERBbeta and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene. 2015;34(20):2597–608. https://doi.org/10.1038/onc.2014.203.
Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythm. 2005;20(5):391–403. https://doi.org/10.1177/0748730405277232.
Cook DN, Kang HS, Jetten AM. Retinoic Acid-Related Orphan Receptors (RORs): Regulatory Functions in Immunity, Development, Circadian Rhythm, and Metabolism. Nucl Recept Res. 2015;2:101185. https://doi.org/10.11131/2015/101185.
Du J, Xu R. RORalpha, a potential tumor suppressor and therapeutic target of breast cancer. Int J Mol Sci. 2012;13(12):15755–66. https://doi.org/10.3390/ijms131215755.
Moretti RM, Marelli MM, Motta M, Polizzi D, Monestiroli S, Pratesi G, et al. Activation of the orphan nuclear receptor RORalpha induces growth arrest in androgen-independent DU 145 prostate cancer cells. Prostate. 2001;46(4):327–35. https://doi.org/10.1002/1097-0045(20010301)46:4<327::AID-PROS1040>3.0.CO;2-6.
Xiong G, Wang C, Evers BM, Zhou BP, Xu R. RORalpha suppresses breast tumor invasion by inducing SEMA3F expression. Cancer Res. 2012;72(7):1728–39. https://doi.org/10.1158/0008-5472.CAN-11-2762.
Moretti RM, Montagnani Marelli M, Sala A, Motta M, Limonta P. Activation of the orphan nuclear receptor RORalpha counteracts the proliferative effect of fatty acids on prostate cancer cells: crucial role of 5-lipoxygenase. Int J Cancer. 2004;112(1):87–93. https://doi.org/10.1002/ijc.20387.
Ueda E, Kurebayashi S, Sakaue M, Backlund M, Koller B, Jetten AM. High incidence of T-cell lymphomas in mice deficient in the retinoid-related orphan receptor RORgamma. Cancer Res. 2002;62(3):901–9.
Risinger JI, Allard J, Chandran U, Day R, Chandramouli GV, Miller C, et al. Gene expression analysis of early stage endometrial cancers reveals unique transcripts associated with grade and histology but not depth of invasion. Front Oncol. 2013;3:139. https://doi.org/10.3389/fonc.2013.00139.
Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25(20):2901–8. https://doi.org/10.1038/sj.onc.1209314.
Fu RD, Qiu CH, Chen HA, Zhang ZG, Lu MQ. Retinoic acid receptor-related receptor alpha (RORalpha) is a prognostic marker for hepatocellular carcinoma. Tumour Biol. 2014;35(8):7603–10. https://doi.org/10.1007/s13277-014-2007-9.
Kottorou AE, Antonacopoulou AG, Dimitrakopoulos FI, Tsamandas AC, Scopa CD, Petsas T, et al. Altered expression of NFY-C and RORA in colorectal adenocarcinomas. Acta Histochem. 2012;114(6):553–61. https://doi.org/10.1016/j.acthis.2011.10.005.
Brozyna AA, Jozwicki W, Skobowiat C, Jetten A, Slominski AT. RORalpha and RORgamma expression inversely correlates with human melanoma progression. Oncotarget. 2016;7(39):63261–82. https://doi.org/10.18632/oncotarget.11211.
Oh TG, Bailey P, Dray E, Smith AG, Goode J, Eriksson N, et al. PRMT2 and RORgamma expression are associated with breast cancer survival outcomes. Mol Endocrinol. 2014;28(7):1166–85. https://doi.org/10.1210/me.2013-1403.
Truong T, Liquet B, Menegaux F, Plancoulaine S, Laurent-Puig P, Mulot C, et al. Breast cancer risk, nightwork, and circadian clock gene polymorphisms. Endocr Relat Cancer. 2014;21(4):629–38. https://doi.org/10.1530/ERC-14-0121.
Kim K, Lee JM, Yu YS, Kim H, Nam HJ, Moon HG, et al. RORalpha2 requires LSD1 to enhance tumor progression in breast cancer. Sci Rep. 2017;7(1):11994. https://doi.org/10.1038/s41598-017-12344-0.
Davidson B, Abeler VM, Forsund M, Holth A, Yang Y, Kobayashi Y, et al. Gene expression signatures of primary and metastatic uterine leiomyosarcoma. Hum Pathol. 2014;45(4):691–700. https://doi.org/10.1016/j.humpath.2013.11.003.
•• Wang J, Zou JX, Xue X, Cai D, Zhang Y, Duan Z, et al. ROR-gamma drives androgen receptor expression and represents a therapeutic target in castration-resistant prostate cancer. Nat Med. 2016;22(5):488–96. https://doi.org/10.1038/nm.4070. This study demonstrated that overexpression of RORγ was required for survival of prostate cancer cells and chemical inhibitors of RORγ reduced tumor growth and metastasis.
Gotter AL. A timeless debate: resolving TIM's noncircadian roles with possible clock function. Neuroreport. 2006;17(12):1229–33. https://doi.org/10.1097/01.wnr.0000233092.90160.92.
Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25(8):3109–16. https://doi.org/10.1128/MCB.25.8.3109-3116.2005.
Engelen E, Janssens RC, Yagita K, Smits VA, van der Horst GT, Tamanini F. Mammalian TIMELESS is involved in period determination and DNA damage-dependent phase advancing of the circadian clock. PLoS One. 2013;8(2):e56623. https://doi.org/10.1371/journal.pone.0056623.
Yang X, Wood PA, Hrushesky WJ. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J Biol Chem. 2010;285(5):3030–4. https://doi.org/10.1074/jbc.M109.050237.
Yoshida K, Sato M, Hase T, Elshazley M, Yamashita R, Usami N, et al. TIMELESS is overexpressed in lung cancer and its expression correlates with poor patient survival. Cancer Sci. 2013;104(2):171–7. https://doi.org/10.1111/cas.12068.
Mao Y, Fu A, Leaderer D, Zheng T, Chen K, Zhu Y. Potential cancer-related role of circadian gene TIMELESS suggested by expression profiling and in vitro analyses. BMC Cancer. 2013;13(1):498. https://doi.org/10.1186/1471-2407-13-498.
Fu A, Leaderer D, Zheng T, Hoffman AE, Stevens RG, Zhu Y. Genetic and epigenetic associations of circadian gene TIMELESS and breast cancer risk. Mol Carcinog. 2012;51(12):923–9. https://doi.org/10.1002/mc.20862.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. https://doi.org/10.1126/science.1260419.
Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science. 2017;356(6340):eaal3321. https://doi.org/10.1126/science.aal3321.
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352):eaan2507. https://doi.org/10.1126/science.aan2507.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anton Shostak declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Sleep and 3D (Cancer, Cardiovascular, Metabolic Diseases)
Rights and permissions
About this article

Cite this article
Shostak, A. Human Clock Genes and Cancer. Curr Sleep Medicine Rep 4, 65–73 (2018). https://doi.org/10.1007/s40675-018-0102-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40675-018-0102-y