
Abstract
Circadian rhythms are patterns of behavior, physiology, and metabolism that occur within a period of approximately 24 h. These rhythms are generated endogenously, but synchronize to external cues, thus enabling organisms to beneficially align physiological processes to the inherently dynamic, yet predictable, seasonal changes in the day–night cycle. The cell autonomous circadian oscillator temporally coordinates cellular processes, including metabolism, proliferation, cell signaling, organelle function, proteostasis, and DNA damage repair to sustain cellular homeostasis. It is hypothesized that the circadian oscillators evolved as a “flight from light” mechanism to minimize UV damage to single stranded DNA by restricting DNA replication to the nighttime. Support for this hypothesis is accumulating with the recent observation that the circadian rhythm and cell cycle are intimately coupled to each other, so that specific phases of cell cycle occur at a defined phase of the circadian oscillator at single-cell level [1]. Furthermore, chronic circadian disruption perturbs cellular homeostasis and predisposes to cancer. Conversely, numerous cancer cell lines display severe circadian alterations, which likely contribute to aggressive proliferation of the tumor. Hence, it is becoming increasingly important to understand the relevance of circadian rhythm for optimizing fitness under natural conditions and its utility and adaptability in the modern world so that the knowledge can be better leveraged for the prevention and treatment of cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Circadian rhythms
- Circadian clock
- Circadian clock cell cycle regulation
- Circadian clock xenobiotic detoxification
- Circadian clock DNA damage response
- Circadian clock energy metabolism
- CLOCK-BMAL1
- Chronodisruption
- Melatonin
Circadian rhythms evolved in a predictable environment of light:dark and the associated daily rhythm in access to food. Accordingly, at the organism level, there are mechanisms to entrain the circadian system to changes in light and food availability in different seasons. However, in the modern anthropogenic world, the use of electrical lighting and abundance of food availability throughout 24 h during the day:night cycle can cause repetitive perturbation of the circadian clock and increase susceptibility to cancer. In this chapter we will first introduce the circadian clock and its molecular mechanisms in mammals, followed by an overview of the epidemiological and experimental evidence linking circadian dysfunction to cancer, especially considering evidence obtained from animal models. Then, we will cover four broadly defined aspects of physiology that are regulated by the clock, and within which we find the mechanisms upon which the hallmarks of cancer arise: (1) cell cycle regulation and DNA damage response, (2) xenobiotic detoxification, (3) endocrine function, and (4) energy metabolism.
These discussions will lay the framework to understand how circadian disruption can increase cancer risk and shed light on novel lifestyle or pharmacological cancer treatments that can build on a better understanding of the circadian cancer link.
Mechanisms of the Clock
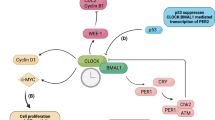
In mammals, the circadian system is based on a cell-autonomous and self-sustaining molecular oscillator. At the molecular level the circadian oscillator is a genetic circuit composed of two interlocking transcription–translation feedback loops that revolve around the transcription factors CLOCK and BMAL1 (and their respective homologs NPAS2 and BMAL2). CLOCK and BMAL1 function as heterodimers that bind to E-box elements at promoter regions of their target genes. CLOCK-BMAL1 drive the expression of the Cryptochrome (Cry1 and Cry2), and Period (Per1, Per2) genes. In turn, CRYs and PERs form complexes that repress CLOCK-BMAL1 activity, ultimately suppressing their own expression. Eventually, CRYs and PERs are degraded, relieving CLOCK-BMAL1 repression and beginning the cycle anew. Essentially, the interplay between CLOCK/NPAS2, BMAL1/2, CRYs and PERs gives rise to the self-sustained, near-24 h, alternating activation–repression cycles that are the proverbial ticking of the clock.
In a second loop, CLOCK and BMAL1 activate the Rev-Erb- and Ror-class nuclear hormone receptors. REV-ERBs and RORs are, respectively, transcriptional repressors and activators that bind to ROR elements (ROREs) present in the Bmal1 gene promoter, and whose interplay enforces rhythmic expression of Bmal1. Additionally, REV-ERBs and RORs also act on fine-tuning the rhythmic expression of additional clock components. These rhythms are then propagated to off-clock genes generating large-scale transcription rhythms (up to 15 % of expressed genes in any given tissue), which eventually manifest as overt physiological rhythms.
These transcription–translation loops form the core of the molecular clock, but the mechanisms that underlie the complete circadian oscillator are far more complex. For instance, the duration of the cycle needs to have a period of completion close to 24 h. One feature of this process is that the translation of circadian mRNAs does not follow immediately after transcription; instead, there is an approximate 2–4 h delay between generation of the mRNA and protein synthesis. This delay is in part regulated via miRNA-dependent mechanisms, consistent with findings that DICER deficient cells exhibited a shortened circadian period length and had an associated acceleration on PER1 and PER2 translation [2]. In addition, the timing of individual steps and of the overall circadian cycle involves the extensive posttranslational regulation of oscillator components. All known oscillator components are subject to posttranslational modifications of which most extensively studied and thus the best understood is protein phosphorylation. For example, casein kinase Iε (CKIε) phosphorylates the period proteins to regulate their degradation by the β-transducin repeat containing protein 1 (β-TrCP1)-SCF complex. In contrast, PER2 stability is enhanced by phosphorylation by casein kinase 2 (CK2). CRYs stability is similarly regulated by phosphorylation by the AMP-activated protein kinase (AMPK), an event that promotes CRY association with the F-box component of the SCF-FBXL3 ubiquitin ligase complex. In addition, phosphorylation also regulates subcellular localization of clock proteins, as well as their activity. For instance, CKIε phosphorylates PER1 and BMAL1, regulating the nuclear entry of the former and the transcriptional activity of the latter. Glycogen synthase kinase 3b (GSK3β) phosphorylates REV-ERBα, BMAL1 and CLOCK, PER2, and CRY2 with differing impacts on stability, activity, and subcellular localization [3–7]. Besides phosphorylation, clock components also undergo acetylation, O-GlcNacylation, ubiquitylation, and sumoylation, which have a range of effects as those mentioned [8, 9].
A fundamental feature of genes that are circadianly expressed is that they exhibit robust rhythms in chromatin regulation, including epigenetic modifications. As such, chromatin regulators such as histone lysine deacetylases, methyltransferases, and demethylases are components of the transcriptional machinery. Altogether, the combined action of signaling pathways, chromatin regulators, and non-clock transcription factors enable the fine-tuning of circadian rhythms and their entrainment by environmental cues.
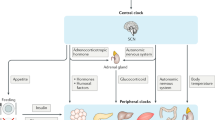
In spite of their cell-autonomous nature, cellular-level oscillators are orchestrated in a tissue-specific manner in order to give rise to tissue- and organ-level physiological rhythms. Interestingly, sustained tissue-level circadian rhythms are dependent on the phase alignment of their individual cellular-level oscillators, yet their synchronization (or entrainment) is not a tissue-autonomous property. Thus, the circadian system is a hierarchical network, composed of peripheral oscillators that are entrained by a master circadian pacemaker: the suprachiasmatic nucleus (SCN) of the hypothalamus [10–12]. The SCN is a neural structure composed of 20,000 cells bilaterally situated above the optic chiasm that has the unique property of being able to impose coordinated self-sustaining circadian oscillations at the tissue level. In addition, the SCN is innervated by the retinohypothalamic tract through which it receives photic cues that enable it to set its circadian phase to match the local geographic time. The SCN then relays this information to peripheral oscillators via neural, humoral, and metabolic signals in order to maintain the appropriate phase relationships between the different organs and the time of day.
Cancer and the Clock
In 2007, the World Health Organization (WHO) and the International Agency for Research on Cancer (IARC) classified shift work as a possible carcinogen, a decision that was largely based on epidemiological data along with results from animal studies.
Chronodisruption, specifically night work and rotating shift work, is associated with increased incidence of certain cancers. Numerous studies have identified a link between shift work and breast cancer. A meta-analysis of 13 independent epidemiological studies conducted between 1995 and 2005 estimated that the risk of breast cancer in shift workers (nurses and flight attendants) was up to 48 % higher than in control populations [13]. Interestingly, a nested case-controlled study of women serving in the Danish military found that chronotype had a significant impact on cancer incidence, with rates being higher in evening-type populations after controlling for shift work [14]. Furthermore, the same study found that morning-type workers subjected to shift work were nearly twice more likely to develop breast cancer than the control group. Similarly, chronodisruption increases the incidences of endometrial (42 % in nonobese and 100 % in obese night-shift workers), colorectal (35 %), and possibly prostate cancers [15–21]. Interestingly, blind subjects with total absence of photoreception, and thus less sensitive to circadian stress, have reduced cancer rates in comparison to those that retain at least some light sensitivity [21, 22]. Consistently, the survival of patients with cancer of the breast, colon, or lung, is associated with marked circadian rhythmicity [23].
The importance of the circadian oscillator to cancer is further supported by epidemiological analysis of circadian genetic variability in human populations, analysis of clock gene expression in cancers in vivo and in cell models, and experimental data. In humans, several SNPs (SNP) that occur in circadian clock genes are associated with cancer incidence, progression, and survival. For example, in a study of Chinese men, individuals that harbored a SNP in Npas2 had a decreased risk of developing prostate cancer, whereas those with an SNP in Cry2 had nearly double the risk of those that did not [24]. Similarly, other circadian polymorphisms have been linked to increased postmenopausal breast cancer risk (Cry2), reduced risk of developing non-Hodgkins lymphoma (Npas2), increased occurrence and development of non-small cell lung cancer, and increased survival in colorectal and hepatocellular carcinoma (Clock and Per3, respectively) [25–30].
In addition to these SNP associations, the expression of clock genes is dysregulated in tumors in vivo and in cancer cell culture models. Per2 and Cry1 gene expression is upregulated in gastric cancer, with Cry1 heightened expression correlating with disease progression [31]. In ovarian cancer cell lines, the Bmal1 gene is silenced via DNA methylation [32], whereas it is down-regulated in chronic lymphocytic leukemia patients along with Per1 and Per2 [33]. Similarly, in colorectal carcinoma Cry1 and Clock are overexpressed with their levels being correlated with progression of the disease, and decreased survival, whereas lower expression of Bmal1, Cry2, Per1, and Per3 was reduced in tumor samples [34–36].
The data derived from genetic animal models is also consistent with human observations. Clock mutant mice, which harbor a mutant gene allele that gives rise to a defective Clock protein, have decreased proliferation and increased apoptosis rates in lymphocytes, although without increased susceptibility to low-dose γ-irradiation induced cancer [37]. Double genetic ablation of Cry1 and Cry2 delays tumorigenesis onset and increases the lifespan of p53 mutant mice [38]. Mice that are homozygote for the Per2 mutant allele (Per2 m/m) have a 100 % increase in [39] the number of intestinal and colonic polyps when occurring in a model of colorectal cancer (Apc (Min/ + ) mice) background [40]. Consistently, siRNA-mediated decreases in Per2 levels in colon cancer cell lines resulted in heightened β-catenin expression and increased proliferation, with similar effects observed with Per1 [41]. Interestingly, mice that carry a transgenic Per2 allele that occurs in familial advanced sleep phase syndrome (FASPS) show increased cancer risk and incidence [42]. Furthermore, Bmal1, Per1, Per2, Cry1, or Cry2 genetic-null mice have predisposition to both spontaneous and irradiated-induced cancer [43].
Finally, nongenetic chronodisruptive paradigms have found links between tumor formation and progression in mice models. Tumor growth was greater in mice with bilateral SCN lesions than in sham control cohorts [44]. Under a jet lag paradigm, in which mice with tumor xenografts were subject to repeated 8 h light shifts, the rate of tumor growth also increased, with a mitigation of the effect occurring when meal time was matched to the light shifts; food-derived cues are dominant peripheral entraining signals, which reinforce the phase relationship between the SCN and the periphery [45]. In addition, chronic jet lag promotes metastases [46]. Similarly, when circadian mutants or wild-type mice are subjected to jet lag, their cancer risk is greatly increased [43]. Finally, rats chronically exposed to light-at-night, a fundamental characteristic of shift work and night work, increased the incidence of spontaneous tumorigenesis [47].
So, what mechanistic link exists between circadian disruption and cancer? For cancer to arise, normal cells must acquire certain characteristics, or “hallmarks,” in order to become tumorigenic. Specifically, these hallmarks are (1) self-sustained proliferation, (2) evasion of growth suppressive mechanisms, (3) apoptosis resistance, (4) induction of angiogenesis, (5) replicative immortality, (6) activation of tissue invasion and metastasis, (7) immune system evasion, and (8) reprogramming of cellular energy metabolism. The mechanisms that underlie all these hallmarks are under circadian control: cell cycle regulation and DNA damage repair, endocrine function, inflammation and apoptosis, xenobiotic detoxification, modulation of immune function, and energy metabolism are under extensive circadian control [48–51]. Thus, by impairing circadian oscillator function, chronodisruptive agents lead to a wide-spread dysregulation of physiological processes that are conducive for cancer development and progression.
The Circadian Clock, Cell Cycle Regulation and DNA Damage Response
The circadian clock and the cell cycle are now known to be extensively interconnected at the molecular level. The first and most obvious link is that many key cell cycle regulators—including wee1, p21-Waf1, p20, cdc2, cyclin B1, cyclin D1, and c-Myc—are, in fact, clock-controlled genes (CCGs). Consistently, the circadian clocks from cyanobacteria to mammals are well-known to gate the different stages of the cell cycle, and thus impose temporal regulation to its progression. In mammals, for instance, S-phase and mitotic index in the oral mucosa, corneal epithelium, digestive tract and even bone marrow exhibit circadian fluctuations [52–56]. In a striking example of cell cycle gating by the clock, the timing of M-phase entry following partial hepatectomy is dictated by the time of day of the procedure [57].
Similarly, the circadian clock impinges on the DNA damage response. PER1 physically associates with the cell cycle checkpoint proteins, ataxia telangiectasia mutated (ATM) and CHK2, sensitizes cells to apoptosis when overexpressed and, conversely, confers protection to irradiation-induced cell death [58]. In humans, the timeless protein (TIM), a putative oscillator component, interacts with CRY2 and CHK1, possibly helping further the crosstalk between circadian and cell cycle regulatory mechanisms [59]. Further, BMAL1 is necessary for p53-mediated activation of p21CIP; BMAL1 shRNA knockdown decreased the induction of both p53 and p21CIP in response to γ-irradiation, and rendered cells insensitive to p19ARF-induced cell cycle arrest [60]. Intriguingly, CLOCK protein is localized to DNA damage sites following UV irradiation [61].
The Circadian Clock and Detoxification
Xenobiotics are environmentally derived molecules and compounds that are taken up by organisms as a result of the processes required for life. Many xenobiotics, as well as endobiotic metabolites, are toxicants that have deleterious effects on the genome and, thus, organisms have evolved mechanisms that enable their removal and/or neutralization. As with other physiological processes, xenobiotic detoxification metabolism is profoundly influenced by the circadian system. For example, the constitutive androstane receptor (CAR), retinoic X receptor (RXR), and small heterodimer partner (SHP) are nuclear hormone receptors that are involved in the activation of detoxification programs and whose expression pattern is robustly circadian. DBP, TEF, and HLF PAR-bZIP are transcription factors that are direct outputs of the circadian oscillator involved in regulating drug metabolism and detoxification. In triple DBP;TEF;HLF genetic-null mice, CAR-targets and CAR itself are expressed at very low levels [62]. Two CAR targets that are downregulated in these mice are 5-aminolevulinic acid synthase 1 (Alas1) gene, which is also regulated by NPAS2, and P450 oxidoreductase (POR). Both ALAS1 and POR are required for CYP activity; ALAS1 is the rate-limiting enzyme in the heme biosynthetic pathway, and POR is required for transfer of electrons to CYPs. This results in oscillations in CYP activity. In addition, the expression of some Cyp genes is controlled by DBP, including CYP3A4, CYP2A4, and CYP2A5, or directly by oscillator components (CYP2E1). In all, this results in circadian regulation of Phase I metabolism, which has maximum function during the night, which is when food consumption peaks. Phases II and III metabolism are also influenced by the clock. For example, Phase II processes are scheduled so that glutathione conjugation occurs at the beginning of the fasting period (daytime), followed by glucuronidation towards the end of the light phase, and sulfation occurring at the day-to-night period. The expression of several phase III metabolism transporters are also regulated by the circadian oscillator, including organic cation transporter 1 (OCT1), organic anion transporters 1, 2, and 3 (OAT1–3), multidrug resistance protein 1 (MDR1), and others [49].
The Circadian Clock and the Endocrine System
Since the endocrine system enables communication and coordinated physiological functions across tissues and organs throughout the body, one might expect circadian influence over those processes that occur with regularity. Indeed, the circulating levels of many hormones and other endocrine factors oscillate strongly over the course of the day/night cycle in normal conditions and chronodisruption interferes with their synthesis, release and sometimes their effect within target cells. The disruption of circadian rhythms certainly means a disruption of endocrine systems, which, in turn, contributes to pathologies, including metabolic syndrome, obesity, type-2 diabetes, and cancer.
Melatonin
Melatonin is a pineal hormone with synthesis and secretion occurring during the night regardless of whether activity occurs during the day as in diurnal organisms or the night: melatonin can thus be considered the chemical signal through which physiology recognizes darkness [63]. Light exposure inhibits the production of melatonin according to its intensity, wavelength, and duration. Accordingly, reduced production is observed following shift work or other light-at-night environments. Additionally, production declines with age along with various other outputs of the circadian system. The pineal gland receives direct innervation from the SCN and, in terms of both synthesis and release, melatonin is under direct regulation of the clock. In a positive feedback loop, melatonin also signals to the SCN, resynchronizing clock gene expression and thereby fortifying rhythmicity. It is also one of the most important signals serving to synchronize peripheral clocks, regulating the phase and period of the transcription/translation cycle of peripheral clock genes. This action supports the segregation of daytime appropriate physiology from that of the night; however, its effects in their entirety extend beyond circadian rhythms with important consequences for overall metabolic function and health. Acting on adipocytes, for instance, melatonin inhibits lipogenesis and increases the production of the satiety hormone, leptin [64]; an effect that is potentiated with rhythmicity of melatonin exposure, as would occur endogenously. Additionally, melatonin functions synergistically with insulin, being important for its proper synthesis and secretion as well as improving insulin sensitivity in target cells. Consequently, pinealectomized animals demonstrate glucose intolerance and insulin resistance which can be reverted by melatonin replacement therapy [65].
Melatonin is known to have powerful oncostatic and oncoprotective effects. It can prevent and reverse tumorigenesis in murine models. Furthermore, circulating melatonin levels are inversely correlated with tumor growth rates in cancer patients, and positively associated with survival [66]. Interestingly, blind subjects with total absence of photoreception have reduced cancer rates in comparison to those that retain at least some light sensitivity, possibly due to a decreased susceptibility to alterations in melatonin secretion [22, 67].
Melatonin is thought to influence cancer occurrence and progression both directly and indirectly. Several receptor-dependent and -independent actions of melatonin within tumors oppose various hallmarks of cancer: reducing proliferation, boosting antioxidant defenses, regulating cellular metabolism, and blocking invasion and metastasis. Alternatively, melatonin’s impact on the circadian system stimulates robust oscillations and coordinates physiology across the body, supporting healthy circadian and metabolic states which in turn are protective.
Glucocorticoids
Glucocorticoids (GCs) are steroid hormones derived from the adrenal gland which function in a wide array of physiological processes, including inflammation, glucose homeostasis, and cell proliferation [68]. Their anti-inflammatory effects are exploited pharmaceutically to combat pain, allergies, arthritis as well as lymphomas and leukemia. They impinge on various other physiological processes as well, with the outcome depending on the target cell type—even positively or negatively affecting cell proliferation, for instance. Glucocorticoids exhibit strong daily oscillations in plasma concentrations. The SCN drives these fluctuations via the hypothalamic-pituitary-adrenal axis and their rhythmic presence participates in the entrainment of peripheral clocks [69–71]. In jet-lag conditions, both the synthesis and release of GCs are disturbed, which, in turn, contribute to desynchrony of peripheral clocks as well as disrupting various other physiological processes that GCs normally regulate [72, 73].
The role of glucocorticoids in cancer varies. The outcome of GC signaling is tissue-specific and likewise the effect of GC signaling in cancerous cells depends on the type; pro-apoptotic in certain cancers, but pro-survival and promoting resistance to cell death in others [68, 74]. In obesity, GCs also have pleiotropic effects but generally promote fat deposition.
Insulin/Insulin-Like Growth Factor
Insulin is produced in the pancreas and regulates energy metabolism throughout the body; its cousin, insulin-like growth factor (IGF) is secreted from the liver and promotes cell proliferation [75]. Although serving some distinct functions, these proteins are very similar in structure and share many components of their signaling pathways. High levels of insulin are observed in the blood of obese and diabetic patients, and insulin/IGF signaling has been implicated in the risk, incidence, and tumorigenic responses of various human cancers in such individuals [76, 77]. Intriguingly, in human breast cancer patients, a disruption to IGF-1 rhythmicity has been observed whereas other rhythms remain intact; it is possible that the timing of release may be as important as the overall levels [78].
Insulin release is responsive to nutrient availability in the blood; however, it is also under circadian control [79, 80]. Many components of the insulin/IGF signaling pathway are regulated in a circadian manner at the level of transcription, and blood levels of both oscillate over 24 h even in the absence of food [81]. Conversely, insulin stimulates circadian gene expression [82]. Additionally, mice fed a high-fat diet (HFD) become obese and hyperinsulinemic and have disrupted feeding rhythms and free-running period length [83].
The Circadian Clock and Energy Metabolism
The rise in obesity and excess body weight rates constitute one of the greatest modern global health challenges. In the USA alone, ~60 % of the population is considered overweight, with half of those considered clinically obese, 8–11 % suffering from type2 diabetes, and a quarter exhibiting metabolic syndrome. Interestingly, this obesity epidemic has been paralleled by the rise of the 24-h society, which is characterized by the proliferation of, and chronic exposure to, light pollution, and altered activity profiles inherent to modern economies (e.g., rotating shift work, jet lag). Indeed, epidemiological studies have consistently linked circadian dysfunction to metabolic syndrome, weight gain, obesity, and type-2 diabetes [84]. A recent study of more than 113,000 women living in the UK found a strong association between increasing levels of light exposure at night with weight gain and obesity [85]. Similarly, rotating shift work, sleep deprivation, and sleep disruption are positively associated with type-2 diabetes and impairment in glucose metabolism [86–88]. Consistently, human volunteers subjected to circadian misalignment that mimic shift work and jet lag had marked increases in blood glucose and insulin levels, increased arterial pressure, decreased leptin levels, and sleep quality [89, 90], whereas disrupted or insufficient sleep increases the risk of developing T2D [91, 92]. Intriguingly, human circadian genetic variability has been found to impact metabolic function. For example, a number of SNPs that occur in human circadian genes are correlated with propensity for obesity and type-2 diabetes (Clock and Bmal1), heightened fasting (NPAS2 and PER2) or otherwise correlated with (CRY2 gene variants) blood glucose levels [93–95].
Importantly, experimental data firmly corroborate human population-based studies, and have helped establish the circadian oscillator as a key regulator of energy homeostasis, from the animal as a whole to the cellular level. For example, whole-body Bmal1 knockout mice present with a number of metabolic phenotypes, including hypoinsulinemia, higher body fat mass content, and fasting hypoglycemia, with the latter also occurring in liver-specific Bmal1 knockouts [96, 97]. In addition, in both Bmal1 knockouts and Clock mutant mice, circadian rhythms in insulin sensitivity and glucose tolerance are eliminated, and gluconeogenesis is impaired [96, 98]. Clock mutant mice, which harbor a dysfunctional allele, are also predisposed to weight gain under both regular and high-fat dietary paradigms and exhibit dyslipidemia, hyperglycemia, and hypoinsulinemia [99]. Cryptochrome-deficient mice exhibit abnormal glucose metabolism. Cry double-null mice (Cry1 ‒/‒ ;Cry2 ‒/‒ ) show heightened postprandial blood glucose levels, are glucose intolerant, and are sensitized to HFD-induced weight gain and hyperinsulinemia [100]. Cry1 ‒/‒ or Cry2 ‒/‒ animals also show glucose intolerance, yet Cry1 but not Cry2 knockouts are protected against HFD-induced obesity [101]. Similarly, Per2 mutant mice have lower body fat content and total body weight, and show aberrations in blood glucose regulation, with fasting hypoglycemia, impaired gluconeogenesis and increased insulin sensitivity despite being hyperinsulinemic [102]. Rev-erbα ‒/‒ animals have increased white adipose tissue, elevated blood glucose, decreased blood lipid levels, and abnormal bile acid levels [103, 104]. More recently, mice with a Per1 gene mutation paralogous to that occurring in humans affected with FASPS were found to be predisposed to HFD-induced obesity [105].
Genomic analyses of tissues such as liver, white and brown adipose tissues, and skeletal muscle have been instrumental in furthering our understanding of how the clock regulates metabolism and why its dysfunction contributes to pathological states. In liver, a key organ in the regulation of systemic energy homeostasis whose function is extensively influenced by the circadian oscillator, many of the direct targets of the hepatic oscillator are essential components of genomic programs and pathways that regulate energy homeostasis. Indeed, circadian clock components (BMAL1, NPAS2, PER1, PER2, CRY, CRY2, NR1D1, and NR1D2) are enriched at the promoters of key regulators of glucose (G6pc, Pck1, Pcx, Pdk1), lipid, and cholesterol homeostasis (Acaca, Hmgcr, Scap, Insig2, Cyp7a1) [106, 107]. Additionally, the circadian oscillator influences metabolism indirectly by imposing strong rhythms in the levels of non-clock transcription factors. Notably, out of the 41 hepatic-expressed NHRs, 20 are rhythmically expressed, including the non-clock NHR components PPARα,δ,γ, thyroid hormone receptor, and ERRs (Estrogen-related receptors) [108, 109]. Likewise, the genes that code for the sterol homeostasis regulators SREBP1 and SREBP2– Srebf1 and Srebf2– are expressed with a circadian pattern that, respectively, peaks at the beginning and end of the dark period [110, 111].
White adipose tissue (WAT) is a widely distributed, complex tissue that is a major site of fat storage and de novo fatty acid synthesis. As with liver, the circadian clock involvement in WAT physiology is extensive, affecting both adult tissue function and adipogenesis. In adult WAT, a major role of the circadian clock is to regulate the balance between lipolysis and lipogenesis needed to store energy for use under fasting conditions, yet prevent dyslipidemia and excess fat accumulation. For example, BMAL1 drives the activation of the lipogenic genes Elovl6, Scd1, Atgl, and Hsl through direct binding to the promoter regions [112, 113]. During adipogenesis, Bmal1 gene expression is enhanced [114]. Consistently, Bmal1 ‒/‒mice exhibit reduced adiposity and low levels of long-chain polyunsaturated fatty acids. Like BMAL!, REV-ERBα is highly expressed during adipocyte differentiation, although its exact role is this process is not clear; as cell-based experiments show REV-ERBα is required for adipocyte differentiation yet Rev-Erb knockout mice show no adipose tissue defects. WAT is also the source of endocrine factors known as adipokines, including adiponectin, leptin, and resistin, all of which are released into the bloodstream in a circadian pattern. In mammals, brown adipose tissue (BAT) is a major site of thermogenesis and a key organ in the regulation of body temperature. In BAT, thermogenesis is achieved via dissipation of the mitochondrial proton gradient through uncoupling protein 1 (UCP1), whose expression has been found to be regulated by Rev-Erbα and Per2 [115, 116]. In skeletal muscle, the circadian clock imposes rhythmicity on the expression of 215 genes, including genes involved in triglyceride hydrolysis (Ces3, Pnpla3), fatty acid oxidation (Pgc1b, Myod1, Ucp3), and synthesis of fatty acids and cholesterol (Pank1, Dbt, Dgat2, Acat2, Idh1, S3–12) [117].
The clock can also fine-tune its control of energy homeostasis through non-transcriptional mechanisms. For instance, cryptochrome proteins can prevent transcription of gluconeogenic genes by preventing activation of the cAMP response element-binding protein (CREB) by glucagon-activated Gsa, as well as by direct repression of the glucocorticoid receptor [118, 119]. Similarly, PER2 can impact glucose metabolism through physical association and modulation of PPARα, REV-ERBα, and possibly HNF4α, and thus regulate their activity [120]. Finally, the clock can influence energy homeostasis through the production of metabolites that activate energetic regulators. The gene for the key enzyme involved in the production of nicotinamide adenine dinucleotide (NAD + ), NAD phosphoribosyltransferase (Nampt), harbors E-boxes in its promoter region that are bound by CLOCK, and are expressed in a circadian fashion [121, 122]. In turn, hepatic NAD + levels oscillate, and this coincides with the activity of Sirt1, an NAD + -dependent histone deacetylase (HDAC) that is one of the central regulators of energy homeostasis [123, 124].
Timing and Quality of Caloric Intake and the Clock
Perhaps the most obvious mechanism by which the circadian oscillator regulates energy metabolism is by orchestrating feeding behavior. Yet, until recently, the role played by the timing of food intake in the maintenance of energy metabolism homeostasis had not been appreciated. The initial cues came from the observation that mice fed a high-fat diet during their inactive period gained more weight than their control counterparts in spite of similar caloric intake and activity levels [125]. This observation was then followed by studies conducted by us and others in which mice were protected from HFD-induced obesity and diabetes by manipulating the feeding time [126, 127]. Specifically, mice in which access to food was limited (temporally restricted feeding; tRF) exclusively during nighttime (active period in mice) did not show the weight gain and metabolic dysfunction characteristic of HFD-fed mice with ad libitum food access, even though the ad libitum and TRF paradigms were isocaloric [126].
Interestingly, the obesoprotective effects of TRF appear to be due to enhancement in circadian oscillator function. Indeed, tRF restored the amplitude in expression of circadian clock components as well as in a number of clock targets and other rhythmic genes. Such improvement is reflective of the intricate relationship between food-derived input and the local circadian oscillator. Under normal conditions, 2997 genes are rhythmically expressed in the livers of wild-type mice, of which only 368 maintain rhythmicity in the absence of food intake [128]. On the other hand, the imposition of a temporally restricted feeding paradigm in cry double-knockout mice, which have a functional circadian clock, restores rhythmicity of 617 genes, whereas such a paradigm in WT mice increases the number of genes with circadian oscillations to 4960.
However, the relationship between metabolism and the circadian clock is bidirectional. As mentioned previously, energy metabolism regulators and signaling pathways impinge on the clock and vice versa. As such, dietary conditions affect the function of the oscillator. Consistently, dietary quality affects the circadian clock. For instance, an HFD and altered behavioral rhythms, including preference for food intake during daytime hours, dampened rhythms in behavior and in the oscillations of circadian clock component gene expression [83]; a high-carbohydrate, high-protein diet phase advances the phase of clock component abundance rhythms and increases the overall levels of BMAL1 and CRY1 [129]; and a high-salt diet similarly results in phase advances of clock component levels [130].
In recent years, the role that metabolic dysfunction plays in tumorigenesis and cancer progression has received increasing attention [131, 132]. Most notably, the Warburg effect, where cancer cells reprogram their metabolism as to favor glycolysis-derived energy production over mitochondrial respiration, irrespective of oxygen availability, has been intensively studied [133]. Although first postulated by Otto Warburg in 1924 as a possible cause of cancer, the Warburg effect is now considered an acquired trait that enables cancer cells to survive in the otherwise proliferation-limiting hypoxic environment that arises as solid tumors grow. Hypoxia leads to the activation of hypoxia-inducible factors (HIF), amongst whose functions are included the promotion of angiogenesis and, importantly, the increase of glycolysis to compensate for loss of oxygen-dependent mitochondrial respiration [134]. Not surprisingly, HIFs are dysregulated in cancer. Interestingly, HIF proteins, like CLOCK, BMAL1, and PERs, are bHLH-PAS transcription factors; HIFs, CLOCK, and BMAL1 can physically associate to form transcriptionally active complexes (HIF) [135, 136]. In addition, key glycolytic regulators and glucose transporters are dysregulated in cancer, including hexokinase, phosphofructo kinase 1 and 2, and GLUT transporters, all of which are expressed in a circadian pattern in several tissues [137–139].
Conclusions
In all, chronodisruption, metabolic dysfunction, and cancer are so intricately linked that it may be futile to establish a cause–effect relationship between them. Instead, the simplest view that emerges is one where the different processes that underlie these pathologies exist in a dynamic equilibrium and that disrupting any one of them triggers a pathological chain reaction that predisposes organisms to the others. Consider the following scenario: gating of the cell cycle by the circadian clock so that DNA replication and mitosis occurs during the fasting period when food intake and associated metabolically and environmentally derived toxins are at a minimum [140]. Circadian disruption arising from obesity could lead to a temporal spreading of cell division, so that more individual cells are replicating under conditions where genomic insults are more abundant. Simultaneously, this circadian disorganization may result in blunted rhythms and overall lower levels and activities of proteins involved in detoxification mechanisms, exacerbating the increased mutation risk. Conversely, certain cancers, such as insulinomas, trigger metabolic changes that affect circadian and non-circadian mechanisms, again highlighting the extensive interrelatedness of these pathological states. Finally, environmental chronodisruption, such as chronic jet lag or rotating shift work, results in a generalized circadian disorganization that results in hormonal imbalances, including suppression of melatonin, glucose, and lipid metabolic dysregulation, and impaired gating of the cell cycle. As metabolism becomes more perturbed, the circadian oscillator is further disrupted, leading to the aforementioned conditions, thus facilitating oncogenesis.
References
Feillet C, et al. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc Natl Acad Sci U S A. 2014;111:9828–33.
Chen R, D’Alessandro M, Lee C. miRNAs are required for generating a time delay critical for the circadian oscillator. Curr Biol. 2013;23:1959–68.
Spengler ML, Kuropatwinski KK, Schumer M, Antoch MP. A serine cluster mediates BMAL1-dependent CLOCK phosphorylation and degradation. Cell Cycle. 2009;8:4138–46.
Yin L, Wang J, Klein PS, Lazar MA. Nuclear receptor Rev-erbalpha is a critical lithiumsensitive component of the circadian clock. Science. 2006;311:1002–5.
Reischl S, Kramer A. Kinases and phosphatases in the mammalian circadian clock. FEBS Lett. 2011;585:1393–9.
Lee J, et al. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol Cell Biol. 2008;28:6056–65.
Cardone L, et al. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309:1390–4.
Asher G, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–28.
Gossan NC, et al. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765–75.
Panda S, Hogenesch JB, Kay SA. Circadian light input in plants, flies and mammals. Novartis Found Symp. 2003;253:73–82 (discussion 82–8, 102–9, 281–4).
Panda S, Hogenesch JB, Kay SA. Circadian rhythms from flies to human. Nature. 2002;417:329–35.
Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003;4:649–61.
Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Night work and breast cancer risk: a systematic review and meta-analysis. Eur J Cancer. 2005;41:2023–32.
Hansen J, Lassen CF. Nested case-control study of night shift work and breast cancer risk among women in the Danish military. Occup Environ Med. 2012;69:551–6.
Viswanathan AN, Hankinson SE, Schernhammer ES. Night shift work and the risk of endometrial cancer. Cancer Res. 2007;67:10618–22.
Pauley SM. Lighting for the human circadian clock: recent research indicates that lighting has become a public health issue. Med Hypotheses. 2004;63:588–96.
Schernhammer ES, et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95:825–8.
Haus EL, Smolensky MH. Shift work and cancer risk: potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med Rev. 2013;17:273–84.
Conlon M, Lightfoot N, Kreiger N. Rotating shift work and risk of prostate cancer. Epidemiology. 2007;18:182–3.
Kubo T, et al. Industry-based retrospective cohort study of the risk of prostate cancer among rotating-shift workers. Int J Urol. 2011;18:206–11.
Buja A, et al. Cancer incidence among male military and civil pilots and flight attendants: an analysis on published data. Toxicol Ind Health. 2005;21:273–82.
Hahn RA. Profound bilateral blindness and the incidence of breast cancer. Epidemiology. 1991;2:208–10.
Mormont MC, et al. Marked 24-h rest/activity rhythms are associated with better quality of life, better response, and longer survival in patients with metastatic colorectal cancer and good performance status. Clin Cancer Res. 2000;6:3038–45.
Chu LW, et al. Variants in circadian genes and prostate cancer risk: a population-based study in China. Prostate Cancer Prostatic Dis. 2008;11:342–8.
Couto P, et al. Association between CLOCK, PER3 and CCRN4 L with nonsmall cell lung cancer in Brazilian patients. Mol Med Rep. 2014;10:435–40.
Hoffman AE, et al. The core circadian gene Cryptochrome 2 influences breast cancer risk, possibly by mediating hormone signaling. Cancer Prev Res (Phila). 2010;3:539–48.
Zhu Y, et al. Ala394Thr polymorphism in the clock gene NPAS2: a circadian modifier for the risk of non-Hodgkin’s lymphoma. Int J Cancer. 2007;120:432–5.
Zhao B, et al. A functional polymorphism in PER3 gene is associated with prognosis in hepatocellular carcinoma. Liver Int. 2012;32:1451–9.
Zhou F, et al. Functional polymorphisms of circadian positive feedback regulation genes and clinical outcome of Chinese patients with resected colorectal cancer. Cancer. 2012;118:937–46.
Kettner NM, Katchy CA, Fu L. Circadian gene variants in cancer. Ann Med. 2014;46:208–20.
Hu ML, et al. Deregulated expression of circadian clock genes in gastric cancer. BMC Gastroenterol. 2014;14:67.
Yeh CM, et al. Epigenetic silencing of ARNTL, a circadian gene and potential tumor suppressor in ovarian cancer. Int J Oncol. 2014;45:2101–7.
Rana S, et al. Deregulated expression of circadian clock and clock-controlled cell cycle genes in chronic lymphocytic leukemia. Mol Biol Rep. 2014;41:95–103.
Mazzoccoli G, et al. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol Int. 2011;28:841–51.
Yu H, et al. Cryptochrome 1 overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS One. 2013;8:e61679.
Wang L, et al. hClock gene expression in human colorectal carcinoma. Mol Med Rep. 2013;8:1017–22.
Antoch MP, et al. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008;7:1197–204.
Ozturk N, Lee JH, Gaddameedhi S, Sancar A Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2841–6.
Jensen LD, Cao Y. Clock controls angiogenesis. Cell Cycle. 2013;12:405–8.
Wood PA, et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol Cancer Res. 2008;6:1786–93.
Yang X, et al. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol Int. 2009;26:1323–39.
Gu X, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19:397–405.
Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One. 2010;5:e10995.
Filipski E, et al. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst. 2002;94:690–7.
Filipski E, et al. Effects of light and food schedules on liver and tumor molecular clocks in mice. J Natl Cancer Inst. 2005;97:507–17.
Wu M, et al. Experimental chronic jet lag promotes growth and lung metastasis of Lewis lung carcinoma in C57BL/6 mice. Oncol Rep. 2012;27:1417–28.
Anisimov VN, Vinogradova IA, Panchenko AV, Popovich IG, Zabezhinski MA. Light-atnight-induced circadian disruption, cancer and aging. Curr Aging Sci. 2012;5:170–7.
Lee JH, Sancar A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc Natl Acad Sci U S A. 2011;108:12036–41.
Zmrzljak UP, Rozman D. Circadian regulation of the hepatic endobiotic and xenobitoic detoxification pathways: the time matters. Chem Res Toxicol. 2012;25:811–24.
Savvidis C, Koutsilieris M. Circadian rhythm disruption in cancer biology. Mol Med. 2012;18:1249–60.
Everett LJ, Lazar MA. Nuclear receptor Rev-erbα: up, down, and all around. Trends Endocrinol Metab. 2014;25(11):586–92.
Pilgrim C, Erb W, Maurer W. Diurnal fluctuations in the numbers of DNA synthesizing nuclei in various mouse tissues. Nature. 1963;199:863.
Gomes JR, et al. Circadian variation of the cell proliferation in the jejunal epithelium of rats at weaning phase. Cell Prolif. 2005;38:147–52.
Scheving LA. Biological clocks and the digestive system. Gastroenterology. 2000;119:536–49
Smaaland R. Circadian rhythm of cell division. Prog Cell Cycle Res. 1996;2:241–66.
Bjarnason GA, Jordan R. Circadian variation of cell proliferation and cell cycle protein expression in man: clinical implications. Prog Cell Cycle Res. 2000;4:193–206.
Matsuo T, et al. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–9.
Gery S, et al. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006;22:375–82.
Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–16.
Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One. 2009;4:e4798.
Cotta-Ramusino C, et al. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–7.
Gachon F, Olela FF, Schaad O, Descombes P, Schibler U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006;4:25–36.
Reiter RJ. Melatonin: the chemical expression of darkness. Mol Cell Endocrinol. 1991;79:C153–8.
Ng TB, Wong CM. Effects of pineal indoles and arginine vasotocin on lipolysis and lipogenesis in isolated adipocytes. J Pineal Res. 1986;3:55–66.
Cipolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ. Melatonin, energy metabolism, and obesity: a review. J Pineal Res. 2014;56:371–81.
Cutando A, Lopez-Valverde A, Arias-Santiago S, DE Vicente J, DE Diego RG. Role of melatonin in cancer treatment. Anticancer Res. 2012;32:2747–53.
Feychting M, Osterlund B, Ahlbom A. Reduced cancer incidence among the blind. Epidemiology. 1998;9:490–4.
Dickmeis T, Foulkes NS. Glucocorticoids and circadian clock control of cell proliferation: at the interface between three dynamic systems. Mol Cell Endocrinol. 2011;331:11–22.
Son GH, Chung S, Kim K. The adrenal peripheral clock: glucocorticoid and the circadian timing system. Front Neuroendocrinol. 2011;32:451–65.
Kalsbeek A, et al. Circadian rhythms in the hypothalamo-pituitary-adrenal (HPA) axis. Mol Cell Endocrinol. 2012;349:20–9.
Kino T, Chrousos GP. Circadian CLOCK-mediated regulation of target-tissue sensitivity to glucocorticoids: implications for cardiometabolic diseases. Endocr Dev. 2011;20:116–26.
Greene MW. Circadian rhythms and tumor growth. Cancer Lett. 2012;318:115–23.
Spiga F, Walker JJ, Terry JR, Lightman SL. HPA axis-rhythms. Compr Physiol. 2014;4:1273–98.
Herr I, Gassler N, Friess H, Buchler MW. Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids. Apoptosis. 2007;12:271–91.
Humbel RE. Insulin-like growth factors I and II. Eur J Biochem. 1990;190:445–62.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38.
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–69.
Haus E, Dumitriu L, Nicolau GY, Bologa S, Sackett-Lundeen L. Circadian rhythms of basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), insulin-like growth factor-1 (IGF-1), insulin-like growth factor binding protein-3 (IGFBP-3), cortisol, and melatonin in women with breast cancer. Chronobiol Int. 2001;18:709–27.
Mejean L, et al. Circadian and ultradian rhythms in blood glucose and plasma insulin of healthy adults. Chronobiol Int. 1988;5:227–36.
Haus E. Chronobiology in the endocrine system. Adv Drug Deliv Rev. 2007;59:985–1014.
Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nat Rev Endocrinol. 2014;10:466–75.
Balsalobre A, Marcacci L, Schibler U. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Curr Biol. 2000;10:1291–4.
Kohsaka A, et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007;6:414–21.
Monk TH, Buysse DJ. Exposure to shift work as a risk factor for diabetes. J Biol Rhythms. 2013;28:356–9.
McFadden E, Jones ME, Schoemaker MJ, Ashworth A, Swerdlow AJ. The relationship between obesity and exposure to light at night: cross-sectional analyses of over 100,000 women in the breakthrough generations study. Am J Epidemiol. 2014;180:245–50.
Kalsbeek A, la Fleur S, Fliers E. Circadian control of glucose metabolism. Mol Metab. 2014;3:372–83.
Suwazono Y, et al. A longitudinal study on the effect of shift work on weight gain in male Japanese workers. Obesity (Silver Spring). 2008;16:1887–93.
Kroenke CH, et al. Work characteristics and incidence of type 2 diabetes in women. Am J Epidemiol. 2007;165:175–83.
Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009;106:4453–8.
Gangwisch JE. Epidemiological evidence for the links between sleep, circadian rhythms and metabolism. Obes Rev. 2009;10(Suppl 2):37–45.
Meisinger C, Heier M, Loewel H, Study MKAC. Sleep disturbance as a predictor of type 2 diabetes mellitus in men and women from the general population. Diabetologia. 2005;48:235–41.
Beihl DA, Liese AD, Haffner SM. Sleep duration as a risk factor for incident type 2 diabetes in a multiethnic cohort. Ann Epidemiol. 2009;19:351–7.
Kelly MA, et al. Circadian gene variants and susceptibility to type 2 diabetes: a pilot study. PLoS One. 2012;7:e32670.
Ruano EG, Canivell S, Vieira E. REV-ERB ALPHA polymorphism is associated with obesity in the Spanish obese male population. PLoS One. 2014;9:e104065.
Woon PY, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci U S A. 2007;104:14412–7.
Marcheva B, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–31.
Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008;105:15172–7.
Rudic RD, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2:e377.
Turek FW, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–5.
Barclay JL, et al. High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am J Physiol Endocrinol Metab. 2013;304:E1053–63.
Griebel G, Ravinet-Trillou C, Beeske S, Avenet P, Pichat P. Mice deficient in cryptochrome 1 (cry1 (‒/‒)) exhibit resistance to obesity induced by a high-fat diet. Front Endocrinol (Lausanne). 2014;5:49.
Carvas JM, et al. Period2 gene mutant mice show compromised insulin-mediated endothelial nitric oxide release and altered glucose homeostasis. Front Physiol. 2012;3:337.
Le Martelot G, et al. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009;7:e1000181.
Cho H, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. 2012;485:123–7.
Liu Z, et al. PER1 phosphorylation specifies feeding rhythm in mice. Cell Rep. 2014;7:1509–20.
Hughes ME, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442.
Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–20.
Yang X, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–10.
Yang X, Lamia K, Evans R. Nuclear receptors, metabolism, and the circadian clock. Cold Spring Harb Symp Quant Biol. 2007;72:387–94.
Masri S, Zocchi L, Katada S, Mora E, Sassone-Corsi P. The circadian clock transcriptional complex: metabolic feedback intersects with epigenetic control. Ann N Y Acad Sci. 2012;1264:103–9.
Masri S, et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158:659–72.
Paschos GK, et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med. 2012;18:1768–77.
Shostak A, Meyer-Kovac J, Oster H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes. 2013;62:2195–203.
Shimba S, et al. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci U S A. 2005;102:12071–6.
Gerhart-Hines Z, et al. The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature. 2013;503:410–3.
Chappuis S, et al. Role of the circadian clock gene Per2 in adaptation to cold temperature. Mol Metab. 2013;2:184–93.
McCarthy JJ, et al. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol Genomics. 2007;31:86–95.
Zhang E, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–6.
Lamia KA, et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–6.
Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–37.
Ramsey KM, et al. Circadian clock feedback cycle through NAMPT-mediated NAD + biosynthesis. Science. 2009;324:651–4.
Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–7.
Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–71.
Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–38.
Arble D, Bass J, Laposky A, Vitaterna M, Turek F. Circadian timing of food intake contributes to weight gain. Obesity (Silver Spring, Md.). 2009;17:2100–2.
Hatori M, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–60.
Sherman H, et al. Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J. 2012;26:3493–502.
Vollmers C, et al. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci U S A. 2009;106:21453–8.
Oishi K, Uchida D, Itoh N. Low-carbohydrate, high-protein diet affects rhythmic expression of gluconeogenic regulatory and circadian clock genes in mouse peripheral tissues. Chronobiol Int. 2012;29:799–809.
Oike H, Nagai K, Fukushima T, Ishida N, Kobori M. High-salt diet advances molecular circadian rhythms in mouse peripheral tissues. Biochem Biophys Res Commun. 2010;402:7–13.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37.
Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13.
Hogenesch JB, et al. The basic helix-loop-helix-PAS protein MOP9 is a brain-specific heterodimeric partner of circadian and hypoxia factors. J Neurosci. 2000;20:RC83.
Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci U S A. 1998;95:5474–9.
Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835:164–9.
Salani B, et al. Metformin, cancer and glucose metabolism. Endocr Relat Cancer. 2014;21:R461–71.
Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8.
Pacha J, Sumova A. Circadian regulation of epithelial functions in the intestine. Acta Physiol (Oxf). 2013;208:11–24.
Acknowledgments
KD and LD are supported in part by NIH grant numbers P20RR021940 and P20GM103549. Research in SP’s lab is supported by NIH grant numbers DK091618, EY016807, P30 CA014195, and American Federation for Aging Research grant number M14322.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
DiTacchio, L., DiTacchio, K., Panda, S. (2015). Relevance of Circadian Rhythm in Cancer. In: Berger, N. (eds) Murine Models, Energy Balance, and Cancer. Energy Balance and Cancer, vol 10. Springer, Cham. https://doi.org/10.1007/978-3-319-16733-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-16733-6_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16732-9
Online ISBN: 978-3-319-16733-6
eBook Packages: MedicineMedicine (R0)