
Abstract
Purpose of Review
Epigenetic changes can be highly influenced by environmental factors and have in turn been proposed to influence chronic disease. Being able to quantify to which extent epigenomic processes are mediators of the association between environmental exposures and diseases is of interest for epidemiologic research. In this review, we summarize the proposed mediation analysis methods with applications to epigenomic data.
Recent Findings
The ultra-high dimensionality and high correlations that characterize omics data have hindered the precise quantification of mediated effects. Several methods have been proposed to deal with mediation in high-dimensional settings, including methods that incorporate dimensionality reduction techniques to the mediation algorithm.
Summary
Although important methodological advances have been conducted in the previous years, key challenges such as the development of sensitivity analyses, dealing with mediator-mediator interactions, including environmental mixtures as exposures, or the integration of different omic data should be the focus of future methodological developments for epigenomic mediation analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Omics data analysis has moved to the spotlight of scientific research in the last years. Genomics, epigenomics, transcriptomics, proteomics and metabolomics complete the study of an organism from its genetic code to the metabolites it generates [1]. The potential shown by these data for early detection of disease and precision medicine, as well as for the understanding of the complex biological processes underlying disease, has attracted interest of many biomedical researchers.
Epigenetic changes, or heritable phenotype changes that do not alter the DNA sequence, have shown to be highly influenced by environmental factors [2], and they might as well influence the subsequent biological processes including gene expression, protein biosynthesis and metabolite formation. Epigenetic modifications have in turn been proposed to influence chronic disease [3, 4]. Thus, being able to quantify to which extent epigenomic processes are mediators of the association between environmental exposures and diseases can provide mechanistic insights into environment-related disease etiology. Mediation analysis aims to disentangle how an intermediate variable, referred to as a mediator, explains the mechanism or pathway through which an exposure or treatment influences an outcome.
The complexity of epigenomic data and the lack of appropriate statistical methods, though, have hindered the precise quantification of the association between epigenetic marks and chronic disease, including the potential intermediate role of epigenetic changes on the well-known association between environmental factors and chronic disease [5]. Several characteristics of omics data challenge the development of appropriate statistical methods for mediation analysis. First, the ultra-high dimensional nature of omics data requires effective dimensionality reduction techniques in order to select the features that are related to the outcome of interest, and focus subsequent extensive statistical analyses in those features. Second, high correlations between features challenge the performance of traditional methods due to multicollinearity. For example, DNA methylation in nearby CpG sites tends to be similar, therefore, spatial correlations are common [6]. Shrinkage methods such as elastic-net [7], or sure screening methods such as Sure Independence Screening (SIS) [8••], have become popular choices for dimensionality reduction in omics data, as they are able to deal with multicollinearity while effectively selecting features that are associated with the outcome. In addition, SIS has shown to mitigate the bias in post-selection inference in mediation analysis introduced when features associated with the exposure, but not with the outcome, are included in the models [9].
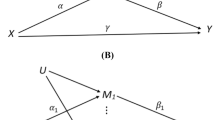
Once the optimal set of omics features associated with the outcome is found, subsequent targeted mediation analyses can be conducted. A directed acyclic graph showing the potential structure of an omics data mediation analysis is shown in Fig. 1. Several multiple mediation analysis methods have been proposed, however, most of them have limitations for applications to epigenomic data. A previous review summarized some of the methods proposed for mediation analysis using high-dimensional data [10••]. However, the high-dimensional mediation analysis field has grown fast and many new methodological developments have been proposed in the last years. In this review, we summarize the state of the art in multiple mediation for epigenomic data analysis, which could be a key statistical tool for the quantification of the intermediate role of epigenetic marks on the association between environmental exposures and disease.

Directed acyclic graph of a mediation analysis with omics markers as mediators. E: Exposure. Mi: Omic mediators. Y: Outcome. Xi: Exposure-mediator, mediator-outcome and exposure-outcome confounders. U: Common causes leading to non-causal correlations
Simple and Multiple Mediation Analysis
Although the product of coefficients and the difference of coefficients methods [11] were the most widely used approaches for mediation analysis in the past, they cannot easily incorporate exposure-mediator interactions. In addition, they might lead to biased estimates for certain effect measures from multiplicative models (such as hazard ratios or odds ratios) due to the non-collapsibility issue, as the measures that are non-collapsible can lead to non-comparable magnitudes when being adjusted for certain variables, even if those variables are unrelated to the outcome [12, 13]. The counterfactual approach is currently the gold standard for mediation analysis [14, 15•]. Let us denote \(E\) as an exposure and \(Y\) as the outcome of interest. Counterfactual outcomes refer to the values \(Y\) would take under each of the potential values of \(E\). Please note that some of those values of \(Y\) will be unobservable, which is the reason why they are called counterfactuals (contrary-to-fact). For example, if the exposure \(E\) is dichotomous (exposed / unexposed), an individual will either be exposed or unexposed, thus, one of the counterfactual outcomes will not be observed. From now on, we will consider dichotomous exposures for simplicity. However, this notation could be easily extended to continuous exposures. Below we summarize the effects of interest for mediation analysis under the counterfactual framework.
Simple Mediation Analysis
Let us denote \(M\) as the mediator, which is dependent on the exposure \(E\); \(X\) as a set of covariates and \(Y\) as the outcome of interest. Let us consider two different values of the exposure, \(e\) and \({e}^{*}.\) Following the counterfactual framework [16], we consider \(Y({e}^{*},M(e))\) as the counterfactual outcome, i.e., the value the outcome would take had the exposure been set to \({e}^{*}\) and the mediator been set to the value it would take when the exposure is set to \(e\). We define the average indirect effect of changing the exposure from \({e}^{*}\) to \(e\) when the covariates are set to \(X = x\) as follows [17, 18•, 19•]:
Similarly, the average direct effect, which refers to the effect of the exposure or treatment on the outcome which does not happen through the mediator, is quantified as:
Last, the average total effect, which denotes the effect of the exposure or treatment on the outcome both through the mediator pathway and through other pathways, is quantified as:
Please also note that, following these definitions, it holds that \(\tau \left(e, {e}^{*}\right)=\zeta \left(e, {e}^{*}\right)+ \delta \left(e, {e}^{*}\right)\), showing that the indirect and direct effects represent an exact decomposition of the total effect.
Multiple Mediation Analysis
Imai and Yamamoto [20•, 21] extended the effect definition for simple mediation analysis to the multiple mediators setting. Let us assume that \(Z = {({M}_{1}, ..., {M}_{K} )}^{T}\) is the vector of all mediators, with \(K\ge 2\). Considering \({M}_{k}\) as the mediator of interest, \(k = 1, ..., K\), let us define \({W}_{k}\) as the vector of all mediators except \({M}_{k}\). We also consider \(Y({e}^{*},{M}_{k}\left(e\right), {W}_{k}({e}^{*}))\) as the counterfactual outcome. In the multiple mediators setting, the average mediated effect of the \(k\)-th mediator is given by:
\({\delta }_{k}\left(e\right)\) is the path-specific effect through \({M}_{k}\), and excludes paths that involve other mediators in addition to \({M}_{k}\). The joint indirect effect of all mediators is defined as:
The direct effect is defined as:
Last, the total effect is defined as:
Sequential Ignorability Assumptions
Importantly, in order for the causal mediation effects to be identifiable, several assumptions need to hold [22]. These assumptions refer to the absence of unmeasured confounding, to having a well-defined treatment or exposure, and to having both exposed and unexposed individuals in each strata of the confounders. Let us define \(Y(e,m,w)\) as the value the outcome would take when the exposure is set to \(e\), the mediator of interest is set to \(m\) and the other mediators are set to \(w\). Below we summarize the sequential ignorability assumptions for multiple mediators:
-
1.
\(\left\{Y\left(e,m,w\right), M\left({e}^{*}\right), W\left({e}^{**}\right)\right\}\perp E\left|X=x\right..\)
-
2.
\(Y\left({e}^{*},m,w\right)\perp \left(M\left(e\right),W\left(e\right)\right)\left|E=e,X=x\right..\)
-
3.
\(Y\left(e,m,w\right)\perp \left(M\left({e}^{*}\right),W\left(e\right)\right)\left|E=e,X=x.\right.\)
Please note that, for path specific effects, assuming a temporal ordering among mediators such that \(W\) precedes \(M\), the sequential ignorability assumptions would be the following:
-
1.
\(\left\{Y\left(e,m,w\right), M\left({e}^{*}\right)\right\}\perp E\left|X=x, W=w\right..\)
-
2.
\(Y\left({e}^{*},m,w\right)\perp M(e)\left|E=e, X=x, W=w\right..\)
In addition, we assume both the positivity assumption: \(P\left(E=e\left|X=x\right.\right)>0\) and \(P\left(M=m,W=w\left|E=e,X=x\right.\right)>0\ \forall x,e,{e}^{*},m,w\); and the Stable Unit Treatment Value Assumption (SUTVA), or no-interference assumption, which implies that:
-
1.
Potential mediator and outcome values of individual \(i\) are not dependent on exposures of other individuals, i.e. \({M}_{ik}\left(e\right)={M}_{ik}\left({e}_{i}\right)\) and \({Y}_{i}\left(e, {M}_{k}\left(e\right), {W}_{k}\left(e\right)\right)= {Y}_{i}\left({e}_{i}, {M}_{ik}\left({e}_{i}\right), {W}_{ik}\left({e}_{i}\right)\right)\).
-
2.
There are no multiple versions of exposures, i.e. \({e}_{i}={e}_{i}^{*}\) implies \({M}_{ik}\left({e}_{i}\right)={M}_{ik}\left({e}_{i}^{*}\right)\) and \({Y}_{i}\left({e}_{i}, {M}_{ik}({e}_{i}), {W}_{ik}({e}_{i}\right))= {Y}_{i}\left({e}_{i}^{*}, {M}_{ik}({e}_{i}^{*}), {W}_{ik}({e}_{i}^{*}\right))\).
-
3.
There are no multiple versions of mediators, i.e. if \({m}_{ik}={m}_{ik}^{*}\) then \({Y}_{i}\left({e}_{i}, {m}_{ik}, {w}_{ik}\right)= {Y}_{i}\left({e}_{i}, {m}_{ik}^{*}, {w}_{ik}\right)\).
Methods for Mediation Analysis with Epigenomic Data
Several methods have been developed for multiple mediation analysis in high-dimensional settings. Some of them rely on univariate mediation analysis, subsequently combining the obtained results to account for the composite nature of the null hypothesis in mediation analysis [23••]. Other more sophisticated methods conduct variable selection (either included in the algorithm or as a previous step) and then apply multiple mediation methods to the reduced set of mediators. Below, we summarize the different methods that have been developed for multiple mediation analysis in the epigenomic data setting, as well as their strengths and limitations. An overview of the described mediation analysis methods is presented in Fig. 2.

Summary of methods for mediation analysis in the omics data setting
One-Marker-at-a-Time Approach for Mediation
The first proposed approach for mediation analysis in the epigenomics data context was to run two separate epigenome-wide association studies (EWAS), one for the exposure and one for the outcome, and select the epigenetic marks that are statistically significant in both sets of models after accounting for multiple comparisons. Subsequently, a simple mediation analysis would be run for each of the methylation sites individually (see, for example, [24]). Although this approach would work well in the setting of independent mediators, it is problematic for the context of correlated mediators, as the approach does not take into account the interrelations between mediators, which leads to the same biological pathways being considered part of the indirect path through epigenetics several times, and therefore to a sum of relative mediated effects greater than 100%, which is not possible.
Permutation tests such as the Causal Inference Test (CIT) [25], which are based on the combination of individual p-values for the associations between each mediator and the outcome and exposure, became popular for mediation analysis several years ago. Improved versions of CIT have lately been proposed. The Multimed R package [26] uses tests that evaluate multiple potential mediators and controls the family-wise error rate. This approach, however, does not provide estimations of indirect, direct and total effects, just p-values and test statistics. In addition, it does not evaluate all mediators simultaneously in the same model, and to our knowledge, it has only been implemented for continuous outcomes.
Similarly, the HDMAX2 method [27] combines latent factor regression models for EWAS with max-squared tests for mediation analysis. Although the latent factor regression models are useful to account for unmeasured confounding factors such as batch effects, this method still relies on the combination of p-values of individual associations to estimate mediated effects. After determining the significant mediators, the mediated effects are calculated using the mediation R package. However, to do so, the causal structure of the mediators (i.e., the sequential order of mediators) needs to be known in advance, which is not generally plausible in the omics data settings. A recent study found a mediated effect of DNA methylation on the association between maternal smoking and birthweight using this method. The study found that a lowering of 44.5 g in birthweight might be attributable to maternal smoking (versus not smoking) mediated by DNA methylation changes in 32 CpGs and 19 genomic regions [27].
Several mediation analysis methods for high dimensions rely on calibrated p-values, which fit one mediator at a time and then borrow information from all mediators to calculate modified p-values that appropriately control for the type I error rate. These methods include JT-comp [28], divide-aggregate composite-null test (DACT) [29], JS-mixture [30] and JTV-comp [31]. Methods based on calibrated p-values have been widely used on DNA methylation mediation analysis. The JT-comp method was used to evaluate the potential mediating role of DNA methylation on the association between socioeconomic status and BMI, with no statistically significant mediated effects found at FDR p-value cut-off of 0.05 [28]. The DACT method was used to identify mediated effects of DNA methylation on the association between smoking and lung function, including the well-known smoking-related genes AHRR and F2RL3 [29]. JS-mixture identified a mediated effect of DNA methylation on the association between genetic regulation of gene expression and prostate cancer [30]. However, all these methods rely on the product of coefficients method, which, as described before, cannot directly accommodate exposure-mediator interactions, and has issues with non-collapsibility for multiplicative models. Also, they are focused on hypothesis testing rather than effect estimation, thus, they only provide p-values. In addition, although these methods borrow information from all mediators, most of them are not able to account for correlations. An approach that considers all mediators together and leverages the effect of each mediator in presence of the others would be desirable for the evaluation of mediated effects through omics data.
Multiple-Markers-at-a-Time Approach for Mediation
The development of statistical methods able to deal with hundreds of thousands or millions of mediators might be challenging. Therefore, most approaches for multiple mediation in the context of omics data still rely on the screening step mentioned in the previous section, which will filter out the variables that are not associated with the exposure or the outcome, as a first step. After this dimensionality reduction, methods that are able to deal with multiple mediators in lower dimensions might be used.
The multimediate R package [19•] fits a quasi-bayesian algorithm that relies on the counterfactual framework. It deals with multiple correlated mediators and is able to accommodate continuous, binary, and survival outcomes [32]. In addition, this method is able to accommodate both exposure-mediator and mediator-mediator interactions. Multimediate was used to identify mediated effects of DNA methylation in three CpG sites on the association between smoking and lung cancer [33]. However, this method assumes that correlations between mediators are independent of the exposure. This could be feasible in the omics data setting, as spatial correlations that do not necessarily depend on the exposure are common. However, it constitutes a strong assumption that is not easy to verify in practice. The mma R package [34] also deals with multiple correlated mediators, however, according to the documentation, this model is only able to deal with exposure-mediator or mediator-mediator interactions in linear models. Even the well-known mediation R package [35], which has been considered the gold standard for simple mediation analysis in the last years and works under many different distributions for both the outcome and the mediator, has been extended to conduct multiple mediation analysis and can deal with interactions. However, this method does not specifically account for the correlated structure of the mediators. The EMAS R package, which uses the methodology of the mediation R package to conduct epigenome-wide mediation analysis, has also been proposed. However, this package performs mediation analysis for each CpG one by one [36].
Some mediation analysis methods even perform variable selection using SIS and mediation analysis in the same algorithm. The R-squared effect size method [9] uses either SIS or the false discovery rate method (FDR) to reduce the dimensionality, and then uses the R2 effect size measure to calculate the joint mediated effect for multiple mediators. By using this method, the authors showed that 38% of the age-related differences in systolic blood pressure might be mediated by gene expression in the Framingham Heart Study [9]. However, this method does not account for exposure-mediator interactions, and the RSqMed R package, which fitted this algorithm, was removed from the CRAN repository several months ago. The high-dimensional mediation analysis (HIMA) method has been used to identify mediated effects of DNA methylation on the association between smoking and lung function. Of 484,548 CpGs tested, two were identified as mediators [37]. This method also uses SIS to select mediators that are associated with the outcome. Subsequently, it uses the Minimax Concave Penalty, a variation of the LASSO that satisfies the oracle property [38], to estimate the coefficients of the mediator and outcome models. The oracle property mathematically ensures that the right subset of variables is selected, and that the estimation rate is optimal. The pathway LASSO [39], another penalization-based high-dimensional mediation method, applies the LASSO penalty, which has shown to be biased in coefficient estimation. In addition, both HIMA and pathway LASSO rely on the product of coefficients method, which presents the previously described drawbacks.
Other mediation methods that perform variable selection and mediation without using SIS have been proposed. The gene-based high-dimensional mediation analysis (gHMA) method [40], for example, has been used in epigenomic data analysis. This method applies kernel principal components analysis, which is a non-linear extension of the regular principal components analysis, to one gene at a time, evaluating all CpG sites annotated to that gene. Then, it performs mediation analysis separately in each gene. However, this method does not consider potential correlations between genomic sites that are annotated to different genes, and it still relies on univariate mediation analysis for each gene. gHMA was proposed to conduct mediation analysis considering DNA methylation marks as mediators, however, the authors did not identify any mediated effects for the associations between alcohol consumption and ovarian cancer risk or between childhood maltreatment and comorbid post-traumatic stress disorder and depression. On the other hand, several PCA-based mediation analysis methods have also been proposed [23••]. PCA methods effectively deal with the problem of correlations between mediators. However, they add the challenge of interpretability, as transformed variables are combinations of the original variables that might not be easy to interpret.
Bayesian Approaches for Mediation
Several Bayesian mediation methods have also been developed. These methods have the advantage that, as in all Bayesian methods, prior information on mediators can be introduced. The BAMA approach, developed in the hdbm R package [41], uses Bayesian variable selection models with sparsity-inducing priors on mediator effects, assuming that only a small proportion of the proposed mediators may mediate the effect of the exposure on the outcome. It applies the Markov chain Monte Carlo (MCMC) algorithm and uses posterior inclusion probabilities (PIP) to select mediators. This method was used to identify DNA methylation sites that might be mediators of the effect of socioeconomic status on cardiometabolic outcomes in the Multi-Ethnic Study of Atherosclerosis [41]. Two improvements of the BAMA method have been proposed, one using four-component Gaussian mixture model (GMM) and the other one based on a product threshold Gaussian (PTG) prior [42]. These methods, however, do not directly incorporate correlations between mediators. The GMM-CorrS method uses Gaussian mixture models while accounting for the composite structure of the null hypothesis [43]. However, all these methods and, in general, most of the Bayesian methods are fitted using MCMC methods, which cannot easily be parallelized as each iteration directly depends on the result of the previous one. Therefore, they present many computational issues.
Discussion
In this article, we present an overview of the different mediation analysis methods that have been developed and could be applied to epigenomic data, highlighting their strengths and limitations. Although extensive research has been conducted to develop mediation methods that account for the particularities of omics data, most of the methods have limitations that should be approached in future work.
Epigenetic marks, and in particular DNA methylation, have shown good predictive ability for several health outcomes [44,45,46,47,48] and have shown evidence of a mediating role between environmental exposures and disease [49,50,51]. However, establishing whether DNA methylation is a biological mediator or, conversely, a biomarker of other disrupted biological processes, is challenging. The no unmeasured confounding assumption, which is essential to identify mediated effects, is impossible to verify in practice for observational studies [52•]. Thus, sensitivity analyses are needed to measure the impact of those potential unmeasured confounders in the mediated effects. Many sensitivity analysis techniques have been developed for mediation analysis, including for survival outcomes [17, 20•, 52•, 53, 54]. Relevant future work should include the adaptation of these sensitivity analysis techniques to the high-dimensional mediation setting. In addition, some methods are able to deal with exposure-mediator interactions, but in most of the methods, mediator-mediator interactions are not addressed.
One important feature when evaluating epigenetic marks as mediators is that, generally, we assume that the correlations between them are non-causal, i.e., that they are not causally ordered. As many epigenetic marks present spatial correlations or correlations due to common influences of environmental factors, this hypothesis might be plausible. However, we cannot discard that some epigenetic marks might influence the activity of others. Even if there was a causal order, it would be very hard to disentangle which epigenetic modifications precede others with current technology. Increasing biological knowledge will hopefully help to shed light on the correlation structure and causal order of epigenetic marks, and will also potentially help with interpretability of results found in mediation analysis of epigenetic data. On the other hand, in cross-sectional studies, the exposure and DNA methylation are often measured at the same time, therefore, it is unclear whether some reverse causation might exist, or whether the exposure actually precedes DNA methylation dysregulations. Collecting longitudinal data on both the exposure and the mediators will help to identify the causal structure and to ensure the correct temporal order. In the meantime, caution needs to be taken to draw conclusions from mediation analysis involving epigenetic marks as mediators.
In principle, many of the described mediation methods could be applied to other omics data types. In fact, one of the main goals of omics data research would be to integrate all omics data together in statistical models, in order to maximize the information they provide. However, each omics data has its particular characteristics. For genomic SNPs data, for example, the dimensionality is sometimes much larger than for microarray epigenomic data (several millions of variables, instead of hundreds of thousands). Therefore, some variable selection or mediation methods might present huge computational times when applied to genomic data. Several efforts have been made to integrate different omics data, such as the Signature Regulatory Clustering (SiRCle) tool [55], which aims to integrate DNA methylation, RNA-seq and proteomics data. The integration of proteomics data, however, constitutes another statistical challenge, as proteomics data generally present a huge number of missing data, sometimes above 90% [56]. Therefore, imputation methods would need to be incorporated to mediation analysis to be able to deal with these data. Future research should focus on disentangling how each omics layer influences the subsequent layer and how to integrate all layers in mediation analysis.
An important limitation of epigenetic studies that also extends to mediation analysis is the lack of generalizability across populations. Although robust and generalizable epigenetic modifications have been identified for some exposures or phenotypes, such as smoking [57] or cancer [58], little overlap has been found across populations for other exposures or endpoints. This could be due to some epigenetic marks being population-specific, due to technical differences on DNA methylation measurement and preprocessing, or due to not being able to appropriately account for confounding, among other reasons. Multi-cohort studies that evaluate the robustness of potential epigenetic mediators across populations are needed. Moreover, given that DNA methylation has shown to be highly tissue specific [59,60,61], DNA methylation measured in different tissues might lead to very different findings. However, DNA methylation measured in blood has shown potential, for example, for early screening of non-hematopoietic cancers such as breast [62] and colorectal [63] cancer. Thus, blood DNA methylation might induce dysregulations of important biological processes in different target tissues.
Importantly, the approach of considering environmental factors as mixtures is more appropriate than considering them as separate exposures, as environmental pollutants are known to co-occur in the environment. Therefore, describing the interrelations and interactions between them in association with health outcomes is a relevant scientific question. However, environmental mixtures have been overlooked for mediation analysis due to the complexity of considering a high-dimensional exposure. The BKMR-causal mediation analysis method [64] deals with mediation analysis in presence of environmental mixtures, however, it has not yet been extended to the setting of multiple mediators. More research is needed to develop methods for mediation analysis in the omics data setting considering environmental mixtures.
Conclusions
The growing interest in evaluating the mediating role of epigenetic marks on the association between environmental factors and chronic disease has led to extensive methodological discovery. Many sophisticated statistical methods that address important challenges of high-dimensional data have been developed in the last years. However, key challenges such as the development of sensitivity analyses, dealing with mediator-mediator interactions, including environmental mixtures as exposures, the integration of different omics data, or the determination of the causal structure of epigenetic marks have not been adequately addressed. Therefore, more research is needed to improve existing methods for epigenomic mediation analysis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Yamada R, Okada D, Wang J, Basak T, Koyama S. Interpretation of omics data analyses. J Hum Genet Nature Publishing Group. 2020;66:93–102.
Toraño EG, García MG, Fernández-Morera JL, Niño-García P, Fernández AF. The impact of external factors on the epigenome: In utero and over lifetime. Biomed Res Int. 2016:2568635.
Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8:9(019505).
Stylianou E. Epigenetics of chronic inflammatory diseases. J Inflamm Res. 2019;12:14.
Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J / National Research Council, Institute of Laboratory Animal Resources. 2012;53:289–305.
Wu X, Choi JM. The impact of spatial correlation on methylation entropy with application to mouse brain methylome. Epigenetics Chromatin. 2023;16:1–12.
An Introduction to `glmnet` • glmnet [Internet]. [cited 2023 Jun 12]. Available from: https://glmnet.stanford.edu/articles/glmnet.html
Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. J R Stat Soc Ser B (Statistical Methodol). 2008;70:849–911. Description of the Sure Independence Screening Method, widely use for dimensionaly reduction prior to mediation analysis in ultra high-dimensional settings.
Yang T, Niu J, Chen H, Wei P. Estimation of total mediation effect for high-dimensional omics mediators. BMC Bioinformatics. 2021;22:414.
Blum MGB, Valeri L, François O, Cadiou S, Siroux V, Lepeule J, et al. Challenges raised by mediation analysis in a high-dimension setting. Environ Health Perspect. 2020;128(5). Summary of several statistical methods developed for high-dimensional mediation analysis by 2020.
MacKinnon DP, Lockwood CM, Hoffman JM, West SG, Sheets V. A Comparison of methods to test mediation and other intervening variable effects. Psychol Methods. 2002;7(1):83–104.
Daniel R, Zhang J, Farewell D. Making apples from oranges: Comparing noncollapsible effect estimators and their standard errors after adjustment for different covariate sets. Biom J. 2021;63(3):528–557.
Martinussen T, Vansteelandt S. On collapsibility and confounding bias in Cox and Aalen regression models. Lifetime Data Anal. 2013;19:279–96.
Pearl J. Direct and Indirect Effects. In Proceedings of the seventeenth conference on uncertainty in artificial intelligence, San Francisco, CA: Morgan Kaufmann, 411–420, 2001.
Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3:143–55. Description of the counterfactual approach for mediation analysis and identifiability conditions.
Imai K, Jo B, Stuart EA. Commentary: using potential outcomes to understand causal mediation analysis. Multivariate Behav Res. 2011;46:842–54.
Albert JM, Wang W. Sensitivity analyses for parametric causal mediation effect estimation. Oxford Acad. 2015;16:339–51.
Lange T, Hansen J V. Direct and indirect effects in a survival context. Epidemiology. 2011;22:575–81. Extension of mediation analysis to a survival outcome setting.
Jérolon A, Baglietto L, Birmelé E, Alarcon F, Perduca V. Causal mediation analysis in presence of multiple mediators uncausally related. Int J Biostat. 2020;17(2):191–221. Extension of multiple mediation analysis to uncausally correlated mediators.
Imai K, Yamamoto T. Identification and sensitivity analysis for multiple causal mechanisms: Revisiting evidence from framing experiments. Polit Anal. 2013;21:141–171. Extension of simple mediation analysis to multiple mediators.
Van Der weele T, Vansteelandt S. Mediation analysis with multiple mediators. Epidemiol Method. 2014;2:95.
Vanderweele TJ, Chan TH. Mediation analysis: a practitioner’s guide. Annu Rev Public Heal. 2016;37:17–32.
Zeng P, Shao Z, Zhou X. Statistical methods for mediation analysis in the era of high-throughput genomics: Current successes and future challenges. ComputStruct Biotechnol J. Elsevier. 2021;19:3209–24. Detailed description of more than ten high-dimensional mediation methods that have been developed in the last years.
Tang Y, Gan H, Wang B. et al. Mediating effects of DNA methylation in the association between sleep quality and infertility among women of childbearing age. BMC Public Health. 2023;23:1802.
Gu T. Power consideration and caveats of Causal Inference Test (CIT) for mediation analysis. 2021 [cited 2023 Jun 26]; Available from: https://digital.lib.washington.edu:443/researchworks/handle/1773/47364
Boca SM, Sinha R, Cross AJ, Moore SC, Sampson JN. Testing multiple biological mediators simultaneously. Bioinformatics. 2014;30:214–20.
Jumentier B, Barrot C-C, Estavoyer M, Tost J, Heude B, François O, et al. High-dimensional mediation analysis: A new method applied to maternal smoking, placental DNA methylation, and birth outcomes. Environ Health Perspect. 2023;131(4):47011.
Huang YT. Genome-wide analyses of sparse mediation effects under composite null hypotheses. Inst Mathe Stat. 2019;13:60–84.
Liu Z, Shen J, Barfield R, Schwartz J, Baccarelli AA, Lin X. Large-scale hypothesis testing for causal mediation effects with applications in genome-wide epigenetic studies. J Am Stat Assoc. 2022;117:67–81.
Dai JY, Stanford JL, LeBlanc M. A multiple-testing procedure for high-dimensional mediation hypotheses. J Am Stat Assoc. 2022;117(537):198–213.
Huang YT. Variance component tests of multivariate mediation effects under composite null hypotheses. Biometrics. 2019;75:1191–204.
GitHub - AllanJe/multimediate. 2023. Available from: https://github.com/AllanJe/multimediate.
Domingo-Relloso A, Joehanes R, Rodriguez-Hernandez Z, Lahousse L, Haack K, Fallin MD, Herreros-Martinez M, Umans JG, Best LG, Huan T, Liu C, Ma J, Yao C, Jerolon A, Bermudez JD, Cole SA, Rhoades DA, Levy D, Navas-Acien A, Tellez-Plaza M. Smoking, blood DNA methylation sites and lung cancer risk. Environ Pollut. 2023;334:122153.
Yu Q, Li B. mma: An R package for mediation analysis with multiple mediators. J Open Res Softw. 2017;5(1):11.
Tingley D, Yamamoto HT, Hirose K, Keele L, Princeton KI. mediation: R Package for Causal Mediation Analysis. [cited 2021 Aug 29]; Available from: http://cran.r-project.org/package=mediation
CRAN - Package EMAS [Internet]. [cited 2023 Jul 10]. Available from: https://cran.r-project.org/web/packages/EMAS/index.html
Zhang H, Zheng Y, Zhang Z, Gao T, Joyce B, Yoon G, et al. Estimating and testing high-dimensional mediation effects in epigenetic studies. Bioinformatics. 2016;32:3150–4.
Wang T, Xu PR, Zhu LX. Non-convex penalized estimation in high-dimensional models with single-index structure. J Multivar Anal. 2012;109:221–35.
Zhao Y, Luo X. Pathway lasso: Pathway estimation and selection with high-dimensional mediators. Stat Interface. 2022;15:39.
Fang R, Yang H, Gao Y, Cao H, Goode EL, Cui Y. Gene-based mediation analysis in epigenetic studies. Brief Bioinform; 2021;22.
Song Y, Zhou X, Zhang M, Zhao W, Liu Y, Kardia SLR, et al. Bayesian shrinkage estimation of high dimensional causal mediation effects in omics studies. bioRxiv [Internet]; 2018 [cited 2023 Jun 26];467399. Available from: https://www.biorxiv.org/content/10.1101/467399v1
Song Y, Zhou X, Kang J, Aung MT, Zhang M, Zhao W, et al. Bayesian sparse mediation analysis with targeted penalization of natural indirect effects. J R Stat Soc Ser C Appl Stat. 2021;70:1391.
Song Y, Zhou X, Kang J, Aung MT, Zhang M, Zhao W, et al. Bayesian hierarchical models for high-dimensional mediation analysis with coordinated selection of correlated mediators. Stat Med. 2021;40:6038–56.
Shanthikumar S, Neeland MR, Maksimovic J, Ranganathan SC, Saffery R. DNA methylation biomarkers of future health outcomes in children. Mol Cell Pediatr. 2020;9;7(1):7.
Wu C, Zhu J, King A, Tong X, Lu Q, Park JY, et al. Novel strategy for disease risk prediction incorporating predicted gene expression and DNA methylation data: a multi-phased study of prostate cancer. Cancer Commun. 2021;41:1387–97.
Li M, Zhu C, Xue Y, Miao C, He R, Li W, et al. A DNA methylation signature for the prediction of tumour recurrence in stage II colorectal cancer. Br J Cancer. 2023;128:1681–9.
Peng Y, Wu Q, Wang L, Wang H, Yin F. A DNA methylation signature to improve survival prediction of gastric cancer. Clin Epigenetics. 2020;12:1–16.
Cappozzo A, McCrory C, Robinson O, FreniSterrantino A, Sacerdote C, Krogh V, et al. A blood DNA methylation biomarker for predicting short-term risk of cardiovascular events. Clin Epigenetics. 2022;14:1–17.
Rutten BPF, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull. 2009;35:1045.
Fujii R, Sato S, Tsuboi Y, Cardenas A, Suzuki K. DNA methylation as a mediator of associations between the environment and chronic diseases: a scoping review on application of mediation analysis. Epigenetics. 2022;17:759.
Poursafa P, Kamali Z, Fraszczyk E, Boezen HM, Vaez A, Snieder H. DNA methylation: a potential mediator between air pollution and metabolic syndrome. Clin Epigenetics. 2022;14:1–13.
Vanderweele TJ, Arah OA. Unmeasured confounding for general outcomes, treatments, and confounders: bias formulas for sensitivity analysis. Epidemiology. 2011;22:42–52. Description of a sensitivity analysis approach for unmeasured confounding in mediation analysis.
Vanderweele TJ, Chiba Y. Sensitivity analysis for direct and indirect effects in the presence of exposure-induced mediator-outcome confounders. Epidemiol Biostat Public Heal. 2014;11:e9027.
Tchetgen Tchetgen EJ. On causal mediation analysis with a survival outcome. Int J Biostat. 2011;7: 33.
Mora A, Schmidt C, Balderson B, Frezza C, Bodén M. SiRCle (Signature Regulatory Clustering) model integration reveals mechanisms of phenotype regulation in renal cancer. bioRxiv 2022.07.02.498058
Jin L, Bi Y, Hu C, Qu J, Shen S, Wang X, et al. A comparative study of evaluating missing value imputation methods in label-free proteomics. Sci Rep. 2021;11:1–11.
Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;5:436–47.
Ibrahim J, Peeters M, Van Camp G, de Beeck KO. Methylation biomarkers for early cancer detection and diagnosis: current and future perspectives. Eur J Cancer. 2023;178:91–113.
Lokk K, Modhukur V, Rajashekar B, Märtens K, Mägi R, Kolde R, Koltšina M, Nilsson TK, Vilo J, Salumets A, Tõnisson N. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15(4):r54.
Slieker RC, Relton CL, Gaunt TR, et al. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin. 2018;11:25.
Cardenas A, Lutz SM, Everson TM, Perron P, Bouchard L, Hivert MF. Mediation by placental DNA methylation of the Association of Prenatal Maternal Smoking and Birth Weight. Am J Epidemiol. 2019;188:1878–86.
Wang T, Li P, Qi Q, Zhang S, Xie Y, Wang J, et al. A multiplex blood-based assay targeting DNA methylation in PBMCs enables early detection of breast cancer. Nat Commun. 2023;14(1):4724.
Cai G, Cai M, Feng Z, Liu R, Liang L, Zhou P, et al. A multilocus blood-based assay targeting circulating tumor DNA methylation enables early detection and early relapse prediction of colorectal cancer. Gastroenterology. 2021;161:2053-2056.e2.
Devick KL, Bobb JF, Mazumdar M, Claus Henn B, Bellinger DC, Christiani DC, et al. Bayesian kernel machine regression–causal mediation analysis. Stat Med. 2022;41:860.
Funding
Drs. Domingo-Relloso and Valeri received funding from the National Institute of Environmental Health Sciences (P42ES033719). Dr. Tellez-Plaza received funding from the Strategic Action for Research in Health sciences (CP12/03080 and PI15/00071), which are initiatives from Instituto de Salud Carlos III and the Spanish Ministry of Science and Innovation and co-funded with European Funds for Regional Development (FEDER) and by the State Agency for Research (PID2019-108973RB- C21). The opinions and views expressed in this article are those of the authors and do not necessarily represent the official position of the Instituto de Salud Carlos III (Spain).
Author information
Authors and Affiliations
Contributions
A.D. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare they have no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Domingo-Relloso, A., Tellez-Plaza, M. & Valeri, L. Methods for the Analysis of Multiple Epigenomic Mediators in Environmental Epidemiology. Curr Envir Health Rpt 11, 109–117 (2024). https://doi.org/10.1007/s40572-024-00436-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-024-00436-9