
Abstract
Purpose of Review
The purpose of the study was to discuss the main mechanisms associated with environmental and genetic factors that contribute to the development of Parkinson’s disease (PD).
Recent Findings
Novel genetic contributors to PD are being identified at a rapid pace in addition to novel environmental factors. The discovery of mutations in alpha-synuclein and leucine-rich repeat kinase 2 causing inherited forms of PD along with epidemiological, in vitro, and in vivo studies identifying herbicides, pesticides, and metals as risk factors have dramatically improved our understanding of mechanisms involved in the development of PD. However, at the same time, these discoveries have also added layers of complexity to the disease.
Summary
Within the last several years, the genetics associated with PD has dominated the field in many ways; however, the majority of PD cases are likely due to different combinations of environmental exposures and genetic susceptibility. The most common toxicants used to model PD including rotenone, paraquat, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine have been shown to interact with many of the genes linked with PD such as alpha-synuclein. Therefore, an understanding of mechanisms common between genetic and environmental factors is essential for early detection and successful translation of potential therapies, which is the ultimate goal.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder with an overall reported prevalence of 1.8 (per 100 population) in people 65 years of age and older [1]. PD is characterized by the loss of dopaminergic (DA) neurons in the substantia nigra and the development of Lewy bodies and Lewy neurites in the brain and periphery. At the time of diagnosis of PD, patients experience DA-related sensorimotor impairments, including bradykinesia, tremor, and rigidity that worsen over time. In addition to motor impairments, non-motor symptoms including gastrointestinal, cardiovascular autonomic, sleep, olfactory, cognitive, and neuropsychiatric dysfunctions can also develop, many of which may precede the onset of the motor symptoms and likely involve neuropathology outside of the substantia nigra [2]. This indicates that PD is a systemic disorder affecting multiple brain regions and peripheral systems.
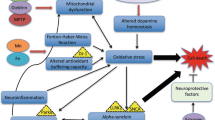
While the cause of the majority of cases is unknown, there are recognized factors that increase the risk of developing PD. These include aging, genetics, and environmental exposures such as metals, pesticides, and solvents [3,4,5]. It is generally considered that gene-environment interactions underlie idiopathic or “sporadic” PD. Although inherited forms of PD account for only 10–15% of all cases, their discovery has had a substantial impact on our knowledge and understanding of the mechanisms involved in gene-environment interactions in PD including mitochondrial function, oxidative stress, protein degradation, and inflammation. In addition, epidemiology studies that emphasize the mechanism of environmental factors have found positive associations between specific pesticides and the development of PD [5]. Currently, there are over 20 PD-related genetic loci (PARK) that have been linked to PD and a growing list of genes that increase risk [6, 7]. These genetic factors, combined with the numerous environmental exposures identified, underscores the complexity, individual variability, and challenge of developing treatments to slow or stop the progression of PD. There are currently no therapeutic interventions that modify disease progression. Thus, identifying common mechanisms between gene-environment interactions is an essential step for improved therapeutics.
Environmental Factors Associated with PD
Environmental factors have long been implicated in PD since James Parkison’s initial description of the disease in 1817 during the industrial revolution [8]. Heavy metals including manganese and iron were linked to Parkinsonian conditions for decades [9, 10]. In the 1980s, the “environmental hypothesis” was revived with the discovery of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its selective toxicity on the nigrostriatal dopamine system in humans [11]. In addition to MPTP, twin studies also suggested a lack of genetic contribution to sporadic PD further supporting an important role for the environment in PD [12]. Since then, pesticides, herbicides, fungicides, and heavy metals have all been linked to the development of PD and PD-like syndromes [4, 5, 9, 10] (Table 1).
Heavy Metals
Environmental exposures to heavy metals are established risk factors for many neurodegenerative conditions including PD. The most studied metals related to PD are manganese and iron. Manganese is an essential metal involved in multiple cellular functions including energy metabolism, development, the immune response, and antioxidant responses [13] It is ubiquitous in the environment, and excessive exposure, especially in occupations such as mining and welding, is a significant health risk and can cause manganism, an age-related neurodegenerative condition partially resembling PD. Manganism is characterized by PD-like motor symptoms and cognitive impairments but is distinct from classical PD as the motor deficits are typically not responsive to levodopa and additional impairments such as dystonia and “cock-walk” develop. Manganese preferentially accumulates in the basal ganglia affecting primarily the globus pallidus. Intracellular manganese toxicity involves multiple mechanisms known to also be involved in PD and other neurodegenerative conditions including mitochondrial dysfunction, oxidative stress, and impaired protein degradation [14].
A link between iron and PD has been known for decades, but its role in PD remains unclear. Epidemiological studies looking at associations between occupational or dietary iron and PD are limited and overall inconclusive. However, the case for iron and PD primarily comes from postmortem and imaging studies. Several postmortem studies show an increase in iron in the substantia nigra in PD cases [10, 15]. Magnetic resonance imaging also reveals iron deposition in the brain in PD [16]. In addition, iron has been found in Lewy bodies in the substantia nigra in postmortem PD cases [17]. Neuromelanin in nigral dopamine neurons is considered a contributing factor to toxicity as it can bind to iron causing oxidative stress that leads to cell death [18]. It still remains unclear though whether iron accumulation is a cause or consequence of PD.
Pesticides
Numerous epidemiology studies over the last two decades link pesticide exposure to an increased risk of developing PD [5, 19, 20]. While it is challenging to single out specific compounds, two pesticides in particular have been identified; these include the herbicide paraquat and pesticide rotenone [5]. Paraquat (1,1′-dimethyl-4,4′-bipyridinium). Several studies show a relationship between long-term paraquat exposure and increased risk of PD [19, 20]. Although structurally similar to MPTP, paraquat’s mechanism of toxicity is distinct from MPTP [21, 22]. MPTP is converted to MPP+ where it is transported by the dopamine transporter into the cell [23, 24]. There, it inhibits complex I of the mitochondrial electron transport chain leading to cell death. In contrast, paraquat induces cytosolic oxidative stress followed by caspase-3-mediated cell death [22]. Studies show that paraquat is toxic to dopaminergic neurons and systemic administration kills a subset of nigrostriatal dopaminergic neurons in mice [25, 26]. Rotenone. Similar to MPTP, rotenone causes mitochondrial dysfunction. Rotenone freely crosses cell membranes and acts as a specific inhibitor of complex I of the mitochondrial electron transport chain [27]. As oxidative stress and mitochondrial dysfunction are key mechanisms underlying PD pathogenesis, both paraquat and rotenone continue to be used in basic science to model PD in in vitro and in vivo systems to study gene-environment interactions [28•, 29,30,31].
Additional Pesticides
More recently, additional groups of pesticides have been associated with PD that will likely be the focus of future gene-environment studies. These include organochlorines, organophosphates, and pyrethroids [32,33,34,35]. Organochlorines are chlorinated hydrocarbons used in agriculture and mosquito control and include dieldrin, DDT, and Β-hexachlorocyclohexane (Β-HCH) [32, 36,37,38]. Although these pesticides have been banned in the USA for some time, they still pose a threat due to their long half-life and lipophilic nature. Organophosphates are the main ingredient in commonly used household pesticides. Increased ambient organophosphate exposure is associated with a higher risk of PD, while variants in PON1, a gene important in the metabolism of organophosphates, combined with organophosphate exposure may modify PD risk [39, 40]. Pyrethroids are a newer class of pesticides widely used in household and agricultural environments and although there is less known about the pyrethroids, they have been shown to affect the dopaminergic system in vivo [35, 41•]. Currently, these pesticides are in the early stages of determining their contribution to gene-environment interactions in PD.
Genetic Factors Associated with PD
Despite their rarity, genes associated with inherited forms of PD have significantly advanced our understanding of the mechanisms involved in cellular dysfunction and cell death in PD. Here, genes associated with inherited forms of PD are grouped according to their broad mechanisms of toxicity-mitochondrial dysfunction, oxidative stress, and impaired protein degradation (Table 2). Notably, these mechanisms are not mutually exclusive and often there is a reciprocal relationship between mechanisms with disruption of one setting into play another. Indeed, several of the genes are involved in multiple mechanisms, highlighting their potential importance in gene-environment interactions .
Mitochondrial Dysfunction
α-Synuclein (aSyn; PARK1) was the first gene to be linked with PD when a missense mutation (A53T) was identified in familial PD [42]. This discovery led to the identification of aSyn as a major component of Lewy bodies and glial cytoplasmic inclusions in sporadic PD, dementia with Lewy bodies, and multiple system atrophy (synucleinopathies). Since then, aSyn dysfunction has dominated the field. Functionally, aSyn is a 140 amino acid presynaptic protein involved in vesicle handling and neurotransmitter release [43, 44]. Pathologically, aSyn is prone to misfolding and aberrant forms of the protein are associated with mitochondrial dysfunction, oxidative stress, impaired autophagy, and cell death [45,46,47]. Inherited forms of PD where there is duplication and triplication of the wildtype aSyn locus suggest that the level of aSyn is an important contributing factor to disease severity and onset indicating a “gene-dosage” toxic effect of aSyn [48, 49]. This also suggests that environmental factors that promote aSyn accumulation could contribute to PD-like pathology. Indeed, MPTP, paraquat, rotenone, and manganese exposure have all been shown to increase aSyn [50,51,52]. Several in vivo studies also show that in combination with aSyn overexpression, MPTP and PQ can potentiate aSyn-induced pathology [53,54,55,56]. More recently, it has been shown that knockdown of aSyn is neuroprotective in a rotenone model of PD indicating that aSyn reduction is an important therapeutic target [30].
Mutations in leucine-rich repeated kinase 2 (LRRK2; PARK8), in particular G2019S, are the most common genetic cause of PD [57, 58]. The G2019S mutation in LRRK2 accounts for approximately 5% of dominantly inherited familial and 1–2% of sporadic PD, and LRRK2-associated PD is indistinguishable from sporadic PD [57,58,59,60]. The function of LRRK2 has been reported to include synaptic vesicle storage, microglial response, and MAPK kinase signaling [61, 62]. LRRK2 is associated with the mitochondrial membrane, and human fibroblasts from LRRK2 G2019S carriers have impaired mitochondria function and morphology [63].
Mutations in the PTEN-induced putative kinase 1 gene (PINK1) are the second most common cause of autosomal recessive PD and are also implicated in sporadic cases [64, 65]. The function of PINK1 remains unclear, but studies implicate PINK1 in mitochondrial function and mitophagy [66]. In humans, PINK1 genetic variants cause progressive functional deficits and nigrostriatal DA cell loss [67, 68]. It has been reported that PINK1 knockout rats develop metabolic and mitochondrial pathogenesis as well as sensorimotor deficits and nigrostriatal DA cell loss [69, 70].
The recently characterized PARK9 gene, also known as ATP13A2, represents a more novel candidate for gene-environment interactions related to neurodegenerative disease. Full loss of function mutations of ATP13A2 in humans are associated with the neurodegenerative conditions Kufor-Rakeb syndrome and neuronal ceroid lipofuscinosis, the former classified as an inherited form of PD. In addition, ATP13A2 polymorphisms have been shown to modify the neurotoxic effect of manganese in an elderly population. In vitro, loss of ATP13A2 is associated with increased manganese and aSyn toxicity and mitochondrial fragmentation. P-type ATPases are a large family of proteins involved in the transport of cations and other substrates across cell membranes through the utilization of energy from ATP hydrolysis [71, 72]. Functionally, they are involved in essential cellular processes including vesicular transport and excitability. P5-type ATPases are only expressed in eukaryotes and are the least characterized of the P-type ATPases. In the human brain, ATP13A2 is highly expressed in neurons in the ventral midbrain but also found in the basal ganglia (globus pallidus and putamen), hippocampus, cortex, and to a lesser extent cerebellum [73]. Given the location in the ventral midbrain, it suggests that reduction of ATP12A2 could create a vulnerability within that region making it susceptible to modest insults. ATP13A2 protein levels were found to be significantly decreased in pure PD and dementia with Lewy bodies, both synucleinopathies [74]. Loss of ATP13A2 expression is associated with mitochondrial fragmentation and dysfunction [75, 76]. The newly identified PD-related genes HTRA2, PLA2G6, CHCHD2, and VPS13C are also associated with mitochondrial function and it will be important to determine the effect of pesticides and metals in models as they are developed [77,78,79,80].
Oxidative Stress
In addition to mitochondrial dysfunction, mutations in aSyn, LRRK2, and ATP13A2 also increase oxidative stress [81,82,83]. Further, deletion or point mutations in DJ-1 have been linked to autosomal recessive early onset parkinsonism [84]. DJ-1 (PARK7) is a 189 amino acid multifunctional protein, initially identified as an oncogene and then implicated in the cellular response to oxidative stress [85, 86]. Downregulation or knockout of DJ-1 in vitro increases the sensitivity to oxidative stress and proteasome inhibition [87]. DJ-1 also has a redox-dependent chaperone function and inhibits the aggregation of aSyn [88]. However, DJ-1 mutations are not that frequent and DJ-1 is only rarely detected in Lewy bodies [89].
Impaired Protein Degradation
Mutations in aSyn involved in inherited forms of PD have been shown to interfere with the autophagy lysosomal degradation pathway in vivo [90]. Since then, many of the PD loci identified are shown to alter aSyn accumulation and implicate mishandling of aSyn as an important contributor to PD in general. LRRK2 has been shown to impair chaperone-mediated autophagy and interfere with aSyn degradation [91]. In addition, aSyn also interacts with metals as it has multiple divalent binding sites [92]. Recent in vitro studies suggest that aSyn may be involved in the regulation of neuronal manganese and actually be neuroprotective against acute manganese exposure [93, 94]. However, chronic manganese exposure has been shown to promote aSyn aggregation and toxicity in dopaminergic cells [94].
Parkin (PARK 2) is a 465 amino acid protein that acts functionally as an E3 ligase and is involved in the ubiquitination of proteins for degredation by the proteasome [95]. It is thought that mutations causing a loss of parkin function can lead to the abnormal accumulation of parkin substrates including glycosolated α-synuclein, synphilin-1, Pael-R, and CDCrel-1 [95]. Knocking out parkin function in mice has been accomplished by deletion of exon 3, exon 7, or exon 2 in the parkin gene.
ATP13A2 also appears to interact with the presynaptic protein aSyn. The role of ATP13A2 in aSyn maintenance is unclear but ATP13A2 is located on lysosomes and lack of function in fibroblasts and dopamine neurons leads to multiple lysosomal defects [96,97,98]. It is also suggested to be involved in the exosomal externalization of aSyn [99] In vivo, loss of ATP13A2 results in age-related sensorimotor deficits, enhanced accumulation of lipofuscin, gliosis, and abnormal accumulation of aSyn [100, 101]. Several of the newly identified PD-associated genes are also linked to protein handling and degradation including PLA2G6, FBXO7, VPS35, DNAJC6, and SYNJ1. It will be interesting to determine the effect of different environmental exposures on these more recent PD-associated mutations. It is also important to note that there are multiple gene-gene interactions that have been discovered such as PINK1/Parkin pathway, suggesting when combined with an environmental toxicant could further contribute to PD risk. An even more recent example is a LRRK2-ATP13A2 interaction adding yet another layer of complexity to gene-environment interactions in PD [102].
Conclusions
Environmental contributions to PD have been recognized for decades; however, not everyone exposed to environmental stressors develops PD. This indicates that gene-environment interactions are likely the most significant contributor to the majority of PD cases. Since the discovery in 1997 of the first inherited form of PD, genetics has had a dominant presence in the PD field. The identification of over 20 genetic loci associated with PD has led to a greater understanding of the mechanisms driving neurodegeneration and substantial proliferation of novel models of PD. Given these genetic advancements, it is essential that environmental factors continue to be included in preclinical and translational studies in order to be successful in developing therapeutics that can modify disease progression.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance
de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54(11 Suppl 5):S21–3.
Goldman JG, Postuma R. Premotor and nonmotor features of Parkinson’s disease. Curr Opin Neurol. 2014;27(4):434–41.
Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12(6):359–66.
Rybicki BA, Johnson CC, Uman J, Gorell JM. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov Disord. 1993;8(1):87–92.
Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119(6):866–72.
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–12.
Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 2016;73(10):1217–24.
Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14(2):223–36.
Smyth LT, Ruhf RC, Whitman NE, Dugan T. Clinical manganism and exposure to manganese in the production and processing of ferromanganese alloy. J Occup Med. 1973;15(2):101–9.
Dexter DT, Wells FR, Agid F, Agid Y, Lees AJ, Jenner P, et al. Increased nigral iron content in postmortem parkinsonian brain. Lancet. 1987;2(8569):1219–20.
Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology. 1985;35(7):949–56.
Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, et al. Parkinson disease in twins: an etiologic study. JAMA. 1999;281(4):341–6.
Roth J, Ponzoni S, Aschner M. Manganese homeostasis and transport. Met Ions Life Sci. 2013;12:169–201.
Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M. Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int J Environ Res Public Health. 2015;12(7):7519–40.
Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74(3):199–205.
Gorell JM, Ordidge RJ, Brown GG, Deniau JC, Buderer NM, Helpern JA. Increased iron-related MRI contrast in the substantia nigra in Parkinson’s disease. Neurology. 1995;45(6):1138–43.
Castellani RJ, Siedlak SL, Perry G, Smith MA. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000;100(2):111–4.
Double KL, Gerlach M, Schünemann V, Trautwein AX, Zecca L, Gallorini M, et al. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem Pharmacol. 2003;66(3):489–94.
Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, et al. Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology. 1997;48(6):1583–8.
Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42(7):1328–35.
Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci. 2005;88(1):193–201.
Ramachandiran S, Hansen JM, Jones DP, Richardson JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicol Sci. 2007;95(1):163–71.
Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69(3):1322–5.
Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82(7):2173–7.
McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, et al. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10(2):119–27.
McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93(4):1030–7.
Talpade DJ, Greene JG, Higgins Jr DS, Greenamyre JT. In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J Neurochem. 2000;75(6):2611–21.
• Nuber S, Tadros D, Fields J, Overk CR, Ettle B, Kosberg K, et al. Environmental neurotoxic challenge of conditional alpha-synuclein transgenic mice predicts a dopaminergic olfactory-striatal interplay in early PD. Acta Neuropathol. 2014;127(4):477–94. This study shows that olfactory dopamine neurons display an enhanced sensitivity to the herbicide paraquat that is dependent on alpha-synuclein. This work has important relevance for early identification of PD and highlights a novel aspect of gene-environment interactions in PD
Richter F, Gabby L, McDowell KA, Mulligan CK, De La Rosa K, Sioshansi PC, et al. Effects of decreased dopamine transporter levels on nigrostriatal neurons and paraquat/maneb toxicity in mice. Neurobiol Aging. 2016;51:54–66.
Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, et al. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest. 2015;125(7):2721–35.
Karuppagounder SS, Xiong Y, Lee Y, Lawless MC, Kim D, Nordquist E, et al. LRRK2 G2019S transgenic mice display increased susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated neurotoxicity. J Chem Neuroanat. 2016;76(Pt B):90–7.
Fleming L, Mann JB, Bean J, Briggle T, Sanchez-Ramos JR. Parkinson’s disease and brain levels of organochlorine pesticides. Ann Neurol. 1994;36(1):100–3.
Akhmedova SN, Yakimovsky AK, Schwartz EI. Paraoxonase 1 Met—Leu 54 polymorphism is associated with Parkinson’s disease. J Neurol Sci. 2001;184(2):179–82.
Pittman JT, Dodd CA, Klein BG. Immunohistochemical changes in the mouse striatum induced by the pyrethroid insecticide permethrin. Int J Toxicol. 2003;22(5):359–70.
Elwan MA, Richardson JR, Guillot TS, Caudle WM, Miller GW. Pyrethroid pesticide-induced alterations in dopamine transporter function. Toxicol Appl Pharmacol. 2006;211(3):188–97.
Corrigan FM, Wienburg CL, Shore RF, Daniel SE, Mann D. Organochlorine insecticides in substantia nigra in Parkinson’s disease. J Toxicol Environ Health A. 2000;59(4):229–34.
Richardson JR, Shalat SL, Buckley B, Winnik B, O’Suilleabhain P, Diaz-Arrastia R, et al. Elevated serum pesticide levels and risk of Parkinson disease. Arch Neurol. 2009;66(7):870–5.
Richardson JR, Roy A, Shalat SL, Buckley B, Winnik B, Gearing M, et al. β-Hexachlorocyclohexane levels in serum and risk of Parkinson’s disease. Neurotoxicology. 2011;32(5):640–5.
Lee PC, Rhodes SL, Sinsheimer JS, Bronstein J, Ritz B. Functional paraoxonase 1 variants modify the risk of Parkinson’s disease due to organophosphate exposure. Environ Int. 2013;56:42–7.
Narayan S, Liew Z, Paul K, Lee PC, Sinsheimer JS, Bronstein JM, et al. Household organophosphorus pesticide use and Parkinson’s disease. Int J Epidemiol. 2013;42(5):1476–85.
• Xiong J, Zhang X, Huang J, Chen C, Chen Z, Liu L, et al. Fenpropathrin, a widely used pesticide, causes dopaminergic degeneration. Mol Neurobiol. 2016;53(2):995–1008. This study shows that a common pyrethroid pesticide, fenpropathrin, can cause alterations in the nigrostriatal dopaminergic pathway in rodents including decreased striatal VMAT2 and dopamine linking this pesticide to PD
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22(20):8797–807.
Yavich L, Tanila H, Vepsäläinen S, Jäkälä P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24(49):11165–70.
Cookson MR. Alpha-synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9.
Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106-107:17–32.
Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, et al. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain. 2013;136(Pt 7):2130–46.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–9.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Vila M, Vukosavic S, Jackson-Lewis V, Neystat M, Jakowec M, Przedborski S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J Neurochem. 2000;74(2):721–9.
Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277(3):1641–4.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–6.
Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186(2):158–72.
Nieto M, Gil-Bea FJ, Dalfó E, Cuadrado M, Cabodevilla F, Sánchez B, et al. Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2006;27(6):848–56.
Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61(12):991–1001.
Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170(2):658–66.
Gandhi PN, Chen SG, Wilson-Delfosse AL. Leucine-rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson’s disease. J Neurosci Res. 2009;87(6):1283–95.
Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365(9457):415–6.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–90.
Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32(5):1602–11.
Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci. 2011;31(6):2225–37.
Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75(22):2017–20.
Houlden H, Singleton AB. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012;124(3):325–38.
Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, et al. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004;56(3):336–41.
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–31.
Bonifati V, Rohé CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65(1):87–95.
Guo JF, Wang L, He D, Yang QH, Duan ZX, Zhang XW, et al. Clinical features and [11C]-CFT PET analysis of PARK2, PARK6, PARK7-linked autosomal recessive early onset parkinsonism. Neurol Sci. 2011;32(1):35–40.
Villeneuve LM, Purnell PR, Boska MD, Fox HS. Early expression of Parkinson’s disease-related mitochondrial abnormalities in PINK1 knockout rats. Mol Neurobiol. 2016;53(1):171–86.
Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis. 2014;70:190–203.
Schultheis PJ, Hagen TT, O’Toole KK, Tachibana A, Burke CR, McGill DL, et al. Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochem Biophys Res Commun. 2004;323(3):731–8.
van Veen S, Sørensen DM, Holemans T, Holen HW, Palmgren MG, Vangheluwe P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front Mol Neurosci. 2014;7:48.
Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38(10):1184–91.
Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. 2013;1:11.
Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33(8):1843. e1-7
Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45(3):962–72.
Casadei N, Sood P, Ulrich T, Fallier-Becker P, Kieper N, Helling S, et al. Mitochondrial defects and neurodegeneration in mice overexpressing wild-type or G399S mutant HtrA2. Hum Mol Genet. 2016;25(3):459–71.
Sumi-Akamaru H, Beck G, Shinzawa K, Kato S, Riku Y, Yoshida M, et al. High expression of α-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol Commun. 2016;4:27.
Ogaki K, Koga S, Heckman MG, Fiesel FC, Ando M, Labbé C, et al. Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology. 2015;85(23):2016–25.
Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. 2016;98(3):500–13.
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157(2):401–10.
Yang D, Li T, Liu Z, Arbez N, Yan J, Moran TH, et al. LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: suppression by curcumin. Neurobiol Dis. 2012;47(3):385–92.
Covy JP, Waxman EA, Giasson BI. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J Neurosci Res. 2012;90(12):2306–16.83.
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–9.
Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic Res. 2001;35(3):301–10.
Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res. 2001;35(6):885–93.
Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun. 2003;312(4):1342–8.
Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004;2(11):e362.
Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain. 2004;127(Pt 2):420–30.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Kuo SJ,SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406.
Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem. 2001;276(47):44284–96.
Dučić T, Carboni E, Lai B, Chen S, Michalke B, Lázaro DF, et al. Alpha-synuclein regulates neuronal levels of manganese and calcium. ACS Chem Neurosci. 2015;6(10):1769–79.
Harischandra DS, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. α-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson’s disease. Toxicol Sci. 2015;143(2):454–68.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302–5.
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109(24):9611–6.
Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, et al. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet. 2012;21(8):1725–43.
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240–6.
Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23(11):2816–33.
Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22(10):2067–82.
Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, et al. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35(14):5724–42.
Lubbe SJ, Escott-Price V, Gibbs JR, Nalls MA, Bras J, Price TR, et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet. 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sheila M. Fleming declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Additional information
This article is part of the Topical Collection on Mechanisms of Toxicity
Rights and permissions
About this article

Cite this article
Fleming, S.M. Mechanisms of Gene-Environment Interactions in Parkinson’s Disease. Curr Envir Health Rpt 4, 192–199 (2017). https://doi.org/10.1007/s40572-017-0143-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-017-0143-2