
Abstract
The review addresses issues pertinent to Mn accumulation and its mechanisms of transport, its neurotoxicity and mechanisms of neurodegeneration. The role of mitochondria and glia in this process is emphasized. We also discuss gene x environment interactions, focusing on the interplay between genes linked to Parkinson’s disease (PD) and sensitivity to Mn.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- divalent metal transporter 1 (DMT1)
- manganese
- mitochondria
- parkin
- transport
- Please cite as: Met. Ions Life Sci. 12 (2013) 169–201
1 Introduction
Manganese is an essential trace metal in all forms of life. In mammalians it is required for normal amino acid, lipid, protein, and carbohydrate metabolism. Mn-dependent enzymes include oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases, to name a few. Mn metalloenzymes include arginase, glutamine synthetase, phosphoenolpyruvate decarboxylase, and Mn superoxide dismutase (MnSOD). Mn plays a critical role in multiple bodily functions including immunity, regulation of blood sugars and cellular energy, blood clotting, reproduction, digestion, and bone growth. Mn-containing polypeptides include arginase, the diphtheria toxin, and MnSOD. The latter is found in eukaryotic as well as bacterial organisms and is likely one of the most ancient enzymes, given that almost all organisms that live in an oxygen environment utilize the enzyme to dismutate superoxide. Several bacteria are an exception; nevertheless, they also use Mn via a non-enzymatic mechanism, involving Mn2+ ions complexed with polyphosphate.
Mn is also essential in plants as it is involved in photosynthesis. Oxygen-evolving complexes (OEC) contained within the thylakoid membranes of chloroplasts are responsible for the terminal photooxidation of water during the light reactions of photosynthesis. The chloroplasts contain a metalloenzyme core with 4 Mn atoms.
Surprisingly, no formal Recommended Dietary Allowance (RDA) exists for Mn, but the U.S. National Research Council (NRC) has established an estimated safe and adequate dietary intake (ESADDI) of 2–5 mg/day for adults [1]. Adequate intakes for newborns (<6 months of age) are 3 μg/day and 600 μg/day for infants at 12 months of age to (NAS, 2001) [2]. Children between 1–3 and 4–8 years of age have adequate daily Mn intakes of 1.2 and 1.5 mg/day, respectively.
The most important source of Mn is diet, with most daily intakes approximating 5 mg Mn/kg. Grain, rice, and nuts are highly enriched with Mn along with tea. Water concentrations of Mn typically range from 1 to 100 μg/L. Only a small fraction (1–5%) of ingested Mn is absorbed in normal conditions, the Mn arriving at the liver in the portal circulation is protein-bound. Within the plasma approximately 80% of Mn in the 2+ oxidation state is bound to globulin and albumin, and a small fraction (<1%) of trivalent (3+) Mn is bound to the iron-carrying protein, transferrin (Tf) [3,4]. Mn deficiency in humans is rare.
Generally, irrespective of intake route (oral, inhalation, dermal), animals maintain stable tissue concentrations given tight homeostatic control of Mn absorption and excretion [5–10]. Yet exposure to high Mn levels may lead to increased body-burden of the metal, resulting in adverse neurological, reproductive, and respiratory effects. The predominant site of Mn-induced damage is the brain, with symptoms commonly manifesting in motor dysfunction and psychological disturbances.
2 Manganese Transport
Since Mn is an essential cofactor for a diverse assortment of enzymes, its cellular concentrations are stringently managed by a variety of processes controlling cellular uptake, retention, and excretion. In general, overall systemic homeostatic levels of Mn are maintained, in vivo, via its rate of transport across enterocytes lining the intestinal wall and by its efficient removal within the liver [11]. Under typical nutritional consumption, the transport processes within the enterocytes lining the intestinal wall and the subsequent down-stream trafficking systems are efficiently balanced and work in harmony to preserve requisite supplies of Mn for the different cells and organelles within the body. This normally proficient system of checks and balances that control Mn levels in vivo, however, appears to fail under conditions of chronic exposure to high atmospheric levels (in various oxidation states; Mn is absorbed though predominantly in the 2+ and 3+ oxidation states) of the metal and thus, vital adjustments in the complex homeostatic processes are no longer adequate to preserve the required status quo. Failure of these systems to adjust to excess exposures infers that the rate limiting step governing systemic levels and ultimately Mn toxicity encompasses the operative biochemical processes necessary for its uptake and elimination within the various compartments of the body.
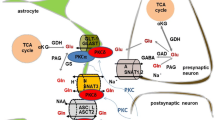
Like other required divalent metals, there are redundant transport systems for cellular uptake of Mn. The functioning system is dependent on both the ionic species present in biological fluids and by the specific transport proteins present within any given cell. Many of these redundant systems are also capable of transporting other metals suggesting that its role in managing the uptake of Mn may not be their primary responsibility. Figure 1 describes the different transport systems responsible for transport of Mn into cells. These include uptake by (i) the voltage-regulated and the ionotropic glutamate receptor Ca2+ channels [12,13], (ii) the transient receptor potential cation channel, subfamily M, member 7 (TRPM7) [14,15], (iii) store-operated Ca2+ channel [16] and a Tf-dependent and independent process via divalent metal transporter 1 (DMT1) [17–19]. Also indicated are members of the Slc39 gene family, ZIP 8 and 14, which have recently been identified as being involved in the transport of Mn [20–23]. Of all these proteins, DMT1 is generally considered to be the predominant transporter for Mn though this does not exclude the possibility that under varying physiological or pathological conditions and in any given cell population that the other transport processes may also contribute significantly to uptake.

Transport mechanisms responsible for the uptake of Mn. Tf – transferrin; TfR transferrin receptor; DMT1- divalent metal transporter 1; VR – voltage-gated Ca2+ channel; SOC – store-operated Ca2+ channel; Glu Rec – glutamic acid ionotropic receptor.
2.1 Divalent Metal Transporter 1
As noted above, under normal conditions, transport of Mn into most cells is generally assumed to preferentially be dependent on DMT1. DMT1 has a broad substrate specificity for a variety of divalent cations including Fe2+, Mn2+, Cd2+, Ni2+, Co2+, and Pb2+ [24–26]. The one exception to the rule is for Cu which may involve transport of the monovalent species [27]. Although DMT1 has a relatively broad substrate specificity, transport of iron is generally considered to be its principal function although, the affinity of Mn for DMT1 is similar to that for iron [17,28]. Smf1p (a homolog of DMT1) has been shown also in yeast to be a high-affinity transporter for Mn. Notably, a range of divalent metals can act as substrates for this transporter and when overexpressed in oocytes, it can increase intracellular concentrations, not only of Mn, but also of copper, cadmium, and iron.
As noted above (Figure 1), two distinct but related mechanisms are responsible for the transport of Mn2+ and Fe2+ by DMT1: a Tf-dependent and a Tf-independent pathway. In the intestines (see Figure 2), both Mn2+ and Fe2+ preferentially utilize the Tf-independent pathway, which is responsible for the direct absorption of the divalent species of both metals on the apical side of the enterocyte [29]. DMT1 is highest in the duodenum and decreases in the subsequent segments of the intestine [30] implying that transport preferentially occurs in the upper intestines. Although Mn2+ has a high affinity for DMT1, equivalent to that of Fe2+, total uptake within the gastrointestinal tract is at best 5% of that present in ingested foods [31–34]. Once inside the enterocytes lining of the microvilli, Mn is transferred to the basolateral surface by a process which has not been adequately defined. Export of Fe2+ on the basal lateral side has been shown to require ferroportin (Fpn), which has recently been suggested to also play a role in the export of Mn [35–37] (see below). Levels of Fpn on the basolateral surface are controlled by the iron-regulated protein, hepcidin, produced in the liver [38,39]. Hepcidin causes internalization of Fpn, which subsequently undergoes ubiquitination before being degraded within the 26S proteasomes.

Mechanism for the transport of Mn across the intestine.
Before or during exit from the enterocyte, Fe2+ and presumably Mn2+ are oxidized to the trivalent state prior to entering the blood stream. Although this has never been directly demonstrated for Mn, it likely occurs, as a major portion of Mn entering the blood stream is tightly bound to Tf [33,40]. In the case of iron, this is catalyzed by either of the copper-containing proteins, hephaestin or ceruloplasmin [41,42], but whether either of the copper-containing ferrooxidases is also responsible for oxidation of Mn is not known, although, Cu does not have the oxidative potential to oxidize Mn from 2+ to 3+. Consistent with this is the recent study [43] suggesting that ceruloplasmin may not function in the loading of Mn onto plasma proteins or in the partitioning of Mn between the plasma and cellular fractions of blood though it does participate in tissue disposition and toxicity of Mn. Nevertheless, a major portion of Mn released from the enterocyte which enters the blood stream is bound tightly to Tf as the trivalent species [31,33,40,44]. Under normal conditions, approximately 30% of the available Tf binding sites in blood are occupied by Fe3+ leaving sufficient sites accessible for binding of Mn3+ [45]. Because Mn3+ forms a stable complex with Tf, hepatic removal is relatively slow and comparable to that for Tf-Fe3+ complex. The availability of these binding sites for Mn, however, may be compromised in several disease states such as hemochromatosis where Tf is saturated with iron. Because Fe, under these abnormal conditions, is in excess and is likely to have the higher affinity for Tf, Mn transport may be limited or carried out by other plasma components. In blood, the divalent species of Mn is preferentially bound to α2-macroglobulin though, because of the significantly greater abundance of albumin in serum, a sizable portion may also be bound to this latter protein [31,33,40,44]. The ligand interaction between Mn2+ and either serum proteins is relatively weak which likely accounts for the rapid hepatic elimination from blood.
In the absence of Fe, the binding sites of Tf can accommodate a number of other metals including gallium, copper, chromium, cobalt, vanadium, aluminum, terbium, and plutonium, raising the possibility that Tf functions in vivo as a transport agent for many of these metals. Fe is taken up by cells after Tf binds to a specific cell surface receptor and the Tf receptor complex is internalized. At its usual concentration in plasma, 3 mg/mL, and with 2 metal ion-binding sites per molecule (Mr 77000) of which only 30% are occupied by Fe3+, transferrin has available 50 μ mole of unoccupied Mn3+ binding sites per liter.
The direct Tf-independent pathway utilizing DMT1 is also likely to be responsible for transport of Mn (in the 2+ oxidation state) into cells within the central nervous system (CNS) despite the fact that the interstitial fluid within the brain contains Tf. This is anticipated based on the fact that Fe levels in the interstitial fluid greatly exceed that of Tf resulting in saturation of the available binding sites causing the exclusion of Mn binding [46]. The actual ionic species of Mn in the extracellular fluid in the CNS and whether some of the Mn is capable of competing with Fe for binding to Tf is not known. Mn2+ at physiological pH is actually quite stable and because the pO2 is low in the brain, little Mn3+ probably exists in the CNS. In addition, it is unknown whether Mn actually undergoes oxidation to the trivalent species upon its exit from endothelial cells comprising the blood-brain barrier (BBB). Based on a recent study by Hernández et al. [47], both Mn2+ or Mn3+ are capable of binding to citrate which is in excess in the brain interstitial fluid (225 to 573 μM) although neuronal toxicity of the Mn/citrate complex is approximately the same for all ionic species. Regardless, transport of the trivalent species of Mn, if it exists, must first be reduced to the divalent state prior to its uptake by either the Tf-dependent or independent pathway or for that matter the ZIP proteins (see below) as well. The role of citrate in this reductive process may be significant in that citrate has been reported to facilitate reduction of Fe3+-bound to Tf as citrate has the ability to displace iron from holo-Tf when present in excess [48]. Reduction of Fe3+- bound citrate, in fact, has been shown to be the rate limiting step in the uptake of Fe in astrocytes [49]. Whether a similar process occurs for Mn3+ in the CNS, however, is not known. Assuming Mn is similar to Fe, it may also bind to ascorbate in the interstitial fluid of brain [50].
The non-Tf-dependent pathway is also most likely to be accountable for the direct entry of Mn into the CNS via retrograde transport within axonal projections impinging on the nasal cavity leading from the neurons within the olfactory bulb [51–56]. Recent studies in the laboratory of Wessling-Resnick [57] have demonstrated that uptake of Mn into the presynaptic endings of these neurons is a DMT1-dependent process. The overall contribution of this pathway to Mn accumulation in the CNS in humans is not known but is likely contingent on its composition within the inspired air with the soluble forms being more efficiently taken up via this process [58]. There is evidence that extremely small ultrafine particles can also be taken up by the nerve endings and subsequently delivered to the CNS via a similar retrograde transport process [59].
For the Tf-dependent pathway, both trafficking and cellular transport of Mn3+ function very much like that for Fe3+, indicative of the fact that a major portion of extracellular Mn3+ utilizes the same biochemical components as that for Fe3+ (Figure 1). The Tf-dependent pathway has been suggested to be the major mechanism within the choroid plexus and blood-brain barrier for transport of Mn into the brain [36,60]. Initially, the Tf-Mn complex attaches to the Tf receptors (TfR) on the cell surface, which is subsequently internalized within endosomal vesicles. Precisely how Mn is delivered to the endosomes is not understood. Furthermore, the specificity to Mn versus Fe has also yet to be delineated in mammalian cells. Next, these vesicles undergo acidification within the endosome via a hydrogen ion ATPase pump causing release of the metal from the Tf/TfR complex. In the case of iron, the released metal is reduced by either duodenal cytochrome b (Dcytb) [61] or a family of ferroreductases referred to as steap2-4 [62,63]. Whether these enzymes are also responsible for the reduction of Mn has not been investigated, although reduction must occur to enable transport via DMT1 and by the fact that the divalent species of Mn2+ is essentially the only form found in cells [64]. As noted above, it is questionable as to whether the Mn3+-Tf complex actually is present in the interstitial fluid with the CNS.
Four different mammalian isoforms of DMT1 have been identified resulting from two autonomous start sites and alternate splicing of a single gene transcript. All are putative 12 membrane-spanning domain proteins [25,65,66] but differ both in their N- and carboxy-terminal amino acid sequences. The gene for DMT1 has two discrete promoter regions (1A and 1B) which independently regulate transcription at the two start sites. mRNA from two of the four species contain an iron response element (IRE) motif in the 3’ UTR and are referred to as the +IRE species whereas those forms lacking the IRE are categorized as the -IRE species. Transcriptionally regulated splice variants, exon 1A and 1B, are present on the proximal N-terminal end for both the ±IRE forms of DMT1 mRNA. Exon 1A extends the N-terminal polypeptide 29 residues in human (31 in rats and 30 in mice) [66,67]. All four species of DMT1 distal from exon 1A share 543 residues in common but differ structurally in the last 18 (+IRE) or 25 (-IRE) carboxy terminal residues. The presence of the IRE in the message provides a site for binding of the iron response proteins, IRP1 and/or 2 [68,69], which potentially stabilize the mRNA when iron levels are low, leading to selective increases in expression of the + IRE isoforms of the transporter. This is consistent with numerous studies demonstrating that Mn disposition is critically dependent on iron levels in vivo [70–73] and with the +IRE isoform of the transporter being present in the apical surface of enterocytes [73]. Mn has also been reported to affect the binding of IRPs to IRE suggesting stabilization of the mRNA to DMT1 [60,74]. In addition to DMT1, the TfR and Fpn similarly contain an IRE in their 3’-UTR and like DMT1 are also regulated by iron via the IRPs [68,75].
The need for four distinct isoforms of DMT1 is not readily apparent especially since all display similar kinetic characteristics in regard to transport of both Mn and Fe [17,28,76]. In all likelihood, the physiological actions of the four isoforms of DMT1 are likely to have distinct roles dependent on the specific requirements within cells to maintain normal homeostatic levels of Fe particularly during fluctuating stress-related conditions. Because Fe is likely to be the primary operational metal transported by DMT1, alterations in expression of the different isoforms caused by changes in Fe content or other stress inducing situations will result in a corresponding adjustment in Mn transport irrespective of the actual in vivo requirements for Mn and thus, potentially stimulating development of Mn toxicity. This is consistent with the observation that DMT1 is elevated in the basal ganglia of iron-deficient rats and that Mn is selectively increased in the globus pallidus in iron-deficient animals exposed to Mn [77,78]. In addition to regulation of the +IRE species of DMT1 by IRPs, the N-terminal isoforms of DMT1 are also independently regulated at the level of transcription by environmental conditions which manifest in various stress-related events and inflammatory processes within the cell [79–83]. Transcription of the 1B isoforms of the transporter is upregulated by both nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and NF-Y (CCAAT promoter) and indirectly by nitric oxide suppression of NF-κB activity. In addition, both the 1A and 1B isoforms have also been reported to be induced by hypoxia implying the presence of an HRE within both promoter sites. Thus, expression of the different isoforms of DMT1 can be independently regulated within cells at both the level of transcription as well as translation.
In yeast and bacteria, Mn is transported from the endosomes to the Golgi apparatus and the mitochondria. Again, precise information on the process in mammalian cells is lacking, but it may involve fusion of Smf2p-containing vesicles with these organelles, as has been previously suggested for iron trafficking to mitochondria. It has also been suggested that Mn chaperones may transport the metals in the cytoplasm. The Tf transport mechanism for Fe is known to deliver both Fe3+ and Mn2+ to the mitochondria for incorporation into hemes [220–224].
2.2 ZIP-Dependent Uptake of Manganese
A number of studies over the past several years have demonstrated that two members of the solute-carrier-39 (SLC39) metal-transporter family, ZIP8 and ZIP14, are also capable of transporting Mn [21,23,84,85]. Unlike DMT1, both ZIP proteins are divalent cation/HCO\( {}_{3}^{-}\)symporters with the HCO\( {}_{3}^{-}\)gradient across the plasma membrane acting as the driving force for divalent metal transport. There is mounting evidence that these membrane proteins may play a significant role in Mn transport but their overall contribution relative to that of DMT1 has not been adequately assessed. Since the interstitial fluid in brain has a pH of approximately 7.3 [86], which is close to the optimum for both ZIP transporters, it is feasible their contribution to Mn uptake may be relatively more significant than anticipated in comparison to DMT1, which maximally functions at around pH 6.0. The significance of this pathway is further accentuated by the fact that Mn2+, which is present in the interstitial fluid of brain, is probably not bound to Tf and thus, is available for transport by these ZIP proteins.
For the ZIP8 transporter, Cd2+ displays the highest affinity, exhibiting a K m of 0.62 μM, though Mn2+ has been posited to be most efficiently transported, with a K m for uptake of 2.2 μM, implying that Mn is probably the physiological substrate for ZIP8 [21]. This value is comparable to the K m for Mn binding to DMT1 suggesting that it has the potential to significantly impact on Mn accumulation in vivo. Unfortunately, there have been few studies that have attempted to quantitate expression of this transporter relative to that of DMT1 and therefore the overall contribution of ZIP8 to Mn uptake in comparison to DMT1 or the other transport systems needs to be examined. One study which examined expression of ZIP8 to that of DMT1 by QPCR in the inner ear found that DMT1 levels were slightly higher than that of ZIP8 [87].
In contrast to ZIP8, its counterpart, ZIP14, has been studied more extensively in regard to both Mn and Fe transport. Of the 14-member ZIP protein family, ZIP14 is most homologous to ZIP8 [23]. Like DMT1, ZIP14 has a relatively broad substrate specificity capable of transporting a wide variety of divalent metals including, Mn, Fe, Co, Ni, Zn, Cd, Ca, and Pb [88]. Two forms of ZIP14 have been identified, A and B, having affinities for Mn2+ of 4.4 and 18.2 μM, respectively, with both being driven by a HCO\( {}_{3}^{-}\)gradient across the membrane [23]. Highest levels of ZIP14A are observed in the lung, testis, and kidney whereas the B isoform of the transporter is relatively evenly distributed. High expression of ZIP14 has also been observed in the intestines where it has the potential to play a significant role in the accumulation of Mn, although direct quantification relative to DMT1 has not been established. Several studies have indicated that like DMT1, expression of ZIP14 is capable of transporting Fe as well as being regulated by Fe levels with high iron decreasing expression whereas low Fe increasing protein levels [72,89]. Transport of Mn by ZIP14 mirrors that of Fe as both are Ca2+-dependent [88].
Expression of the hemochromatosis protein, HFE, promotes a decrease in both ZIP14-dependent Tf bound and non-Tf-bound iron uptake as well as protein levels of ZIP14 with no apparent change in the mRNA level, thus suggesting that HFE decreases the stability of ZIP14 [89]. Since Tf is saturated with Fe3+ in the interstitial fluid in brain, appreciable amounts of both non-Tf bound Fe2+ and Mn2+ exist which may be transported by ZIP14 [90]. Like DMT1, ZIP14 also resides on the plasma membrane and within endosomal vesicles. Although greatest activity is observed at pH 7.5, ZIP14 can still function, although suboptimally, at pH 6.5, a value associated with release of divalent metals within endosomal vesicles. Nevertheless, transport of divalent metals by DMT1, which is H+-coupled and preferentially stimulated at low pH, may serve as the predominant route of Fe2+ and Mn2+ uptake from late endosomes and lysosomes. Thus, the significance of ZIP14 in maintenance of Mn homeostasis and toxicity remains to be determined.
2.3 Calcium(II) Channel-Dependent Transport of Manganese
In addition to cellular uptake by the different isoforms of DMT1 and the ZIP proteins, there is also evidence for transport of Mn via several divalent metal channels which include, (i) the voltage regulated, (ii) the ionotropic glutamate receptor Ca2+ channels [12,13,91], (iii) TRPM7 [14,15] and (iv) the store operated Ca2+ channel (Figure 1) [16,92]. Evidence for the role of these ion channels in transporting Mn comes from a number of studies many of which primarily utilize Mn as a tool to measure functionality of the channel and therefore, it is not known, under normal resting conditions, what the overall contribution of each is to the total accumulation of Mn relative to the other known transport processes.
The potential significance of the voltage-gated Ca2+ comes from studies demonstrating that depolarization of cell membranes leads to increased uptake of Mn which can be prevented by Ca2+ channel blockers [12,91]. In regard to store-operated Ca2+ channels (SOC), Crossgrove and Yokel [93] have speculated this pathway may be responsible for the transport of Mn across the blood-brain barrier, but direct supportive evidence for this is lacking. Whether this also involves the SOC-dependent interaction of the Ca2+ sensor protein, STIM1, with that of calcium release-activated calcium channel protein 1 (ORAI1) at the plasma membrane is not known though the sarcoplasmic reticulum Ca-ATPase (SERCA) inhibitor, thapsigargin, has been reported to induce Mn2+ uptake which is inhibited by Ca2+ [94]. Demonstration of the direct involvement of TRPM7 in the transport of Mn was shown in studies assessing changes in transport in MCF-7 cells generated by overexpression or down-regulation of the protein [95]. Although Mn was shown to be transported by these channels, their overall contribution to neuronal toxicity within the CNS is unknown.
In contrast to the channels described above, the ionotropic glutamate receptor Ca2+ channel may directly impact on Mn toxicity due to the fact that neurons within the globus pallidus receive glutaminergic input from axonal projections leading from the subthalamic nuclei [96,97]. Several studies have demonstrated that Mn toxicity is attenuated by the glutamate post-synaptic ionotropic receptor inhibitor, MK801 [98,99], implying that glutamate participates in the neurotoxic actions of Mn. Similarly, the antiglutaminergic drug, riluzole, has also been shown to attenuate Mn toxicity in rats [100]. It is important to recognize that the cytotoxic events provoking Mn toxicity, to a large extent, parallel similar pathways for the excitatory neurotransmitter, glutamate, as both involve collapse of mitochondrial function initiated by excess sequestration of calcium [101,102]. The extent to which Mn uptake is increased upon glutamate binding within the γ-aminobutyric acid (GABAergic) neurons of the globus pallidus and the role this potentially plays in potentiating neurotoxicity, however, has not been directly investigated.
2.4 Manganese Efflux
Ferroportin (Fpn) [also referred to as IREG1 (iron-regulated protein 1) or MTP1 (metal tolerance protein 1)] is the cytoplamic Fe exporter. It is expressed in all cells, including neurons playing a key role in maintaining optimal Fe-homeostasis. Mutations in Fpn lead to type VI hemochromatosis, commonly known as Fpn disease, which is predominantly characterized by Fe accumulation in reticuloendothelial macrophages. The protein is transcriptionally, translationally, and posttranslationally regulated. Considering the shared uptake and characteristics of Mn and Fe (see above), it is surprising that only a few studies have attempted to delineate the role of Fpn in Mn efflux. Yin et al. [35] have recently reported that Fpn induction in human embryonic kidney cells (HEK293T) led to a reduction in Mn-induced toxicity, concomitant with decreased intracellular Mn accumulation. Notably, cerebellar and cortical Fpn was also increased in mice in vivo exposed to Mn [35]. More recently, Madejczyk and Ballatori [37] examined the role of Fpn in exporting Mn in Xenopus laevis oocytes demonstrating lower Mn accumulation in oocytes expressing Fpn. Furthermore, the efflux was inhibited by Fe, as well as other divalent metals. Collectively, these studies suggest Mn exposure in addition to promoting Fpn protein expression also reduces intracellular Mn levels and cytotoxicity.
3 Manganese: Toxic Mechanisms
Manganism and Parkinson’s disease (PD) are two distinct neurological entities that impair basal ganglia function; the globus pallidus is predominantly damaged in the former and nigral dopaminergic (DAergic) neurons in the latter. DAergic cells from mesostriatal circuitry are also vulnerable to the toxic effects of Mn, making this metal a potential environmental risk factor in the etiology of PD. Cases of Mn poisoning have been reported in patients with chronic liver failure and long-term parenteral nutrition [103] and in individuals chronically exposed to the Mn-containing fungicide (manganese ethylene-bis-dithiocarbamate) [104] as well as other occupational cohorts (smelters, welders, etc.). Mn-induced disturbances of cellular homeostatic mechanisms include reactive oxidative species (ROS) production [105], impairment of antioxidant cellular defenses [106], mitochondrial damage [107], endoplasmatic reticulum stress [108], DNA damage [109], and inflammatory reactions [110], just to name a few. These are common features in a plethora of neurodegenerative diseases making Mn neurotoxicity studies an important tool in the understanding of the etiology of neurodegenerative processes. In this section, we discuss experimental data highlighting novel approaches to elucidate the vulnerability of DAergic nigral cells to the transition metal, Mn.
The DAergic cells that integrate the basal ganglia circuitry reside in the substantia nigra pars compacta (SNpc), a brain region with high oxidative activity. High content of oxidative enzymes and a high metabolic rate leads these cells to produce large quantities of damaging ROS [111]. Notably, catecholamine catabolism, a process which takes place in these cells, involves hydrogen peroxide production by the monoamine oxidase (MAO) and quinones by autoxidation [112]. In vitro assays have demonstrated that Mn increases dopamine (DA) and L-dopa autoxidation, leading to ROS and quinone production (see below); the Mn valence represents an important factor in the metal’s DA oxidizing capacity, with manganic ion (Mn3+) being more efficient than Mn2+ in potentiating DA autoxidation [113,114]. Nevertheless, whether Mn3+ oxidizes DA has been recently questioned, given the inability to locate Mn3+ within various cell types [64]. A schematic representation of Mn-induced DA autoxidation is shown in Figure 3.

Schematic representation of some cellular mechanisms vulnerable to the toxic effects of Mn. Pathway 1 represents the mitochondrial toxic action with decreased ATP production and glutathione levels (GSH) leading to cytochrome c release and caspase activation and apoptosis. Pathway 2 represents endoplasmic reticulum stress with increased expression of glucose-regulated protein (GRP78) and pro-apoptotic protein C/EBP homologous protein (CHOP). Pathway 3 represents Mn clastogenic effect. Pathway 4 represents fibrillation of α-synuclein protein leading to the formation of cytoplasmatic inclusions. Pathway 5 represents microglial activation with the release of pro inflammatory mediators such as interleukin-6 (IL6), interleukin-1β (IL1β) and tumor necrosis factor α (TNF α).
3.1 Mitochondria, Dopamine, and Manganese-Induced Neurotoxicity
The mitochondria are critical organelles in mediating Mn-induced neurotoxicity [64]. Mn preferentially accumulates within these organelles [115] and mitochondrial superoxide dismutase 2 (SOD2) requires Mn as cofactor (there are other SODs in mitochondria, such as CuZn SOD1). Mn mitochondrial overload is toxic, secondary to its ability to impair ATP production and antioxidant defense mechanisms. Mitochondrial Mn2+ neurotoxicity could be related to inhibition of Ca2+ activation of ATP production [64]. Mn2+ attenuates brain mitochondrial ATP formation in two independent inhibitory sites; the primary site, complex II or fumarase, where succinate is the substrate, and the secondary site, the glutamate/aspartate exchanger, where glutamate plus malate are the substrates [107].
The ability of Mn to generate excess ROS has been observed both in cell culture and in vivo experiments. Cell mortality was increased when Mn was added to human fibroblast cultures, an effect that was potentiated in the presence of DA and antagonized by catalase and MnSOD2 [105]. Catechol isoquinolines, 1-methyl-6, 7-dihydroxy-1, 2, 3, 4-tetrahydroquinoline (salsolinol), and N-methyl-salsolinol induced apoptosis and increased levels of malondialdehyde (MDA) in a concentration-dependent manner in PC12 cells exposed to Mn [116]. Worley et al. [117] demonstrated in catecholaminergic CATH.a cells that Mn alone failed to increase ROS production; however, when followed by hydrogen peroxide exposure, ROS levels significantly increased, indicating that in addition to intracellular Mn concentrations, exposure duration, oxidative challenges post Mn exposure and cellular redox capacity represent important factors in mediating Mn-induced stress related effects. Mn-induced cytotoxicity in DAergic cells was also dependent on glutathione (GSH) levels in CATH.a cells [118]. This raises the possibility that susceptibility to Mn-induced ROS-mediated toxicity may be dependent upon the redox status of the cell, with those expressing high GSH levels perhaps being more resistant versus those with a low GSH complement.
In vivo, oral administration of Mn in the drinking water for 30 days decreased rat striatal DA content and increased striatal MAO activity, suggesting a potential increase in ROS production [119]. The ability of Mn to autoxidize DA was addressed by Sloot et al. [120] by demonstrating that intrastriatal Mn2+ injection resulted in depletion of striatal DA, which preceded ROS formation. In addition, reserpine pre-treatment failed to alter both DA depletion and ROS formation. Accordingly, reduced redox potential and impaired cellular antioxidant defense mechanisms likely play a key role in cellular susceptibility to develop Mn toxicity. Decreased levels of mitochondrial glutathione-peroxidase activity, catalase, and GSH were observed in striatum of Mn-treated rats, consistent with Mn-induced impairment of intracellular ROS defense mechanisms [106]. As shown in Figure 3, pathway 1 represents schematically the main effects of Mn on mitochondria. Decreased striatal and pallidal cell numbers expressing glutamine synthetase (astrocyte marker) and Mn-superoxide dismutase were observed in rats treated chronically for 13 weeks with high Mn concentrations in drinking water [121]. Aged and young rats exposed for 8 days to Mn in drinking water showed heightened susceptibility to the toxic effects of Mn concomitant with decreased GSH (also see previous paragraph) and uric acid levels in the striatum and in striatal synaptosomes [122]. Aged rats treated intraperitoneally for 30 days with Mn showed decreased mitochondrial complex II succinate dehydrogenase activity (∼30%) in striatum and nigra, and increased tyrosine hydroxylase (TH) mRNA and protein levels [123]. Subcutaneous Mn administration in C57B1/C6 mice caused persistent striatal accumulation of the metal, observed at 21 days after cessation of the treatment; decreased DA release was observed at 7 and 21 days post treatment and attenuation in potassium stimulated increases in extracellular DA was noted at 1, 7, and 21 days post treatment, suggesting a long-term effect of Mn on striatal DAergic transmission [124]. Taken together these data are consistent with the preferential accumulation of Mn in basal ganglia and altered dopamine homeostasis in response to Mn exposure in various animal models.
3.2 Age-Related Effects of Manganese
The long-term effects of Mn in animals are helpful in delineating mechanisms of toxicity resulting from occupational exposures to this metal. Notably, a link between Mn and PD has been suggested, with Mn exposure exceeding twenty years, correlating with increased risk for PD [125]. Furthermore, chronic exposure to the fungicide Mn ethylene-bis-dithiocarbamate (Maneb) also increases the risk for Parkinsonism [104]. Though PD manifests in mid-life (familial PD) or senescence (idiopathic PD), it needs to be considered that its etiology evolves over decades with environmental exposures heightening the risk for the disease.
Several studies have addressed the developmental effects of Mn. For example, mice exposed to paraquat (PQ) and Maneb during the perinatal period and re-challenged as adults showed 62% reduction in striatal DA levels [126]. Juvenile mice exposed to Mn showed increased susceptibility, characterized by reduced striatal DA concentration in adulthood, induction of nitric oxide synthase (NOS), increased neuronal protein nitration and glial reactivity compared with adult mice that were not subjected to early-life Mn exposure or exposed to it only as adults [127,128]. These epidemiological and experimental findings point to an impairment in cellular antioxidant defenses induced by long-term or early-life exposure to this transition metal that diminishes the ability of the cells to cope with added oxidative challenges. The major susceptibility of aged rats to Mn neurotoxic effects likely reflects impairment in ROS defense mechanisms as a consequence of the aging process. The long-term and persistent cytotoxic effect of Mn and alterations in DAergic transmission suggest that chronic exposure to low metal concentrations in early life may facilitate the development of neurodegenerative processes later in life.
3.3 Manganese and Apoptosis
Mn-induced ROS production may also be a trigger for other processes that lead to cells demise, such as apoptosis. For example, in PC12 cells, Mn not only induces apoptosis, but also enhances L-dopa-induced apoptosis. Both effects were inhibited by antioxidants [129–131]. In the same cells, Mn was also shown to induce caspase 3-dependent proteolytic activation of protein kinase Cd (PKCd), which in turn contributes to apoptosis [132]. Increased intracellular calcium transient, decreased Na+/K+ ATPase and Ca2+ ATPase activities as well as increased apoptosis rates and impairment of N-methyl-D-aspartate (NMDA) receptor subunits synthesis were observed in Mn exposed primary neuronal cultures [133]. In SN4741 cells, a DAergic neuronal cell line, Mn-induced endoplasmic reticulum (ER) stress and activation of caspase-12 and apoptosis; the latter effect was reduced in Bcl-2-overexpressing DAergic cell lines [108].
Concentration-dependent Mn-induced ER stress response was observed in SK-N-MC human neuroblastoma cells, accompanied by increased expression of glucose-regulated protein 78 (GRP78), pro-apoptotic protein C/EBP homologous protein (CHOP) and p-eIF2α, concomitant with reduced mitochondrial complex I, II, III, and IV activities [134]. As shown in Figure 3, pathway 2 represents the ER stress response, which is also linked to pathway 1. Mn also effectively induced apoptosis in cultured mesencephalic cells, and this effect was accelerated by pre-treatment with DA. Cells were protected by blockade of NO synthesis, inhibition of NF-κB activation and pre-treatment with vitamin E. Induction of NF-κB and nitric oxide synthase activation by ROS were proposed as plausible mechanisms for Mn-induced neurotoxicity [135]. NF-κB would also lead to increased uptake of Mn as it has been reported to augment expression of DMT1, the major transport protein of Mn [80]. Treatment of neuronal stem cells with Mn was also shown to stimulate apoptosis via mitochondrial-mediated pathways, concomitant with cytochrome c release, caspase-3 activation, and ROS generation [136]. In immortalized DAergic cells, MES 23.5, Mn was shown to up regulate mitochondrial Bcl2/E1B 19 KDa interacting protein (BNIP3), which correlated with mitochondrially-mediated apoptosis [137]. Finally, in rat astrocytoma C6 cells Mn led to ROS-mediated apoptosis, involving caspase-8 and mitochondrial-mediated pathways, with both the total and mitochondrial levels of Bcl-2 and Bax shifting to favor the apoptotic process [138]. Taken together, these studies establish the propensity of Mn to lead to cell demise by activation of apoptotic mechanisms.
3.4 Effects of Manganese on DNA
Mn also possesses clastogenic activities making DNA a potential target for its toxic action, which could result in impaired gene expression. For example, Chinese hamster V79 cells treated with L-dopa or DA showed decreased proliferative potential and elevated micronuclei frequency. The addition of Mn to the cell culture enhanced the antiproliferative and clastogenic effects of DA and its precursor [109].
Concentration-dependent DNA fragmentation was seen in Mn-treated PC12 cells; apoptosis was preceded by mitochondrial dysfunction and activation of c-Jun N-terminal kinases [139]. In striatal neurons, Mn treatment caused concentration-dependent loss of mitochondrial membrane potential, complex II activity, DNA fragmentation, and decreased microtubule-associated protein (MAP2) [140]. DA-induced oxidative DNA damage was shown to increase in Mn-treated PC12 cells, likely secondary to enhancement in DA autoxidation with semiquinone radical production. The DA-induced DNA damage in the presence of Cu2+ and NADH was further potentiated by Mn [141]. Mn increased the formation of mitochondrial DNA single strand breaks (SSB) probably caused by increased oxidative stress in cultured liver cells [142].
Mn is normally present in the heterochromatin, nucleolus, cytoplasm, and mitochondria of rat striatal astrocytes and neurons. Chronic Mn treatment leads to increased Mn levels in the mitochondria and nuclei, serving as a possible explanation for its combined ability to impair mitochondrial oxidative metabolism, increase ROS production and damage to DNA [143]. Nuclear accumulation of Mn was also observed in cultured PC12 cells [144]. The DNA damage is shown by pathway 3 in Figure 3 (see above). Taken together, research to date indicates that excessive Mn levels may disturb cellular DNA.
3.5 Manganese and Neurotransmitter Homeostasis
Alterations in biosynthetic neurotransmitter pathways, which lead to changes in the activity of rate-limiting enzymes or synaptic receptors may also be responsible for Mn-induced alterations in synaptic transmission and consequently behavior. Mn alters the activity of the rate-limiting enzyme in DA synthesis, tyrosine hydroxylase (TH). In DAergic neural cell lineage, N27 cells, Mn was shown to lead to distinct alterations in TH upon both acute and chronic exposures. In acute conditions TH activity increased, whereas in chronic conditions it was decreased [145]. Decreased striatal DA content was preceded by an initial phase of increased TH activity and striatal DA content in rats chronically treated with Mn in drinking water [146,147]. Manganism is characterized by two phases, commencing with a psychiatric syndrome (bizarre behavior and madness) followed by impaired motor function. Changes in TH activity inherent to the acute or chronic Mn exposure may be related to these two phases, the initial phase corresponding to increased TH activity, which is probably linked to increased DA release, and the second phase related to decrease TH activity and probably linked to reduced DA release or DA cellular content.
The psychiatric phase of manganism could be also a consequence of Mn-induced inhibition of glutamatergic corticostriatal signaling mediated by presynaptic D2-like DA receptors. For example, corticostriatal slices of rats chronically treated with Mn, showed enhanced glutamate-mediated synaptic transmission in the striatum, an effect associated with increased locomotor activity in these rats [148].
3.6 Manganese Nanoparticles
A recent concern on human health is associated with the potential effects associated with exposure to Mn-containing nanoparticles. Nanotechnology is focused on creation and manipulation of particles with dimensions ranging from 1 to 100 nm, leading to the production of new materials that exhibit novel physicochemical properties and functions [149]. Mn has a large industrial use involving steel and non-steel alloy production, colorants, battery manufacture, pigments, and fuel additives, among others [150]. With the availability of nanosized particles, the replacement of macro-sized Mn particles has rapidly evolved [151]. As indicated above, olfactory neuronal pathway has been shown to be efficient in translocating inhaled Mn oxide into the CNS [152]. Some experimental data obtained with nanosized Mn particles will be presented herein, focusing attention on their safety and potential health concerns.
In PC12 Mn oxide nanoparticles (40 nm) were shown to be internalized and to lead to a concentration-dependent depletion of DA and its metabolites, increased ROS production [153] and down-regulation of TH gene expression [149]. Mn nanoparticles (25–900 nm) were also shown to be internalized in N27 DAergic neuronal cells, leading to time-dependent up-regulation of the protein transporter Tf, increased ROS production, and activation of caspase-3-mediated apoptosis and pro apoptotic protein kinase Cd (PKCd). In addition, Mn nanoparticles caused autophagy characterized by decreased levels of the native form of Bcl-2-interacting protein-1, Beclin 1, and increased cleavage of microtubule-associated protein 1 light chain 3, LC3, both of which are associated with autophagosome formation [151].
3.7 Manganese and Cytoplasmic Inclusions
The pathological hallmark of idiopathic PD is the presence of cytoplasmatic inclusions of α-synuclein fibrils, known as Lewy bodies and Lewy neurites; their production can be affected by both genetic and environmental factors [154,155]. α-Synuclein is a heat stable protein that is soluble and natively unfolded. In the CNS it is expressed in neurons where it localizes in pre-synaptic terminals in the vicinity to synaptic vesicles. The precise function of α-synuclein is unknown; a role as modulator in synaptic and neural plasticity has been advanced [156].
In vitro assay showed that pesticides, such as rotenone, PQ and Maneb, as well as metals, such as Al3+, Fe3+, Co3+, Cd2+, and Mn2+, induce a conformational change in α-synuclein accelerating its fibrillation rate; the effect of pesticides and metals when simultaneously present is synergistic [157] (see pathway 4 in Figure 3). Cultured DAergic neuronal cells, MES 23.5, exposed to the Mn-containing pesticide Maneb showed inhibition of proteasomal chymotrypsin-like and postglutamyl peptidase activities. Proteasomal dysfunction was accompanied by cytoplasmatic inclusions that were positive for α-synuclein immunostaining [158]. Metals such as arsenic, copper, zinc, mercury, cadmium, nickel, and lead as well as the metalloid selenium, have been shown to increase α-synuclein-like immunoreactivity aggregates in the CNS of white sucker fish, Catostomus commersoni, sampled from highly contaminated water with metal ions secondary to mining activity [159].
The propensity of Mn to increase cytoplasmic inclusions has also been shown in vitro. For example, Mn decreased the viability of SK-N-MC neuroblastoma cells expressing human DA transporter and α-synuclein [160], and increased expression of α-synuclein in PC12 cells, via ERK1/2 MAPK activation [161]. In SH-SY5Y neuroblastoma cells up-regulation of α-synuclein expression and α-synuclein mRNA by Mn preceded the apoptotic response [162], suggesting up-regulation of α-synuclein may be an early effect.
Similar effects have been noted in vivo. For example, non-human primates exposed to Mn showed up-regulation of another type of cytoplasmic inclusions, namely amyloid-b (A-b) precursor-like protein-1; its gene expression, increased along with the number of A-b diffuse plaques. In the same brains, Mn also led to α-synuclein aggregation particularly within the frontal cortex [163]. While still debatable, these findings raise the plausibility that in addition to its well-established link to PD, Mn may play a role in the etiology of Alzheimer’s disease (AD).
3.8 Manganese and Dopaminergic Circuitry
DA is a universal neurotransmitter present in the animal kingdom from invertebrates to vertebrates. The interaction of Mn with DA has been noted in other species, such as mollusks where exposure to Mn was shown to impair the cilio-inhibitory system of the lateral cilia of the gill, and decreased endogenous DA levels in cerebral and visceral ganglia and gills of Crassostrea virginica [164].
In vertebrate animals, microinjection of Mn into the SNpc or striatum allows for the evaluation of its action on mesostriatal DAergic circuitry. For example, unilateral intra-nigral Mn microinjection in rats led to ipsilateral striatal DA loss in a dose-dependent fashion and a rotational behavior towards the lesion side in response to systemic apomorphine. Systemic administration of L-dopa plus carbidopa or pargyline increased the DAergic striatal loss in these animals [165]. Intra-nigral Mn microinjection in rats decreased nigral and ipsilateral striatal DA and TH cofactor, (6R)-L-erythro-5,6,7,8 tetrahydrobiopterin (BH4) levels. Maximal decrease in BH4 was observed at 60 days, with complete recovery at 90 days after the Mn microinjection [166]. Mn microinjected into rat SNpc decreased ipsilateral striatal DA concentrations, the number of TH-positive cells and dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP-32) expression, and increased rotational behavior in response to systemic apomorphine administration, while systemic L-dopa plus carbidopa treatment worsened these effects [167].
Rats intra-nigrally microinjected with Mn showed rotational behavior in response to apomorphine and increased number of NADPH-d positive neurons in the ipsilateral SNpc as well as ipsi- and contralateral striatum. NO synthesis inhibition by NG-nitro-L-arginine (L-NOARG) reversed the NADPH-diaphorase increase, but worsened the rotational behavior response to systemic apomorphine, suggesting a protective role for NO in the Mn-induced neurodegenerative process [168]. Paradkar and Roth have reported that NO can inhibit NF-κB activity and the subsequent up regulation of DMT1 which may, in part, account for the protective effects of NO [80]. Mn is slowly cleared from the substantia nigra (SN), with a 50% decrease noted at 72 hours post microinjection [169]. Notably, the apomorphine-induced rotational behavior was detected at 24 hours after intra-nigral Mn microinjection reaching its maximum at 72 hours post injection. The time course of striatal TH immunostaining loss observed in Mn-treated rats followed the time course of rotational behavior, suggesting that cellular mechanisms induced by Mn, which lead to DAergic cell death occurred shortly after injection. It also suggests that threshold metal concentration is required in order to induce neurotoxic effects [169]. The slow brain clearance of Mn could be explained by its propensity to accumulate within brain mitochondria as shown by Gavin et al. [102].
Mn-induced DAergic neurodegeneration may be a consequence of its ability to produce an indirect excitotoxic process, as demonstrated by Brouillet et al. [98]. These authors have shown that Mn microinjected into rat striatum caused a dose-dependent decrease in DA, γ-aminobutyric acid, and substance P concentrations in the striatum. Such effects were blocked by prior removal of cortico-striatal glutamatergic inputs or by treatment with MK-801, a non-competitive NMDA antagonist. Striatal slices of rats treated chronically with Mn showed an enhancement in cortical glutamate-mediated synaptic transmission in the striatum, suggesting impairment in corticostriatal glutamatergic transmission and increased excitotoxic damage [170].
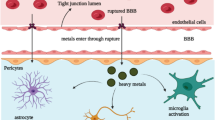
3.9 Manganese and Microglia
Reactive microglia are present in the substantia nigra of PD patients, suggesting an inflammatory component in the neurodegenerative process. CNS inflammatory responses are predominantly mediated by microglial cells which represent the first line of defense. Microglial activation has two stages; the first one is associated with release of neurotrophins leading to increased neuronal survival, and the second phase associated with release of pro-inflammatory mediators potentially leading to impaired neuronal survival [171]. Several studies suggest a potential role for microglia in mediating Mn-induced neurodegeneration. In vivo, intrastriatal Mn microinjections in rats were shown (7 days post injection) to reduce the number of TH+ cells and increase the number of activated microglia in the substantia nigra; up regulation of inducible nitric oxide synthase (iNOS) and tumor necrosis factor α gene expression was paralleled by increased protein levels of iNOS and interleukin-1β in the substantia nigra [172]. The microglial participation in Mn-induced neurodegeneration response is shown in pathway 5 in Figure 3 (see above). Non-human primates chronically treated with Mn also displayed increased microglial number in SNpc and reticulate concomitant with increased expression of iNOS, L-ferritin, and intracellular ferric ion in reactive microglial cells [173]. The decrease in TH+ cells observed in animals treated with intrastriatal Mn microinjection could be the consequence of a microglial defense response, which depending upon the duration and intensity could lead to inflammation and cellular degeneration.
Consistent with the above, mouse N9 microglial cells treated in vitro with bacterial lipopolysaccharide (LPS) and challenged with Mn showed significant induction of NO production, with a concomitant increase in iNOS gene transcription [110]. The same cells, when exposed to Mn showed increased production of interleukin-6 and TNFα. In the presence of lipopolysaccharide, this response was significantly potentiated. Co-exposure to LPS and Mn also increased NO production and iNOS expression [174]. It is noteworthy that Mn regulates iNOS expression at the transcriptional level in BV2 microglial cells, increasing iNOS protein expression by the activation of JNK-ERK MAPK and PI3/Akt signaling pathways [175].
More recently, it has been shown in rat primary neuron-glia co-cultures that simultaneous treatment with Mn and LPS increased production of pro-inflammatory cytokines, including TNFα and IL-1β, ROS as well as reactive nitrogen species (RNS). Minocycline (an antibiotic) pre-treatment effectively reduced the pro-inflammatory cytokine production [176]. These experimental data suggest that Mn can potentiate the effects of bacterial toxins and that ensuing inflammatory reactions in basal ganglia thus exacerbating the neurodegenerative process.
4 Manganese and Genetics
The clinical features of the extrapyramidal symptoms of manganism can be quite diverse reflecting the complex nature of the basal ganglia which is associated with a variety of integrated inhibitory and excitatory neurochemical pathways. The degree to which Mn disrupts any of these interdependent processes promotes an imbalance of output from the basal ganglia which potentially result in capricious disparities in onset, severity, and specific symptoms expressed as well as the progression of the disorder. The source which contributes to deviations in the characteristics of the expressed symptoms and neurological lesions generated may include exposure to other noxious environmental toxins, nutrition status, state of health and, most importantly, underlying genetic variability.
All of these reasons to varying degrees have the potential to alter the biochemical processes by which Mn imbalance can occur. Of these, genetic polymorphisms have to be considered as potentially playing a dominant role in regulating alterations in specific signaling pathways controlling Mn-induced cell death. This is best exemplified by the publication by Sadek et al. [177] describing a patient who worked as a welder for a total of three years. This individual acquired the progressive symptoms of Mn toxicity within one year after beginning employment making his clinical history quite remarkable as development of manganism after only one year of welding is highly unusual as onset usually requires considerably longer exposure times. The reason for the rapid development of the neurological deficits is unknown but, most likely, reflects a genetic predisposition in this individual. In addition, within several years of the initial diagnosis, he developed a unilateral tremor in his right hand more characteristic of Parkinsonism. The prominence of a genetic component contributing to manganism is further substantiated by the reality that not all welders or Mn miners develop manganism yet exposures are comparable to their fellow workers that acquire the disorder. Clearly, this underscores the potential impact of genetic polymorphisms that likely contribute to the variability in development of Mn toxicity. It is essential to stress that although chronic exposure to Mn is not the causative agent provoking Parkinson’s disease, there is compelling evidence in the literature that it may be one of the most influential metals correlating with increased susceptibility to develop this condition [178–182].
Manganism is a disorder anatomically and functionally distinct from that of Parkinson’s disease, at least in regard to the critical initial site of injury, leaving us with the issue as to why many of the observed symptoms overlap between the two disorders and whether chronic exposure to Mn can eventually provoke idiopathic Parkinsonism. Some of the similarities between the two disorders simply reflect the fact that Mn can influence DAergic transmission by inhibiting dopamine release and decreasing dopamine transporter (DAT) levels in the striatum [124,183–187]. Although this may clarify why some of the symptoms of Mn toxicity overlap with that of Parkinson’s disease, it fails to explain individual differences in susceptibility to develop manganism.
Over the past 15 years, a number of gene variants have been identified as being linked to early (α-synuclein, parkin, PINK1, DJ-1, and ATP13A2) and late (LRRK2) onset of Parkinson’s disease [188–190]. Because many of the symptoms and neurodegenerative features between the two neurological disorders appear to be interrelated, it is feasible that polymorphisms in some of these genes may also be associated with the susceptibility to develop manganism. If a relationship does exist, then it may also explain, in a reciprocal fashion, why excess exposure to Mn may provoke early onset of Parkinson’s disease. Of the six genes linked to Parkinsonism, two of these, parkin and ATP13A2, have already been reported to influence Mn toxicity in cell culture systems [191–194]. Thus, one-third of the known polymorphisms in genes linked to onset of Parkinsonism are suggested to promote increased susceptibility to acquire manganism.
The two genes, parkin and ATP13A2, are both linked to early onset of Parkinsonism but differ in their enzymatic activity though both have several features in common including alteration of α-synuclein activity and protein degradation as well as mitochondrial dysfunction [195–197]. Parkin is one of over 600 identified E3 ligases responsible for the conjugation of ubiquitin to a variety of proteins [198,199]. Of the known genes correlating with early onset of Parkinson’s disease, those involving parkin are the most prevalent encompassing approximately 50% of all recessive cases [200,201]. Although considered to be an autosomal recessive gene, there is evidence in the literature suggesting that mutations in a single allele may exert sufficient imbalance in dopaminergic activity to cause subclinical features of Parkinsonism [202] and thus, even in the heterozygous state, may play a significant role in development of idiopathic Parkinsonism [203,204]. Two studies [191,205] have reported that overexpression of parkin can protect cells against Mn toxicity. Recent studies [191] have further demonstrated that parkin is responsible for the ubiquitination of DMT1, the major transport protein for Mn (see above) [191]. This process is relatively specific as it is responsible for degradation of only the 1B species of the transporter which predominates in the CNS [206,207]. Thus, mutations in parkin, which lead to its inactivation would likely increase DMT1 levels and the subsequent accumulation of Mn within the CNS.
Like parkin, ATP13A2 is also associated with the pathogenesis of both familial and sporadic Parkinson’s disease as well as Kufor-Rakeb syndrome (KRS), a rare juvenile-onset autosomal recessive disease characterized by progressive Parkinsonism [190,208]. ATP13A2 encodes a large membrane-bound lysosomal P-type ATPase and when mutated acts, presumably, to disrupt its lysosomal localization, thus, linking lysosomal degradation with Parkinson’s disease. A recent paper by Gitler et al. [193] demonstrated a strong genetic interaction between α-synuclein and PARK9, a yeast ortholog of human Parkinson’s disease-linked gene, ATP13A2. Coexpression of PARK9 (ATP13A2) in animals overexpressing α-synuclein was shown to protect against dopaminergic neuron loss whereas knockdown of the ATP13A2 ortholog in Caenorhabditis elegans enhance α-synuclein misfolding. It was also reported that wild-type ATP13A2, but not the KRS pathogenic ATP13A2 mutant, protected cells from Mn-induced cell death in mammalian cell lines and primary rat neuronal cultures. In addition, wild-type ATP13A2 reduced intracellular Mn and prevented cytochrome c release from mitochondria when compared to the mutant cells. Based on these findings, these authors suggested that PARK9 is involved in Mn transport though direct evidence for this is lacking.
Thus, one-third of the known Parkinson-linked polymorphisms are linked to Mn toxicity leaving us with the question as to the role of the other four genes, DJ-1, PINK1, α-synuclein, and LRRK2, in the development of manganism [192,193,209, 210]. The proposed mechanisms for each of these proteins in inducing Parkinsonism are relatively complex and often overlap in that each may mutually influence the others activity. Based on the predicted mechanisms, there is compelling evidence to justify the hypothesis that mutations in these genes will augment Mn toxicity as polymorphisms produce gene products that disrupt mitochondrial function and thus, potentiate mitochondrial-induced oxidative stress which, independent of other toxic mechanisms, will facilitate Mn-induced toxicity.
Whereas the aforementioned genes are autosomal recessive, LRRK2 is an autosomal dominant gene and the only gene associated with late onset of the disorder [211]. Thus, a mutation in a single allele may be sufficient to elicit effects on Mn toxicity prior to the appearance of the symptoms of Parkinsonism. LRRK2 is a relatively complex gene which encodes a large multidomain protein that includes a Rho/Ras-like GTPase domain (termed Roc, for Ras) and a protein kinase domain. Because of this complexity, the mechanism by which mutations in LRRK2 elicits Parkinsonism is likely to be multifaceted and are anticipated to influence a number of signaling pathways [212]. In regard to dopamine function, LRRK2 can cause a decrease in the dopamine transporter, DAT [213,214], as well as impair dopamine-stimulated neurotransmission [215] both of which are also seen with overexposure to Mn [183,184,186,187]. Relevant to manganism, studies have reported that Mn can increase phosphorylation activity of the most predominant polymorphic species of LRRK2, the G2019S mutant, while inhibiting the wild-type kinase activity activated by Mg [216,217]. Thus, LRRK2 displays a number of critical functions many of which can potentially affect or be affected by Mn.
Support for a potential role for DMT1 in neurodegeneration associated with PD was recently suggested by several researchers. Salazar et al. [218] have recently shown that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication in mice, a well established PD model, caused increased DMT1 expression in the ventral mesencephalon concomitant with iron accumulation, oxidative stress, and DAergic cell loss. More recently He et al. [219], direct sequencing identified two single-nucleotide polymorphisms in the DMT1 gene (CC haplotype) as a risk factor for PD in the Han Chinese population. The above studies have not directly assessed whether polymorphisms in DMT1 in PD patients also alter CNS Mn levels. Given this gene’s role in regulating intracellular Mn levels (see above), it is essential that future studies in suitable cohorts also address the association between DMT1 polymorphisms and Mn accumulation.
5 Concluding Remarks
As described above, uptake of Mn in the intestines and the various processes controlling its delivery and access to the CNS represent the rate limiting steps regulating Mn levels and toxicity in vivo. Multiple systems are involved in the maintenance of Mn levels, all of which must function, to some extent, in the preservation of its homeostatic levels. These systems routinely work in harmony to sustain required levels of Mn even though Mn may not be an operational cation these systems were designed for. As a consequence, conditions that upset this delicate balance result in a failure of the various components to adjust to the practical needs of Mn in order to support the functional demands of the other essential cations, the most important of which is Fe. Thus, conditions of excess exposures to Mn or a variety stress inducing or inflammatory conditions are likely to provoke an imbalance in the various transport systems for Mn resulting in increased CNS toxicity.
The synopsis provided above establishes that Mn exerts neurotoxicity specifically in DAergic nigral cells. However, its aberrant effects may be much broader given recent evidence on its ability to perturb other cell types and neurotransmitters, as well as its propensity to increase cytoplasmic inclusions. Taken together, a broad array of mechanisms appears to mediate Mn-induced DAergic neurodegeneration. Considering the essentiality of Mn, future studies should continue to focus on its role both in health and disease. Given that the safety margin for Mn appears to be narrow, it is incumbent upon the research community to further delineate its toxicity, considering its ubiquitous presence in the environment and anthropogenic usage. This novel information will be highly relevant for future considerations on its safety and in delineating risk assessments for exposure and safety.
Finally, the findings to date support a possible genetic link between the manifestations of manganism with genes associated with onset of Parkinsonism. This is not totally surprising as mutations in these genes, in many respects, display similar aberrant neurological activities within the basal ganglia, which provoke the distinctive dystonic movements associated with both disorders. It remains to be determined whether the other genes linked to Parkinsonism will also be associated with the development of Mn toxicity.
Abbreviations
- AD:
-
Alzheimer’s disease
- A-b:
-
amyloid-b
- BBB:
-
blood-brain barrier
- BH4 :
-
tetrahydrobiopterin
- CHOP:
-
C/EBP homologous protein
- CNS:
-
central nervous system
- DA:
-
dopamine
- DAergic:
-
dopaminergic
- DARPP-32:
-
dopamine- and cAMP-regulated neuronal phosphoprotein
- DAT:
-
dopamine transporter
- Dcytb:
-
duodenal cytochrome B
- DMT1:
-
divalent metal transporter 1
- ER:
-
endoplasmic reticulum
- ESADDI:
-
estimated safe and adequate dietary intake
- Fpn:
-
ferroportin
- GABA:
-
γ-aminobutyric acid
- GRP78:
-
glucose-regulated protein 78
- GSH:
-
glutathione
- HEK293:
-
human embryonic kidney cells 293
- HFE:
-
hemochromatosis protein
- HRE:
-
hypoxia response element
- IL-1β:
-
interleukin-1β
- IL-6:
-
interleukin-6
- iNOS:
-
inducible nitric oxide
- IRE:
-
iron response element
- IREG1:
-
iron-regulated protein 1
- IRP:
-
iron-responsive protein
- KRS:
-
Kufor-Rakeb syndrome
- l-dopa:
-
L-3,4-dihydroxyphenylalanine
- l-NOARG:
-
NG-nitro-l-arginine
- LPS:
-
lipopolysaccharide
- Maneb:
-
manganese ethylene-bis-dithiocarbamate
- MAO:
-
monoamine oxidase
- MAP2:
-
microtubule-associated protein
- MDA:
-
malondialdehyde
- MnSOD:
-
Mn superoxide dismutase
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTP1:
-
metal tolerance protein 1
- NADH:
-
nicotinamide adenine dinucleotide (reduced)
- NF:
-
necrosis factor
- NMDA:
-
N-methyl-D-aspartate
- NOS:
-
nitric oxide synthase
- NRC:
-
National Research Council
- OEC:
-
oxygen-evolving complex
- ORAI1:
-
calcium release-activated calcium channel protein 1
- PD:
-
Parkinson’s disease
- PKCd:
-
protein kinase Cd
- PQ:
-
paraquat
- QPCR:
-
real-time polymerase chain reaction
- RDA:
-
Recommended Dietary Allowance
- RNS:
-
reactive nitrogen species
- ROS:
-
reactive oxygen species
- SERCA:
-
sarcoplasmic reticulum Ca-ATPase
- SLC39:
-
solute-carrier-39
- SNpc:
-
substantia nigra pars compacta
- SOC:
-
store-operated Ca2+ channels
- SOD:
-
superoxide dismutase
- SOD2:
-
mitochondrial superoxide dismutase
- SSB:
-
single strand breaks
- STIM1:
-
Ca2+ sensor protein
- Tf:
-
transferrin
- TfR:
-
transferrin receptor
- TH:
-
tyrosine hydroxylase
- TNFα:
-
tumor necrosis factor α
- TRPM7:
-
transient receptor potential cation channel, subfamily M, member 7
- Ub:
-
ubiquitin
- UTR:
-
untranslated region
- ZIP:
-
zinc transporter
References
J. L. Greger, J. Nutr. 1998, 128, 368S–371S.
National Academy of Sciences (NAS). Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Panel on Micronutrients, Subcommittees on Upper Reference Levels of Nutrients and of Interpretation and Use of Dietary Reference Intakes, and the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. 2001, available from: <www.nap.edu/books/ 0309072794/html/>
P. Aisen, R. Aasa, A. G. Redfield, J. Biol. Chem. 1969, 244, 4628–4633.
J. W. Critchfield, C. L. Keen, Metabolism 1992, 41, 1087–1092.
C. L. Keen, J. G. Bell, B. Lonnerdal, J. Nutr. 1986, 116, 395–402.
C. L. Keen, S. Zidenberg-Cherr, Manganese Toxicity in Humans and Experimental Animals, in Manganese in Health and Disease, Ed D. J. Klimis-Tavantzis, CRC Press, Boca Raton, FL, 1994, pp 193–205.
C. L. Keen, B. Lonnerdal, M. Clegg, L. S. Hurley, J. Nutr. 1981, 111, 226–230.
C. L. Keen, J. L. Ensunsa, M. H. Watson, D. L. Baly, S. M. Donovan, M. H. Monaco, M. S. Clegg, Neurotoxicology 1999, 20, 213–223.
J. L. Greger, J. Nutr. 1998, 128, 368S–371S.
G. L. Rehnberg, J. F. Hein, S. D. Carter, R. S. Linko, J. W. Laskey, J. Toxicol. Environ. Health 1982, 6, 217–226.
P. S. Papavasiliou, S. T. Miller, G. C. Cotzias, Am. J. Physiol. 1966, 211, 211–216.
C. M. Lucaciu, C. Dragu, L. Copaescu, V. V. Morariu, Biochim. Biophys. Acta 1997, 1328, 90–98.
S. S. Kannurpatti, P. G. Joshi, N. B. Joshi, Neurochem. Res. 2000, 25, 1527–1536.
C. Grimm, R. Kraft, S. Sauerbruch, G. Schultz, C. Harteneck, J. Biol. Chem. 2003, 278, 21493–21501.
A. Riccio, C. Mattei, R. E. Kelsell, A. D. Medhurst, A. R. Calver, A. D. Randall, J. B. Davis, C. D. Benham, M. N. Pangalos, J. Biol. Chem. 2002, 277, 12302–12309.
R. A. Yokel, Neuromol. Med. 2009, 11, 297–310.
J. A. Roth, M. D. Garrick, Biochem. Pharmacol. 2003, 66, 1–13.
M. Aschner, D. C. Dorman, Toxicol. Rev. 2006, 25, 147–154.
M. D. Garrick, S. T. Singleton, F. Vargas, H. C. Kuo, L. Zhao, M. Knopfel, T. Davidson, M. Costa, P. Paradkar, J. A. Roth, L. M. Garrick, Biol. Res. 2006, 39, 79–85.
H. Fujishiro, K. Kubota, D. Inoue, A. Inoue, T. Yanagiya, S. Enomoto, S. Himeno, Toxicology 2011, 280, 118–125.
L. He, K. Girijashanker, T. P. Dalton, J. Reed, H. Li, M. Soleimani, D. W. Nebert, Mol. Pharmacol. 2006, 70, 171–180.
S. Himeno, T. Yanagiya, H. Fujishiro, Biochimie 2009, 91, 1218–1222.
K. Girijashanker, L. He, M. Soleimani, J. M. Reed, H. Li, Z. Liu, B. Wang, T. P. Dalton, D. W. Nebert, Mol. Pharmacol. 2008, 73, 1413–1423.
B. Mackenzie, H. Takanaga, N. Hubert, A. Rolfs, M. A. Hediger, Biochem. J. 2007, 403, 59–69.
H. Gunshin, B. Mackenzie, U. V. Berger, Y. Gunshin, M. F. Romero, W. F. Boron, S. Nussberger, J. L. Gollan, M. A. Hediger, Nature 1997, 388, 482–488.
J. A. Roth, Biol. Res. 2006, 39, 45–57.
M. Arredondo, P. Munoz, C. V. Mura, M. T. Nunez, Am. J. Physiol. Cell Physiol. 2003, 284, C1525–1530.
M. D. Garrick, S. T. Singleton, F. Vargas, H. C. Kuo, L. Zhao, M. Knopfel, T. Davidson, M. Costa, P. Paradkar, J. A. Roth, L. M. Garrick, Biol. Res. 2006, 39, 79–85.
B. Mackenzie, M. D. Garrick, Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G981–986.
J. F. Collins, Biol. Res. 2006, 39, 25–37.
L. Davidsson, A. Cederblad, B. Lonnerdal, B. Sandstrom, Am. J. Dis. Child. 1989, 143, 823–827.
L. Davidsson, A. Cederblad, B. Lonnerdal, B. Sandstrom, Am. J. Clin. Nutr. 1991, 54, 1065–1070.
R. A. Gibbons, S. N. Dixon, K. Hallis, A. M. Russell, B. F. Sansom, H. W. Symonds, Biochim. Biophys. Acta 1976, 444, 1–10.
P. E. Johnson, G. I. Lykken, E. D. Korynta, J. Nutr. 1991, 121, 711–717.
Z. Yin, H. Jiang, E.S. Lee, M. Ni, K. M. Erikson, D. Milatovic, A. B. Bowman, M. Aschner, J. Neurochem. 2010, 112, 1190–1198.
X. Wang, D. S. Miller, W. Zheng, Toxicol. Appl. Pharmacol. 2008, 230, 167–174.
M. S. Madejczyk, N. Ballatori, Biochim. Biophys. Acta 2012, 1818, 651–657.
J. F. Collins, M. Wessling-Resnick, M. D. Knutson, J. Nutr. 2008, 138, 2284–2288.
T. Ganz, Blood 2011, 117, 4425–4433.
L. Davidsson, B. Lonnerdal, B. Sandstrom, C. Kunz, C. L. Keen, J. Nutr. 1989, 119, 1461–1464.
G.J. Anderson, D. M. Frazer, A. T. McKie, C. D. Vulpe, Blood Cells Mol. Dis. 2002, 29, 367–375.
S. Cherukuri, R. Potla, J. Sarkar, S. Nurko, Z. L. Harris, P. L. Fox, Cell Metab. 2005, 2, 309–319.
T. Jursa, D. R. Smith, Toxicol. Sci. 2009, 107, 182–193.
R. A. Gibbons, S. N. Dixon, A. M. Russell, B. F. Sansom, Biochim. Biophys. Acta 1976, 437, 301–304.
G. J. Anderson, C. D. Vulpe, Cell Mol. Life Sci. 2009, 66, 3241–3261.
T. Moos, E. H. Morgan, J. Neurosci. Res. 1998, 54, 486–494.
R. B. Hernández, M. Farina, B. P. Esposito, N. C. Souza-Pinto, F. Barbosa, Jr., C. Sunol, Toxicol. Sci. 2011, 124, 414–423.
G. W. Bates, C. Billups, P. Saltman, J. Biol. Chem. 1967, 242, 2816–2821.
K. Tulpule, S. R. Robinson, G. M. Bishop, R. Dringen, J. Neurosci. Res. 2010, 88, 563–571.
Y. Ke, Z. M. Qian, Prog. Neurobiol. 2007, 83, 149–173.
J. Henriksson, J. Tallkvist, H. Tjalve, Toxicol. Appl. Pharmacol. 1999, 156, 119–128.
D. Vitarella, B. A. Wong, O. R. Moss, D. C. Dorman, Toxicol. Appl. Pharmacol. 2000, 163, 279–285.
L. D. Fechter, D. L. Johnson, R. A. Lynch, Neurotoxicology 2002, 23, 177–183.
A. W. Dobson, S. Weber, D. C. Dorman, L. K. Lash, K. M. Erikson, M. Aschner, Biol. Trace Elem. Res. 2003, 93, 113–126.
D. C. Dorman, B. E. McManus, C. U. Parkinson, C. A. Manuel, A. M. McElveen, J. I. Everitt, Inhal. Toxicol. 2004, 16, 481–488.
L. Normandin, L. Ann Beaupre, F. Salehi, A. St-Pierre, G. Kennedy, D. Mergler, R. F. Butterworth, S. Philippe, J. Zayed, Neurotoxicology 2004, 25, 433–441.
K. Thompson, R. M. Molina, T. Donaghey, J. E. Schwob, J. D. Brain, M. Wessling-Resnick, FASEB J. 2007, 21, 223–230.
D. C. Dorman, M. F. Struve, R. A. James, M. W. Marshall, C. U. Parkinson, B. A. Wong, Toxicol. Appl. Pharmacol. 2001, 170, 79–87.
G. Oberdorster, Z. Sharp, V. Atudorei, A. Elder, R. Gelein, W. Kreyling, C. Cox, Inhal. Toxicol. 2004, 16, 437–445.
X. Wang, G. J. Li, W. Zheng, Brain Res. 2006, 1097, 1–10.
A. T. McKie, Biochem. Soc. Trans. 2008, 36, 1239–1241.
R. S. Ohgami, D. R. Campagna, E. L. Greer, B. Antiochos, A. McDonald, J. Chen, J. J. Sharp, Y. Fujiwara, J. E. Barker, M. D. Fleming, Nat. Genet. 2005, 37, 1264–1269.
R. S. Ohgami, D. R. Campagna, A. McDonald, M. D. Fleming, Blood 2006, 108, 1388–1394.
T. E. Gunter, C. E. Gavin, M. Aschner, K. K. Gunter, Neurotoxicology 2006, 27, 765–776.
M. D. Fleming, M. A. Romano, M. A. Su, L. M. Garrick, M. D. Garrick, N. C. Andrews, Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153.
N. Hubert, M. W. Hentze, Proc. Natl. Acad. Sci. USA 2002, 99, 12345–12350.
P. L. Lee, T. Gelbart, C. West, C. Halloran, E. Beutler, Blood Cells Mol. Dis. 1998, 24, 199–215.
J. Kato, M. Kobune, S. Ohkubo, K. Fujikawa, M. Tanaka, R. Takimoto, K. Takada, D. Takahari, Y. Kawano, Y. Kohgo, Y. Niitsu, Exp. Hematol. 2007, 35, 879–887.
J. A. Roth, C. Horbinski, D. Higgins, P. Lein, M. D. Garrick, Neurotoxicology 2002, 23, 147–157.
S. J. Garcia, K. Gellein, T. Syversen, M. Aschner, Toxicol. Sci. 2007, 95, 205–214.
V. A. Fitsanakis, N. Zhang, J. G. Anderson, K. M. Erikson, M. J. Avison, J. C. Gore, M. Aschner, Toxicol. Sci. 2008, 103, 116–124.
S. L. Hansen, N. Trakooljul, H. C. Liu, A. J. Moeser, J. W. Spears, J. Nutr. 2009, 139, 1474–1479.
M. T. Nunez, V. Tapia, A. Rojas, P. Aguirre, F. Gomez, F. Nualart, Am. J. Physiol. Cell Physiol. 2010, 298, C477–485.
C. Kwik-Uribe, D. R. Smith, J. Neurosci. Res. 2006, 83, 1601–1610.
E. C. Theil, J. Nutr. 2011, 141, 724S–728S.
J. A. Roth, L. Feng, K. G. Dolan, A. Lis, M. D. Garrick, J. Neurosci. Res. 2002, 68, 76–83.
K. M. Erikson, T. Syversen, E. Steinnes, M. Aschner, J. Nutr. Biochem. 2004, 15, 335–341.
K. M. Erikson, Z. K. Shihabi, J. L. Aschner, M. Aschner, Biol. Trace Elem. Res. 2002, 87, 143–156.
P. N. Paradkar, J. A. Roth, Biochem. J. 2006, 394, 173–183.
P. N. Paradkar, J. A. Roth, J. Neurochem. 2006, 96, 1768–1777.
P. N. Paradkar, J. A. Roth, J. Cell Physiol. 2007, 211, 183–188.
A. Lis, P. N. Paradkar, S. Singleton, H. C. Kuo, M. D. Garrick, J.A. Roth, Biochem. Pharmacol. 2005, 69, 1647–1655.
D. Wang, L. H. Wang, Y. Zhao, Y. P. Lu, L. Zhu, IUBMB Life 2010, 62, 629–636.
H. Fujishiro, M. Doi, S. Enomoto, S. Himeno, Metallomics 2011, 3, 710–718.
J. P. Liuzzi, F. Aydemir, H. Nam, M. D. Knutson, R. J. Cousins, Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617.
M. Chesler, Physiol. Rev. 2003, 83, 1183–1221.
C. Ma, S. N. Schneider, M. Miller, D. W. Nebert, C. Lind, S. M. Roda, S. E. Afton, J. A. Caruso, M. B. Genter, J. Biochem. Mol. Toxicol. 2008, 22, 305–310.
J. J. Pinilla-Tenas, B. K. Sparkman, A. Shawki, A. C. Illing, C. J. Mitchell, N. Zhao, J. P. Liuzzi, R. J. Cousins, M. D. Knutson, B. Mackenzie, Am. J. Physiol. Cell Physiol. 2011, 301, C862–871.
J. Gao, N. Zhao, M. D. Knutson, C. A. Enns, J. Biol. Chem. 2008, 283, 21462–21468.
N. Zhao, J. Gao, C. A. Enns, M. D. Knutson, J. Biol. Chem. 2010, 285, 32141–32150.
K. Loutzenhiser, R. Loutzenhiser, Circ. Res. 2000, 87, 551–557.
R. A. Yokel, J. S. Crossgrove, Res. Rep. Health Eff. Inst. 2004, 7–58; discussion 59–73.
J. S. Crossgrove, R. A. Yokel, Neurotoxicology 2005, 26, 297–307.
I. Jardin, L. J. Gomez, G. M. Salido, J. A. Rosado, Biochem. J. 2009, 420, 267–276.
A. Guilbert, M. Gautier, I. Dhennin-Duthille, N. Haren, H. Sevestre, H. Ouadid-Ahidouch, Am. J. Physiol. Cell Physiol. 2009, 297, C493–502.
D. Plenz, M. Herrera-Marschitz, S. T. Kitai, J. Comp. Neurol. 1998, 397, 437–457.
S. T. Rouse, M. J. Marino, S. R. Bradley, H. Awad, M. Wittmann, P. J. Conn, Pharmacol. Ther. 2000, 88, 427–435.
E. P. Brouillet, L. Shinobu, U. McGarvey, F. Hochberg, M. F. Beal, Exp. Neurol. 1993, 120, 89–94.
Z. Xu, K. Jia, B. Xu, A. He, J. Li, Y. Deng, F. Zhang, Toxicol. Ind. Health 2010, 26, 55–60.
Y. Deng, Z. Xu, B. Xu, Y. Tian, X. Xin, X. Deng, J. Gao, Brain Res. 2009, 1289, 106–117.
E. V. Stelmashook, E. R. Lozier, E. S. Goryacheva, P. Mergenthaler, S. V. Novikova, D. B. Zorov, N. K. Isaev, Neurosci. Lett. 2010, 482, 151–155.
C. E. Gavin, K. K. Gunter, T. E. Gunter, Neurotoxicology 1999, 20, 445–453.
A. Takeda, N. Sotogaku, N. Oku, Neuroscience 2002, 114, 669–674.
G. Meco, V. Bonifati, N. Vanacore, E. Fabrizio, Scand. J. Work Environ. Health 1994, 20, 301–305.
M. Parenti, L. Rusconi, V. Cappabianca, E. A. Parati, A. Groppetti, Brain Res. 1988, 473, 236–240.
J. J. Liccione, M. D. Maines, J. Pharmacol. Exp. Ther. 1988, 247, 156–161.
T. E. Gunter, B. Gerstner, T. Lester, A. P. Wojtovich, J. Malecki, S. G. Swarts, P. S. Brookes, C. E. Gavin, K. K. Gunter, Toxicol. Appl. Pharmacol. 2010, 249, 65–75.
H. S. Chun, H. Lee, J. H. Son, Neurosci. Lett. 2001, 316, 5–8.
R. D. Snyder, M. B. Friedman, Mutat. Res. 1998, 405, 1–8.
J. Y. Chang, L. Z. Liu, Brain Res. Mol. Brain Res. 1999, 68, 22–28.
A. Barbeau, Neurotoxicology 1984, 5, 13–35.
D. G. Graham, Mol. Pharmacol. 1978, 14, 633–643.
F. S. Archibald, C. Tyree, Arch. Biochem. Biophys. 1987, 256, 638–650.
J. Donaldson, D. McGregor, F. LaBella, Can. J. Physiol. Pharmacol. 1982, 60, 1398–1405.
L. S. Maynard, G. C. Cotzias, J. Biol. Chem. 1955, 214, 489–495.
Y. Deng, Y. Luan, H. Qing, H. Xie, J. Lu, J. Zhou, Neurosci. Lett. 2008, 444, 122–126.
C. G. Worley, D. Bombick, J. W. Allen, R. L. Suber, M. Aschner, Neurotoxicology 2002, 23, 159–164.
D. L. Stredrick, A. H. Stokes, T. J. Worst, W. M. Freeman, E. A. Johnson, L. H. Lash, M. Aschner, K. E. Vrana, Neurotoxicology 2004, 25, 543–553.
M. N. Subhash, T. S. Padmashree, Food Chem. Toxicol. 1990, 28, 567–570.
W. N. Sloot, J. Korf, J. F. Koster, L. E. De Wit, J. B. Gramsbergen, Exp Neurol. 1996, 138, 236–245.
M. Morello, P. Zatta, P. Zambenedetti, A. Martorana, V. D’Angelo, G. Melchiorri, G. Bernardi, G. Sancesario, Brain Res. Bull. 2007, 74, 406–415.
M. S. Desole, G. Esposito, R. Migheli, L. Fresu, S. Sircana, D. Zangani, M. Miele, E. Miele, Neuropharmacology 1994, 34, 289–285.
M. Tomas-Camardiel, A. J. Herrera, J. L. Venero, M. Cruz Sanchez-Hidalgo, J. Cano, A. Machado, Brain Res. Mol. Brain Res. 2002, 103, 116–129.
M. Khalid, R. A. Aoun, T. A. Mathews, J. Neurosci. Methods 2011, 202, 182–191.
J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown, R. J. Richardson, Neurology 1997, 48, 650–658.
M. Thiruchelvam, E. K. Richfield, B. M. Goodman, R. B. Baggs, D. A. Cory-Slechta, Neurotoxicology 2002, 23, 621–633.
J. A. Moreno, K. M. Streifel, K. A. Sullivan, M. E. Legare, R. B. Tjalkens, Toxicol. Sci. 2009, 112, 405–415.
J. A. Moreno, E. C. Yeomans, K. M. Streifel, B. L. Brattin, R. J. Taylor, R. B. Tjalkens, Toxicol. Sci. 2009, 112, 394–404.
M. S. Desole, L. Sciola, M. R. Delogu, S. Sircana, R. Migheli, Neurosci. Lett. 1996, 209, 193–196.
M. S. Desole, L. Sciola, M. R. Delogu, S. Sircana, R. Migheli, E. Miele, Neurochem. Int. 1997, 31, 169–176.
R. Migheli, C. Godani, L. Sciola, M. R. Delogu, P. A. Serra, D. Zangani, G. De Natale, E. Miele, M. S. Desole, J. Neurochem. 1999, 73, 1155–1163.
M. Kitazawa, V. Anantharam, Y. Yang, Y. Hirata, A. Kanthasamy, A. G. Kanthasamy, Biochem. Pharmacol. 2005, 69, 133–146.
B. Xu, Z. F. Xu, Y. Deng, Neurotoxicology 2009, 30, 941–949.
H. Yoon, G. H. Lee, D. S. Kim, K. W. Kim, H. R. Kim, H. J. Chae, Toxicol. In Vitro. 2011, 25, 1259–1268.
K. Prabhakaran, D. Ghosh, G. D. Chapman, P. G. Gunasekar, Brain Res. Bull. 2008, 76, 361–367.
C. Tamm, F. Sabri, S. Ceccatelli, Toxicol. Sci. 2008, 101, 310–320.
K. Prabhakaran, G. D. Chapman, P. G. Gunasekar, Neurotoxicology 2009, 30, 414–422.
A. Alaimo, R. M. Gorojod, M. L. Kotler, Neurochem. Int. 2011, 59, 297–308.
Y. Hirata, K. Adachi, K. Kiuchi, J. Neurochem. 1998, 71, 1607–1615.
E. A. Malecki, Brain Res. Bull. 2001, 55, 225–228.
S. Oikawa, I. Hirosawa, S. Tada-Oikawa, A. Furukawa, K. Nishiura, S. Kawanishi, Free Radic. Biol. Med. 2006, 41, 748–756.
J. Jiao, Y. Qi, J. Fu, Z. Zhou, Environ. Toxicol. Pharmacol. 2008, 26, 123–127.
M. Morello, A. Canini, P. Mattioli, R. P. Sorge, A. Alimonti, B. Bocca, G. Forte, A. Martorana, G. Bernardi, G. Sancesario, Neurotoxicology 2008, 29, 60–72.
K. Kalia, W. Jiang, W. Zheng, Neurotoxicology 2008, 29, 466–470.
D. Zhang, A. Kanthasamy, V. Anantharam, Toxicol. Appl. Pharmacol. 2011, 254, 65–71.
E. Bonilla, Neurobehav. Toxicol. 1980, 2, 37–41.
S. V. Chandra, G. S. Shukla, J. Neurochem. 1981, 36, 683–687.
P. Calabresi, M. Ammassari-Teule, P. Gubellini, G. Sancesario, M. Morello, D. Centonze, G. A. Marfia, E. Saulle, E. Passino, B. Picconi, G. Bernardi, Neurobiol. Dis. 2001, 8, 419–432.
J. Wang, M. F. Rahman, H. M. Duhart, G. D. Newport, T. A. Patterson, R. C. Murdock, S. M. Hussain, J. J. Schlager, S. F. Ali, Neurotoxicology 2009, 30, 926–933.
M. J. Han, T. Ozaki, J. Yu, J. Chem. Phys. 2005, 123, 34306.
H. Afeseh Ngwa, A. Kanthasamy, Y. Gu, N. Fang, V. Anantharam, A. G. Kanthasamy, Toxicol. Appl. Pharmacol. 2011, 256, 227–240.
A. Elder, R. Gelein, V. Silva, T. Feikert, L. Opanashuk, J. Carter, R. Potter, A. Maynard, Y. Ito, J. Finkelstein, G. Oberdorster, Environ. Health Perspect. 2006, 114, 1172–1178.
S. M. Hussain, A. K. Javorina, A. M. Schrand, H. M. Duhart, S. F. Ali, J. J. Schlager, Toxicol. Sci. 2006, 92, 456–463.
A. B. Bowman, G. F. Kwakye, E. Herrero Hernandez, M. Aschner, J. Trace Elem. Med. Biol. 2011, 25, 191–203.
J. Lotharius, P. Brundin, Nat. Rev. Neurosci. 2002, 3, 932–942.
J. P. Covy, B. I. Giasson, Neurotoxicology 2011, 32, 622–629.
V. N. Uversky, J. Li, K. Bower, A. L. Fink, Neurotoxicology 2002, 23, 527–536.
Y. Zhou, F. S. Shie, P. Piccardo, T. J. Montine, J. Zhang, Neuroscience 2004, 128, 281–291.
H. S. Boudreau, K. M. Krol, J. K. Eibl, L. D. Williams, J. P. Rossiter, V. P. Palace, G. M. Ross, Aquat. Toxicol. 2009, 92, 258–263.
C. Pifl, M. Khorchide, A. Kattinger, H. Reither, J. Hardy, O. Hornykiewicz, Neurosci. Lett. 2004, 354, 34–37.
T. Cai, T. Yao, G. Zheng, Y. Chen, K. Du, Y. Cao, X. Shen, J. Chen, W. Luo, Brain Res. 2010, 1359, 201–207.
Y. Li, L. Sun, T. Cai, Y. Zhang, S. Lv, Y. Wang, L. Ye, Brain Res Bull. 2010, 81, 428–433.
T. R. Guilarte, Neurotoxicology 2010, 31, 572–574.
K. Martin, T. Huggins, C. King, M. A. Carroll, E. J. Catapane, Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2008, 148, 152–159.
M. Parenti, C. Flauto, E. Parati, A. Vescovi, A. Groppetti, Brain Res. 1986, 367, 8–13.
A. J. Daniels, J. Abarca, Neurotoxicol. Teratol. 1991, 13, 483–487.
G. Biagini, F. Ferraguti, S. Ponzoni, M. Zoli, L. Alboni, G. Toffano, K. Fuxe, L.F. Agnati, 1994.
S. Ponzoni, F. S. Guimaraes, E. A. Del Bel, N. Garcia-Cairasco, Prog. Neuropsychopharmacol. Biol. Psychiatry 2000, 24, 307–325.
S. Ponzoni, L. C. Gaziri, L. R. Britto, W. J. Barreto, D. Blum, Neurosci. Lett. 2002, 328, 170–174.
D. Centonze, P. Gubellini, G. Bernardi, P. Calabresi, Exp. Neurol. 2001, 172, 469–476.
E. Polazzi, B. Monti, Prog. Neurobiol. 2010, 92, 293–315.
F. Zhao, T. Cai, M. Liu, G. Zheng, W. Luo, J. Chen, Toxicol. Sci. 2009, 107, 156–164.
T. Verina, S. F. Kiihl, J. S. Schneider, T.R. Guilarte, Neurotoxicology 2011, 32, 215–226.
N. M. Filipov, R. F. Seegal, D. A. Lawrence, Toxicol. Sci. 2005, 84, 139–148.
J. H. Bae, B. C. Jang, S. I. Suh, E. Ha, H. H. Baik, S. S. Kim, M. Y. Lee, D. H. Shin, Neurosci. Lett. 2006, 398, 151–154.
P. Zhang, K. M. Lokuta, D. E. Turner, B. Liu, J. Neurochem. 2010, 112, 434–443.
A. H. Sadek, R. Rauch, P. E. Schulz, Int. J. Toxicol. 2003, 22, 393–401.
P. K. Pal, A. Samii, D. B. Calne, Neurotoxicology 1999, 20, 227–238.
J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown, R. J. Richardson, Neurotoxicology 1999, 20, 239–247.
H. K. Hudnell, Neurotoxicology 1999, 20, 379–397.
Y. Kim, J. M. Kim, J. W. Kim, C. I. Yoo, C. R. Lee, J. H. Lee, H. K. Kim, S. O. Yang, H. K. Chung, D. S. Lee, B. Jeon, Mov. Disord. 2002, 17, 568–575.
B. A. Racette, L. McGee-Minnich, S. M. Moerlein, J. W. Mink, T. O. Videen, J. S. Perlmutter, Neurology 2001, 56, 8–13.
T. R. Guilarte, Environ. Health Perspect. 2010, 118, 1071–1080.
T. R. Guilarte, N. C. Burton, J. L. McGlothan, T. Verina, Y. Zhou, M. Alexander, L. Pham, M. Griswold, D. F. Wong, T. Syversen, J. S. Schneider, J. Neurochem. 2008, 107, 1236–1247.
T. R. Guilarte, M. K. Chen, J. L. McGlothan, T. Verina, D. F. Wong, Y. Zhou, M. Alexander, C. A. Rohde, T. Syversen, E. Decamp, A. J. Koser, S. Fritz, H. Gonczi, D. W. Anderson, J. S. Schneider, Exp. Neurol. 2006, 202, 381–390.
T. M. Peneder, P. Scholze, M. L. Berger, H. Reither, G. Heinze, J. Bertl, J. Bauer, E. K. Richfield, O. Hornykiewicz, C. Pifl, Neuroscience 2011, 180, 280–292.
K. Sriram, G. X. Lin, A. M. Jefferson, J. R. Roberts, R. S. Chapman, B. T. Chen, J. M. Soukup, A. J. Ghio, J. M. Antonini, Arch. Toxicol. 2010, 84, 521–540.
C. Wider, Z. K. Wszolek, Parkinsonism Relat. Disord. 2007, 13, Suppl 3, S229–232.
Y. X. Yang, N. W. Wood, D. S. Latchman, Neuroreport 2009, 20, 150–156.
S. Lesage, A. Brice, Hum. Mol. Genet. 2009, 18, R48–59.
J. A. Roth, S. Singleton, J. Feng, M. Garrick, P. N. Paradkar, J. Neurochem. 2010, 113, 454–464.
J. A. Roth, Neuromol. Med. 2009, 11, 281–296.
A. D. Gitler, A. Chesi, M. L. Geddie, K. E. Strathearn, S. Hamamichi, K. J. Hill, K. A. Caldwell, G. A. Caldwell, A. A. Cooper, J. C. Rochet, S. Lindquist, Nat. Genet. 2009, 41, 308–315.
J. Tan, T. Zhang, L. Jiang, J. Chi, D. Hu, Q. Pan, D. Wang, Z. Zhang, J. Biol. Chem. 2011, 286, 29654–29662.
T. Pan, S. Kondo, W. Le, J. Jankovic, Brain 2008, 131, 1969–1978.
A. Ramirez, A. Heimbach, J. Grundemann, B. Stiller, D. Hampshire, L. P. Cid, I. Goebel, A. F. Mubaidin, A. L. Wriekat, J. Roeper, A. Al-Din, A. M. Hillmer, M. Karsak, B. Liss, C. G. Woods, M. I. Behrens, C. Kubisch, Nat. Genet. 2006, 38, 1184–1191.
J. B. Schulz, J. Neurol. 2008, 255, Suppl 5, 3–7.
K. K. Dev, H. van der Putten, B. Sommer, G. Rovelli, Neuropharmacology 2003, 45, 1–13.
K. L. Lim, K. C. Chew, J. M. Tan, C. Wang, K. K. Chung, Y. Zhang, Y. Tanaka, W. Smith, S. Engelender, C. A. Ross, V. L. Dawson, T. M. Dawson, J. Neurosci. 2005, 25, 2002–2009.
Y. Mizuno, N. Hattori, H. Mori, T. Suzuki, K. Tanaka, Curr. Opin. Neurol. 2001, 14, 477–482.
A. B. West, D. Maraganore, J. Crook, T. Lesnick, P. J. Lockhart, K. M. Wilkes, G. Kapatos, J. A. Hardy, M. J. Farrer, Hum. Mol. Genet. 2002, 11, 2787–2792.
R. Hilker, C. Klein, M. Ghaemi, B. Kis, T. Strotmann, L. J. Ozelius, O. Lenz, P. Vieregge, K. Herholz, W. D. Heiss, P. P. Pramstaller, Ann. Neurol. 2001, 49, 367–376.
S. A. Oliveira, W. K. Scott, M. A. Nance, R. L. Watts, J. P. Hubble, W. C. Koller, K. E. Lyons, R. Pahwa, M. B. Stern, B. C. Hiner, J. Jankovic, W. G. Ondo, F. H. Allen, Jr., B. L. Scott, C. G. Goetz, G. W. Small, F. L. Mastaglia, J. M. Stajich, F. Zhang, M. W. Booze, J. A. Reaves, L.T. Middleton, J. L. Haines, M. A. Pericak-Vance, J. M. Vance, E. R. Martin, Arch. Neurol. 2003, 60, 975–980.
M. Periquet, M. Latouche, E. Lohmann, N. Rawal, G. De Michele, S. Ricard, H. Teive, V. Fraix, M. Vidailhet, D. Nicholl, P. Barone, N. W. Wood, S. Raskin, J. F. Deleuze, Y. Agid, A. Durr, A. Brice, Brain 2003, 126, 1271–1278.
Y. Higashi, M. Asanuma, I. Miyazaki, N. Hattori, Y. Mizuno, N. Ogawa, J. Neurochem. 2004, 89, 1490–1497.
P. Haeger, A. Alvarez, N. Leal, T. Adasme, M. T. Nunez, C. Hidalgo, Neurotox. Res. 2010, 17, 238–247.
I. Pelizzoni, D. Zacchetti, C. P. Smith, F. Grohovaz, F. Codazzi, J. Neurochem. 2012, 120, 269–278.
M. I. Behrens, N. Bruggemann, P. Chana, P. Venegas, M. Kagi, T. Parrao, P. Orellana, C. Garrido, C. V. Rojas, J. Hauke, E. Hahnen, R. Gonzalez, N. Seleme, V. Fernandez, A. Schmidt, F. Binkofski, D. Kompf, C. Kubisch, J. Hagenah, C. Klein, A. Ramirez, Mov. Disord. 2010, 25, 1929–1937.
F. Kamp, N. Exner, A. K. Lutz, N. Wender, J. Hegermann, B. Brunner, B. Nuscher, T. Bartels, A. Giese, K. Beyer, S. Eimer, K. F. Winklhofer, C. Haass, EMBO J. 2010, 29, 3571–3589.
S. J. Chung, S. M. Armasu, J. M. Biernacka, T. G. Lesnick, D. N. Rider, S. J. Lincoln, A. I. Ortolaza, M. J. Farrer, J. M. Cunningham, W. A. Rocca, D. M. Maraganore, Mov. Disord. 2010, 26, 280–288.
S. Lesage, C. Condroyer, A. Lannuzel, E. Lohmann, A. Troiano, F. Tison, P. Damier, S. Thobois, A. M. Ouvrard-Hernandez, S. Rivaud-Pechoux, C. Brefel-Courbon, A. Destee, C. Tranchant, M. Romana, L. Leclere, A. Durr, A. Brice, J. Med. Genet. 2009, 46, 458–464.
S. Saha, M. D. Guillily, A. Ferree, J. Lanceta, D. Chan, J. Ghosh, C. H. Hsu, L. Segal, K. Raghavan, K. Matsumoto, N. Hisamoto, T. Kuwahara, T. Iwatsubo, L. Moore, L. Goldstein, M. Cookson, B. Wolozin, J. Neurosci. 2009, 29, 9210–9218.
J. R. Adams, H. van Netten, M. Schulzer, E. Mak, J. McKenzie, A. Strongosky, V. Sossi, T. J. Ruth, C. S. Lee, M. Farrer, T. Gasser, R. J. Uitti, D. B. Calne, Z. K. Wszolek, A. J. Stoessl, Brain 2005, 128, 2777–2785.
R. Nandhagopal, E. Mak, M. Schulzer, J. McKenzie, S. McCormick, V. Sossi, T.J. Ruth, A. Strongosky, M. J. Farrer, Z. K. Wszolek, A.J. Stoessl, Neurology 2008, 71, 1790–1795.
Y. Tong, A. Pisani, G. Martella, M. Karouani, H. Yamaguchi, E. N. Pothos, J. Shen, Proc. Natl. Acad. Sci. USA 2009, 106, 14622–14627.
B. Lovitt, E. C. Vanderporten, Z. Sheng, H. Zhu, J. Drummond, Y. Liu, Biochemistry 2010, 49, 3092–3100.
J. P. Covy, B. I. Giasson, J. Neurochem. 2010, 115, 36–46.
J. Salazar, N. Mena, S. Hunot, A. Prigent, D. Alvarez-Fischer, M. Arredondo, C. Duyckaerts, V. Sazdovitch, L. Zhao, L. M. Garrick, M. T. Nunez, M. D. Garrick, R. Raisman-Vozari, E. C. Hirsch, Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583.
Q. He, T. Du, X. Yu, A. Xie, N. Song, Q. Kang, J. Yu, L. Tan, J. Xie, H. Jiang, Neurosci. Lett. 2011, 501, 128–131.
S. J. Lippard, J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, Ca, 1995.
The Biological Chemistry of Magnesium, VCH Publishers, Inc., New York, 1995.
T. Pan, D. M. Long, O. C. Uhlenbeck, Divalent Metal Ions in RNA Folding and Catalysis, in The RNA World, Eds R. Gesteland, J. Atkins, Cold Spring Harbor Press, Cold Spring Harbor, 1993, pp. 271–302.
L. A. Finney, T. V. O’Halloran, Science 2003, 300, 931–936.
M. Pechlaner, R. K. O. Sigel, Met. Ions. Life Sci. 2012, 10, 1–42.
Acknowledgment
This review was supported in part by grants from NIH, ES R01 10563 and ES P32 000267.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Roth, J., Ponzoni, S., Aschner, M. (2013). Manganese Homeostasis and Transport. In: Banci, L. (eds) Metallomics and the Cell. Metal Ions in Life Sciences, vol 12. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5561-1_6
Download citation
DOI: https://doi.org/10.1007/978-94-007-5561-1_6
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5560-4
Online ISBN: 978-94-007-5561-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)