
Abstract
Neurodegenerative diseases affect millions of people globally and are characterized by their poor prognosis and decreased quality of life. In the quest to shed light on the development and progression of neurodegenerative diseases, researchers have examined a plethora of potential instigating factors and implicated many of them in disease pathogenesis. Increasing evidence has shown that environmental exposure plays a major role in the onset and outcome of neurodegenerative disorders. Furthermore, strong associations have been found between genetic variants/mutations and neurodegenerative disease development and prognosis. Yet, most importantly, research has shown that there exists a delicate yet complex interplay between genetic factors and environmental exposures in neurodegenerative pathologies. In this chapter, we outline potential interactions between genetic and environmental elements in the neurodegenerative landscape highlighting the need for a holistic consideration of both. Furthermore, we examine some of the previously established interplays between genetics and exposure in diseases such as Alzheimer’s and Parkinson’s diseases. Finally, we describe potential study designs to identify and evaluate gene-environment nexus interactions discussing the strengths and drawbacks of each.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

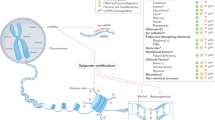

2.1 Introduction
Neurodegenerative diseases are one of the biggest health burdens worldwide; they affect millions of people and often have poor prognosis and decreased quality of life (Mayeux 2003). This group of diseases is characterized by the fact that they are degenerative conditions that affect areas of the central nervous system leading to cognitive or motor impairments depending on the type of nervous cells that are affected by the degeneration (Minghetti 2005). The majority of these diseases have a strong correlation with age and are thus becoming a problem especially in aging populations as their frequency increases with the increase of life expectancy (Logroscino and Tortelli 2015). The reason why this group of diseases is characterized by its heterogeneity is the fact that some of them are complex and multifactorial in nature such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, while others result from single-gene mutations such as Huntington disease and finally others such as prion disease that can be either hereditary, sporadic, or transmitted (Pihlstrøm et al. 2018). Research has shown that certain causative genes can account for inherited forms of ND diseases and that susceptibility genes can also play a major role in sporadic development of the diseases. Time and time again, research has shown that the environment also plays a major part in the development of such diseases as exposure to metals and pesticides, nutrition, head trauma, and certain infectious diseases that can highly increase the risk of developing indeed disorders (Brown et al. 2005). Furthermore, the interaction between environmental exposure and preexisting genetic variants has been established as one of the risk factors as well. Studies performed on Twins (Gatz and Pedersen 2013) as well as animal models are also highly suggestive that the gene-environment nexus can actually strongly impact the way newer degenerative diseases present, manifest, and progress (Hannan 2004). In addition, the role of the mitochondria and oxidative damage in such diseases has been repeatedly demonstrated, and various studies have attempted to clarify whether there is an association between mitochondrial DNA variants and susceptibility to development of ND (Reddy 2009).
2.2 Genetics and Neurodegenerative Diseases
The development in the insights regarding the link between genetics and neurodegenerative disorders reflects the rapid progress of techniques in the field of molecular biology. In fact, the identification of the genes causing Huntington disease in 1983 was one of the pioneering endeavors in identifying the relationship between genetics and disease (Gusella et al. 1983). This success was based on an old technique called restriction fragment length polymorphism (RFLP) that showed that Huntington’s patients had a certain marker on their chromosomes (huntingtin gene, p arm of chromosome 4). With further advancements in the field of molecular biology, scientists were able to identify that this marker was actually due to an increase in the number of trinucleotide repeats in the HTT gene. This process of identifying the exact mutation took around 10 years, showcasing the difficulty of mapping mutations back then. However, this opened up the way for scientists to later on identify many causative/associated genes and mutations for various illnesses (Bates 2005). The technique used for decades later on was called linkage analysis which aims to determine the association between a certain disease and various chromosomal markers. After this linkage, researchers would narrow down the areas of genes that were actually linked with the disease. Thanks to this technique, a multitude of disease-causing mutations was identified for diseases such as prion disease, familial Alzheimer’s disease, and spinocerebellar ataxia 1. This identification of disease-causing genes allowed for screening of a large number of patients with similar phenotype for the presence of the same genes, allowing researchers to begin testing hypothesis regarding etiology, frequency of said mutations, and the degree to which they can be linked to the disease (Pihlstrøm et al. 2018). However, these early strategies, although successful for model diseases that follow mendelian rules of inheritance, were not enough to identify causative mutations in complex sporadic disorders. Not all cases, for example, followed the clear-cut influence of the apolipoprotein E variant whose presence greatly increases the risk for familial Alzheimer’s disease (2–3 times). This variant was fairly common and largely influential which is not the case for most variants that are associated with other complex disorders. The study of such variants required large-scale research studies, computational power, and breakthroughs in the field which luckily enough began appearing as we progressed to the twenty-first century (Hirschhorn and Daly 2005).
2.2.1 Monogenic Forms of Neurodegenerative Diseases: Alzheimer’s and Parkinson’s Diseases as Examples
Alzheimer’s disease: Although most AD cases are complex and sporadic in nature, there exists three familial forms of the disease that follow the rules of Mendelian inheritance. These familial forms are autosomal dominant and result from the inheritance of mutations affecting the formation of amyloid b which leads to the formation of amyloid plaques. The amyloid precursor protein (APP) is expressed and found on the transmembranes of neurons. This protein is later on cleaved and released through the action of a group of enzymes known as secretases (mostly a-secretase). When cleaved, fragments known as Ab40 and Ab42 are produced, and these fragments are highly prone to aggregation. Disease-causing point mutations tip the balance in the Ab40:Ab42 ratio, causing the aggregation of Ab42. Furthermore, duplication of the gene encoding APP has been linked to susceptibility to AD with cerebral amyloid angiopathy. Mutations in genes other than APP, like PSEN1 and PSEN2 (encoding g-secretase), have also been implicated in familial AD, with PSEN1 being the most common mutation followed by APP and then PSEN2 (Pilotto et al. 2013).
Parkinson’s disease: Similarly to Alzheimer’s disease, mutations in the SNCA gene encoding alpha synuclein have been shown to cause familial autosomal-dominant Parkinson’s disease. This was first demonstrated in 1997, and since then a multitude of mutations in this gene have been identified and demonstrated to cause early-onset poor prognosis parkinsonism. The protein, which is the subject of these mutations, has been shown to be a component of intracellular Lewy bodies which play a major role in the development of Parkinson’s disease. The identified point mutations increase the protein’s capacity to oligomerize, causing the formation of fibrils. Similar to Alzheimer’s disease, the multiplication of the SNCA gene also causes familial autosomal-dominant Parkinson’s disease. It has been shown that duplications of the gene cause a familial phenotype that resembles the sporadic one, while the presence of the gene in triplicate form causes early onset and strong cognitive impairments, showing that the amount of the wild-type protein can cause poor prognosis (Alkanli and Ay 2019). Other genes have also been linked to the disease both in autosomal-dominant and autosomal-recessive inheritance patterns. These genes include LRRK2 and VPS35; their exact mechanism of action has not been fully elucidated; however, they have been linked to various critical pathways such as autophagy and protein degradation (Klein and Westenberger 2012).
By looking at the two monogenic forms of Alzheimer’s and Parkinson’s disease, scientists have been able to infer important insights. Firstly, a mutation in a single gene often produces a disease phenotype that is not distinguishable from sporadic illness. Furthermore, even the familial diseases that present with a somewhat universal clinical presentation and inheritance often are the result of different mutations in different genes that affect related pathways. To further add to the complexity, a single gene has the capacity of inducing various phenotypic presentations of a disease. The studies that were discussed previously showed that protein aggregation is a strong deterministic factor in the case of neurodegenerative diseases. The two examples that were discussed show that neurodegenerative diseases can result from structural mutations or expression changes in proteins. These monogenic studies have paved the way for later on mechanistic studies to elucidate the exact role of said genes in pathogenesis (Bekris et al. 2010).
2.3 The Environment and Its Effect
The belief that environmental exposures are strongly implicated in neurodegeneration is not a new one; environmental exposures such as certain types of food, metals, contaminants, pathogenic, and nonpathogenic microorganisms as well as lifestyle can strongly affect a person’s risk in developing neurodegenerative disorders either directly or indirectly.
2.3.1 Effect of Environmental Exposures: The In Utero Example
One such example is how the environment and exposures surrounding a fetus during pregnancy can actually affect its adult life (Gluckman et al. 2008). It has been proven that inadequate environmental conditions during pregnancy as well as infancy can strongly increase the risk of neurodegenerative disorders. The maternal environment can strongly impact the future of her child as during the in utero phase; the fetus is only exposed to the environment through its mother. This is even evident as the growth of the fetus is constrained by the mother’s body size, stature, and parity. During pregnancy, a human fetus can be exposed to hypoxia, metals, hormones, and other substances (Charalambous et al. 2007). It has been proven that exposure to hypoxenima during pregnancy can lead to synaptic dysfunctions in the fetus, causing neurodegeneration (Morales et al. 2008). As for hormonal disturbances, in the case that the placental barrier is compromised, certain hormones such as glucocorticoids can travel through the placenta and affect the fetus, leading to altered programming of hypothalamus-pituitary-adrenal (HPA) (Kapoor et al. 2008). Human studies that follow the effect of environmental exposures on fetal development and neurodegeneration are limited by either their retrospective nature or difficulty of their implementation in the case of longitudinal studies. In animals, a model of prenatal asphyxia has demonstrated that retinal development is impaired due to the reduction of ganglion cells as a result of degeneration which is anticipated to lead to long-term complications (Piscopo et al. 2008). In another animal study on rabbits, placental insufficiency was directly associated with brain damage through the impairment of metabolic processes (van Vliet et al. 2013). Studies have repeatedly shown that environmental exposures on the developing brain have far more detrimental effects than those on the adult brain. In yet another interesting animal study, it was found that when the mother was subjected to lipopolysaccharide treatment during pregnancy, the pups turned out to suffer from loss of dopaminergic neurons, giving insight into how exposure to LPS during pregnancy can increase susceptibility to parkinsonism (Ling et al. 2002). In addition, exposure to metal toxins was found to induce altered levels of antioxidants in rats accompanied by increases in oxidative stress (Erikson et al. 2006). Collectively, these studies showcase how vital is to decipher the role of environmental exposures in utero in determining fetal as well as adult brain health.
2.4 The Gene-Environment Nexus
Research has shown that neurological diseases can be caused by the interaction between genetic and environmental factors. With the advancement in the arsenal of tools of genomic sciences, more evidence points to the fact that genetic modulators can strongly affect the risk and susceptibility to neurodegenerative diseases (Patel 2016). With these advancements, the goal now is to begin standardizing the definition of environmental exposure and how to analyze it. Newer types of studies include exposome studies that analyze the entirety of environmental exposures throughout the individual’s lifespan (Wild 2005). Huge amounts of data are currently being generated both with regard to genetics and genomics as well as exposure; the major challenge remains, including how to study and integrate this data to begin elucidating how certain gene variants can control a human’s response to environmental factors, hence altering their susceptibility to diseases including neurodegenerative ones (Rappaport 2011). This nexus of interaction may actually prove to be incredibly useful in the case of diseases of aging as the accumulation of a lifetime of exposures can strongly alter a populations risk overdeveloping a certain disease (Shulman et al. 2011). Two major examples of this are Alzheimer’s disease and Parkinson’s disease which have been proven to have strong genetic components yet can strongly be affected by certain environmental exposures during a person’s life (Barnes and Yaffe 2011). The interaction between these factors, however, is yet to be elucidated.
The extrapolation of data and the identification of points of interaction between gene and environment are very challenging, and even when this task is done correctly, the challenge remains on identifying how this interaction affects this disease development and progression (Ahmad et al. 2013). For example, the effect of this interaction may actually be very small, and in order to be well observed, it would require expensive studies that integrate a large number of research subjects to acquire their genomic data. Even if such studies are implemented, the acquiring of exposure data especially retrospectively is yet another challenge that needs to be overcome. Retrospective data often suffers from poor quality and this is evident in environment-wide association studies whose challenges include incomplete or biased recall, various confounding variables, difficulty of ascertaining the exact timing and duration of exposure, and finally sampling issues. The challenging nature of such studies suffers from yet another layer of added complexity when we consider that the environmental factors and exposures themselves evolve as human population and industries evolve. Certain environmental factors that are very prominent nowadays were not prominent a few decades ago and vice versa. These challenges can be circumvented to a certain extent by performing animal studies under controlled conditions and the added benefit of the shortened life span of laboratory animals. However, such studies are preliminary at best and can only serve as pointers to potential interactions. These studies are useful when studying already identified variants which cannot allow for the de novo identification of human polymorphisms (Ermann and Glimcher 2012).
Another way to circumvent these challenges is genome-wide association and interaction studies that have robust analysis algorithms allowing for strong predictive power and relatively smaller sample sizes (C. Lin et al. 2015). Furthermore, a lot of interest is currently directed towards the observation of epigenetic changes occurring in response to environmental exposure as potential hints to gene-environment interactions (Maloney et al. 2012). Advances in technologies that can be used for tracking environmental exposure data are also strongly overcoming the challenges of inaccurate reporting, such technologies include wearables and smartphone technologies (Ueberham and Schlink 2018). The interest of governmental agencies in funding research in this area has allowed for the implementation of large-scale prospective studies that are currently providing large amounts of data both on the genetic and environment fronts, allowing for a true exploration of gene-environment interactions (McAllister et al. 2017).
Animal studies are also progressing by the enhancement of animal strains by diversifying their genetic makeup allowing for more complex studies under a controlled lab environment (Schoenrock et al. 2018; Williams et al. 2016). This is exceptionally useful in the area of gene environment interaction studies as mouse models allow for validation of findings found in humans as well as potential identification of novel interactions going hand in hand with large-scale human studies (Jones et al. 2013).
2.5 How to Approach Gene-Environment Interaction Studies in the Neurodegenerative Context
2.5.1 Approach 1: One Gene-One Exposure
In an attempt to elucidate whether certain disease variance can affect individual susceptibility to an environmental exposure, various studies have adopted the one gene-one exposure model (Fig. 2.1) (Dunn et al. 2019). In this type of studies, researchers compare between those who are variant positive and those who are negative and evaluate their odds ratios once exposed to a certain environmental factor. In these studies, it is possible that the accumulation of the exposure along with the presence of the disease variant can both strongly lead to the development of neurodegenerative diseases (Lill 2016). One such example is studies evaluating the effect of dietary interventions in those carrying certain variants in the APOE gene on cholesterol levels and susceptibility to neurodegenerative diseases (Head et al. 2012). Both human and animal studies have shown that a person’s APOE genotype affects their response to various environmental risk factors such as poor diet, obesity, stress, poor lifestyle, and heavy metal exposure by making them more susceptible to the development of neurodegenerative disorders (Pankratz and Foroud 2004). Collectively this evidence shows that certain risk factors that are genetic in nature can act together with environmental exposures and strongly affect this risk. Some other studies have shown that certain APOE genotypes can actually be protective against the effects of certain environmental factors (Wirth et al. 2014). One example is how APOE for carriers benefit from cognitive activity and are relatively protected from Alzheimer’s disease one compared to APOE 2 and 3 carriers (Rajan et al. 2014).

Candidate gene analyses ask whether an exposure or exposures result in differential disease risk in carriers (e.g., “T/T” individuals) or noncarriers (e.g., “A/A” individuals) of a particular known disease risk allele (Dunn et al. 2019)
2.5.2 Approach 2: Gene Datasets—Multiple Exposures
The complexity of the human genome and environmental exposures guarantees that the one gene-one exposure approach is potentially faulty and may not be the most accurate way to begin inferring relevant gene-environment interactions. Several studies have attempted to go beyond this model as an intermediary approach between the previous one and genome-wide associations. For example, a study observing the effect of a multitude of lifestyle choices on a panel on 27 genes associated with Alzheimer’s found that one genotype (SLC24A4) was strongly affected by whether the person was a smoker or a drinker and in turn impacted the person’s risk of developing the disease (E. Lin et al. 2017). In a similar approach, another group looked at the relationship between cardiovascular disease risk factors and another panel of Alzheimer’s-associated genes. They found an interesting correlation between the ABCA7 genotype and right parietal volume indicating a potential relationship between this genotype and susceptibility to cognitive impairment induced by cardiovascular-associated factors (Wang et al. 2017). This type of studies does offer significant advantages to those conferred by one gene-one exposure-type studies as it eliminates certain biases and provides insights as to which pathways may be affected by environmental exposures, thus mediating disease. Yet, despite their usefulness, they do not enable de novo identification of relevant genes/pathways (Holmans et al. 2013).
2.5.3 Approach 3: Observing the Genome
As previously discussed, approaches that start with a pre-identified list of candidate genes and exposures is minimally useful in the de novo identification of gene-environment interactions that affect disease susceptibility. In the case of mapping environmental exposure, the best study design remains to be genome-wide association and interaction studies (GWAIS) (Fig. 2.2). When conducted in the realm of neurobiology, these studies have strikingly identified risk genes and exposures that were previously implicated in diseases of different nature, hinting at the importance of expanding neurobiology research even beyond the genome-wide model. GWAIS have identified interactions between novel genetic modulators and environmental exposures in Alzheimer’s and Parkinson’s (W.-Y. Lin et al. 2019). For example, one of these studies found that glutamate receptor genes and their variants are deterministic factors in a person’s response to caffeine in Parkinson’s disease. One interesting variant was the rs4998386 SNP of GRIN2A which was found to be strongly associated with a 70–80% caffeine-induced reduction in Parkinson’s Disease risk of development (Hamza et al. 2011). These findings were identical in two studies and irreproducible in a third, raising the doubt that this relationship may count on another unidentified factor, further adding to the complexity of the gene-environment interaction in this scenario (Ahmed et al. 2014; Yamada-Fowler et al. 2014). Another GWAIS approach shed light on the relationship between smoking and the risk of Parkinson’s showing that according to the individual genetic landscape, smoking may actually confer protective properties from the disease. Implicated genes included variants of gene known as the synaptic vesicle glycoprotein 2 (SV2C) gene. Homozygous major allele individuals showed a reduction in Parkinson’s disease risk associated with smoking, while those homozygous for the minor alleles showed an increased risk induced by smoking (Hill-Burns et al. 2013). These results were also validated in animal studies where SV2C knockout mice showed drastically different dopamine responses to nicotine than wild-types (Dunn et al. 2017). Despite the promise of GWAIS-type studies, our understanding of the gene-environment nexus will also remain predominantly hindered by the incomplete nature of exposure data in human studies.

GWAIS ask which genetic variants and genomic loci correlate with disease risk given individuals’ exposure to a known disease-relevant environmental factor (Dunn et al. 2019)
2.6 What the Future Holds for the Gene-Environment Nexus in Neurodegenerative Disorders
Evidence has repeatedly shown that genetics have a strong bearing on the risk and development of neurodegenerative diseases and so does environmental exposure. Furthermore, evidence strongly suggests that certain genetic variants modulate a person’s response to environmental exposures and have the capacity of greatly affecting their susceptibility to disease and its prognosis. GWAIS approaches are currently under constant refinement to allow for expanded genome-wide investigations of gene-environment interactions. However, contrary to common belief, the real challenge lies in the tracking and reporting of exposure data that does not suffer from the effects of recall bias and inaccurate reporting.
The apparent solution is large-scale prospective longitudinal studies where the researchers collect exposure data in a timely and accurate manner (Fig. 2.3). Indeed, various studies of this nature have been initiated such as the UK Biobank study which is collecting general exposure data and genomic sequences of approximately 50,000 participants (Sudlow et al. 2015). A similar study is found in the USA under the name “All of Us,” an NIH-funded program collecting massive amounts of genomic, environmental, and health data from a million participants (Lyles et al. 2018). These studies are achieving what previous studies couldn’t in terms of the sheer amounts of data being collected as well as the diversity of their participants and sample size. Furthermore, they are heavily backed with the most recent advances in analysis tools in the field of bioinformatics (Bradley et al. 2018).

GxEWAS take an integrated approach of measuring and determining which exposome-wide and genome-wide factors contribute to the risk of a variety of diseases and how these factors interact across time in order to determine individual risk for any number of diseases (Dunn et al. 2019)
It is inevitable, however, that these studies will be affected by some of the hurdles that affected their predecessors like selection and survival bias or attrition or different confounding variables. This is complemented by the ongoing research and enhancement in the quality of animal studies as a means of evaluating and validating the associations revealed by these studies. Furthermore, a multitude of other factors needs to be considered including duration and age at exposure (C. Lin et al. 2015). The data to be collected from these large-scale studies as well as other cohorts of both human and animal studies are bound to provide valuable insights with regard to the role of gene-environment interactions in neurodegenerative diseases.
References
Ahmad S, Rukh G, Varga TV, Ali A, Kurbasic A, Shungin D, Ericson U, Koivula RW, Chu AY, Rose LM, Ganna A, Qi Q, Stančáková A, Sandholt CH, Elks CE, Curhan G, Jensen MK, Tamimi RM, Allin KH et al (2013) Gene × physical activity interactions in obesity: combined analysis of 111,421 individuals of European ancestry. PLoS Genet 9(7):e1003607. https://doi.org/10.1371/journal.pgen.1003607
Ahmed I, Lee P-C, Lill CM, Nielsen SS, Artaud F, Gallagher LG, Loriot M-A, Mulot C, Nacfer M, Liu T, Biernacka JM, Armasu S, Anderson K, Farin FM, Lassen CF, Hansen J, Olsen JH, Bertram L, Maraganore DM et al (2014) Lack of replication of the GRIN2A-by-coffee interaction in Parkinson disease. PLoS Genet 10(11):e1004788. https://doi.org/10.1371/journal.pgen.1004788
Alkanli N, Ay A (2019) The relationship between alpha-synuclein (SNCA) gene polymorphisms and development risk of Parkinson’s disease. In: Synucleins—biochemistry and role in diseases. https://doi.org/10.5772/intechopen.82808
Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10(9):819–828. https://doi.org/10.1016/S1474-4422(11)70072-2
Bates GP (2005) The molecular genetics of Huntington disease—a history. Nat Rev Genet 6(10):766–773. https://doi.org/10.1038/nrg1686
Bekris LM, Yu C-E, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23(4):213–227. https://doi.org/10.1177/0891988710383571
Bradley WG, Andrew AS, Traynor BJ, Chiò A, Butt TH, Stommel EW (2018) Gene-environment-time interactions in neurodegenerative diseases: hypotheses and research approaches. Ann Neurosci 25(4):261–267. https://doi.org/10.1159/000495321
Brown RC, Lockwood AH, Sonawane BR (2005) Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect 113(9):1250–1256. https://doi.org/10.1289/ehp.7567
Charalambous M, da Rocha ST, Ferguson-Smith AC (2007) Genomic imprinting, growth control and the allocation of nutritional resources: consequences for postnatal life. Curr Opin Endocrinol Diabetes Obes 14(1):3–12. https://doi.org/10.1097/MED.0b013e328013daa2
Dunn AR, Stout KA, Ozawa M, Lohr KM, Hoffman CA, Bernstein AI, Li Y, Wang M, Sgobio C, Sastry N, Cai H, Caudle WM, Miller GW (2017) Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci U S A 114(11):E2253–E2262. https://doi.org/10.1073/pnas.1616892114
Dunn AR, O’Connell KMS, Kaczorowski CC (2019) Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci Biobehav Rev 103:73–80. https://doi.org/10.1016/j.neubiorev.2019.06.018
Erikson KM, Dorman DC, Fitsanakis V, Lash LH, Aschner M (2006) Alterations of oxidative stress biomarkers due to in utero and neonatal exposures of airborne manganese. Biol Trace Elem Res 111(1–3):199–215. https://doi.org/10.1385/BTER:111:1:199
Ermann J, Glimcher LH (2012) After GWAS: mice to the rescue? Curr Opin Immunol 24(5):564–570. https://doi.org/10.1016/j.coi.2012.09.005
Gatz M, Pedersen NL (2013) Study of dementia in Swedish twins. Twin Res Hum Genet 16(1). https://doi.org/10.1017/thg.2012.68
Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359(1):61–73. https://doi.org/10.1056/NEJMra0708473
Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shoulson I, Bonilla E, Martin JB (1983) A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306(5940):234–238. https://doi.org/10.1038/306234a0
Hamza TH, Chen H, Hill-Burns EM, Rhodes SL, Montimurro J, Kay DM, Tenesa A, Kusel VI, Sheehan P, Eaaswarkhanth M, Yearout D, Samii A, Roberts JW, Agarwal P, Bordelon Y, Park Y, Wang L, Gao J, Vance JM et al (2011) Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet 7(8):e1002237. https://doi.org/10.1371/journal.pgen.1002237
Hannan AJ (2004) Molecular mediators, environmental modulators and experience-dependent synaptic dysfunction in Huntington’s disease. Acta Biochim Pol 51(2):415–430. https://doi.org/035001415
Head D, Bugg JM, Goate AM, Fagan AM, Mintun MA, Benzinger T, Holtzman DM, Morris JC (2012) Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol 69(5):636–643. https://doi.org/10.1001/archneurol.2011.845
Hill-Burns EM, Singh N, Ganguly P, Hamza TH, Montimurro J, Kay DM, Yearout D, Sheehan P, Frodey K, Mclear JA, Feany MB, Hanes SD, Wolfgang WJ, Zabetian CP, Factor SA, Payami H (2013) A genetic basis for the variable effect of smoking/nicotine on Parkinson’s disease. Pharmacogenomics J 13(6):530–537. https://doi.org/10.1038/tpj.2012.38
Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6(2):95–108. https://doi.org/10.1038/nrg1521
Holmans P, Moskvina V, Jones L, Sharma M, The International Parkinson’s Disease Genomics Consortium (IPDGC), Vedernikov A, Buchel F, Sadd M, Bras JM, Bettella F, Nicolaou N, Simón-Sánchez J, Mittag F, Gibbs JR, Schulte C, Durr A, Guerreiro R, Hernandez D, Brice A et al (2013) A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum Mol Genet 22(5):1039–1049. https://doi.org/10.1093/hmg/dds492
Jones BC, Miller DB, O’Callaghan JP, Lu L, Unger EL, Alam G, Williams RW (2013) Systems analysis of genetic variation in MPTP neurotoxicity in mice. Neurotoxicology 37:26–34. https://doi.org/10.1016/j.neuro.2013.03.010
Kapoor A, Petropoulos S, Matthews SG (2008) Fetal programming of hypothalamic-pituitary-adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Res Rev 57(2):586–595. https://doi.org/10.1016/j.brainresrev.2007.06.013
Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2(1). https://doi.org/10.1101/cshperspect.a008888
Lill CM (2016) Genetics of Parkinson’s disease. Mol Cell Probes 30(6):386–396. https://doi.org/10.1016/j.mcp.2016.11.001
Lin C, Chu C-M, Lin J, Yang H-Y, Su S-L (2015) Gene-gene and gene-environment interactions in meta-analysis of genetic association studies. PLoS One 10(4):e0124967. https://doi.org/10.1371/journal.pone.0124967
Lin E, Tsai S-J, Kuo P-H, Liu Y-L, Yang AC, Kao C-F (2017) Association and interaction effects of Alzheimer’s disease-associated genes and lifestyle on cognitive aging in older adults in a Taiwanese population. Oncotarget 8(15):24077–24087. https://doi.org/10.18632/oncotarget.15269
Lin W-Y, Huang C-C, Liu Y-L, Tsai S-J, Kuo P-H (2019) Genome-wide gene-environment interaction analysis using set-based association tests. Front Genet 9. https://doi.org/10.3389/fgene.2018.00715
Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong J-S, Carvey PM (2002) In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord 17(1):116–124. https://doi.org/10.1002/mds.10078
Logroscino G, Tortelli R (2015) Epidemiology of neurodegenerative diseases. In: Imaging in neurodegenerative disorders. Oxford University Press. https://oxfordmedicine.com/view/10.1093/med/9780199671618.001.0001/med-9780199671618-chapter-1
Lyles CR, Lunn MR, Obedin-Maliver J, Bibbins-Domingo K (2018) The new era of precision population health: insights for the all of us research program and beyond. J Transl Med 16(1):211. https://doi.org/10.1186/s12967-018-1585-5
Maloney B, Sambamurti K, Zawia N, Lahiri DK (2012) Applying epigenetics to Alzheimer’s disease via the latent early–life associated regulation (LEARn) model. Curr Alzheimer Res 9(5):589–599. https://www.eurekaselect.com/97893/article
Mayeux R (2003) Epidemiology of neurodegeneration. Annu Rev Neurosci 26:81–104. https://doi.org/10.1146/annurev.neuro.26.043002.094919
McAllister K, Mechanic LE, Amos C, Aschard H, Blair IA, Chatterjee N, Conti D, Gauderman WJ, Hsu L, Hutter CM, Jankowska MM, Kerr J, Kraft P, Montgomery SB, Mukherjee B, Papanicolaou GJ, Patel CJ, Ritchie MD, Ritz BR et al (2017) Current challenges and new opportunities for gene-environment interaction studies of complex diseases. Am J Epidemiol 186(7):753–761. https://doi.org/10.1093/aje/kwx227
Minghetti L (2005) Role of inflammation in neurodegenerative diseases. Curr Opin Neurol 18(3):315–321. https://doi.org/10.1097/01.wco.0000169752.54191.97
Morales P, Fiedler JL, Andrés S, Berrios C, Huaiquín P, Bustamante D, Cardenas S, Parra E, Herrera-Marschitz M (2008) Plasticity of hippocampus following perinatal asphyxia: effects on postnatal apoptosis and neurogenesis. J Neurosci Res 86(12):2650–2662. https://doi.org/10.1002/jnr.21715
Pankratz N, Foroud T (2004) Genetics of Parkinson disease. NeuroRx 1(2):235–242
Patel CJ (2016) Analytical complexity in detection of gene variant-by-environment exposure interactions in high-throughput genomic and exposomic research. Curr Environ Health Rep 3(1):64–72. https://doi.org/10.1007/s40572-016-0080-5
Pihlstrøm L, Wiethoff S, Houlden H (2018) Chapter 22—genetics of neurodegenerative diseases: an overview. In: Kovacs GG, Alafuzoff I (eds) Handbook of clinical neurology, vol 145. Elsevier, pp 309–323. https://doi.org/10.1016/B978-0-12-802395-2.00022-5
Pilotto A, Padovani A, Borroni B (2013) Clinical, biological, and imaging features of monogenic Alzheimer’s disease. Biomed Res Int 2013. https://doi.org/10.1155/2013/689591
Piscopo P, Bernardo A, Calamandrei G, Venerosi A, Valanzano A, Bianchi D, Confaloni A, Minghetti L (2008) Altered expression of cyclooxygenase-2, presenilins and oxygen radical scavenging enzymes in a rat model of global perinatal asphyxia. Exp Neurol 209(1):192–198. https://doi.org/10.1016/j.expneurol.2007.09.014
Rajan KB, Skarupski KA, Rasmussen HE, Evans DA (2014) Gene-environment interaction of body mass index and apolipoprotein E ε4 allele on cognitive decline. Alzheimer Dis Assoc Disord 28(2):134–140. https://doi.org/10.1097/WAD.0000000000000013
Rappaport SM (2011) Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol 21(1):5–9. https://doi.org/10.1038/jes.2010.50
Reddy PH (2009) The role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr 14(8 Suppl 7):8–18
Schoenrock SA, Oreper D, Farrington J, McMullan RC, Ervin R, Miller DR, Pardo-Manuel de Villena F, Valdar W, Tarantino LM (2018) Perinatal nutrition interacts with genetic background to alter behavior in a parent-of-origin-dependent manner in adult collaborative cross mice. Genes Brain Behav 17(7):e12438. https://doi.org/10.1111/gbb.12438
Shulman JM, De Jager PL, Feany MB (2011) Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol 6(1):193–222. https://doi.org/10.1146/annurev-pathol-011110-130242
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12(3):e1001779. https://doi.org/10.1371/journal.pmed.1001779
Ueberham M, Schlink U (2018) Wearable sensors for multifactorial personal exposure measurements—a ranking study. Environ Int 121:130–138. https://doi.org/10.1016/j.envint.2018.08.057
van Vliet E, Eixarch E, Illa M, Arbat-Plana A, González-Tendero A, Hogberg HT, Zhao L, Hartung T, Gratacos E (2013) Metabolomics reveals metabolic alterations by intrauterine growth restriction in the Fetal rabbit brain. PLoS One 8(5):e64545. https://doi.org/10.1371/journal.pone.0064545
Wang C, Sun J, Guillaume B, Ge T, Hibar DP, Greenwood CMT, Qiu A, Initiative, The A. D. N (2017) A set-based mixed effect model for gene-environment interaction and its application to neuroimaging phenotypes. Front Neurosci 11. https://doi.org/10.3389/fnins.2017.00191
Wild CP (2005) Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Prev Biomark 14(8):1847–1850. https://doi.org/10.1158/1055-9965.EPI-05-0456
Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, Aebersold R, Auwerx J (2016) Systems proteomics of liver mitochondria function. Science (New York, NY) 352(6291):aad0189. https://doi.org/10.1126/science.aad0189
Wirth M, Villeneuve S, Joie RL, Marks SM, Jagust WJ (2014) Gene–environment interactions: lifetime cognitive activity, APOE genotype, and beta-amyloid burden. J Neurosci 34(25):8612–8617. https://doi.org/10.1523/JNEUROSCI.4612-13.2014
Yamada-Fowler N, Fredrikson M, Söderkvist P (2014) Caffeine interaction with glutamate receptor gene GRIN2A: Parkinson’s disease in Swedish population. PLoS One 9(6):e99294. https://doi.org/10.1371/journal.pone.0099294
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Abdelzaher, H. (2022). The Gene-Environment Nexus: A Holistic Approach to Neurodegenerative Diseases. In: Salama, M. (eds) Nutrigenomics and the Brain. Nutritional Neurosciences. Springer, Singapore. https://doi.org/10.1007/978-981-16-9205-5_2
Download citation
DOI: https://doi.org/10.1007/978-981-16-9205-5_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-9204-8
Online ISBN: 978-981-16-9205-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)