
Abstract
It is crucial to analyze the population structure and genetic diversity of the samples to be studied before a breeding program can be launched. Thirty-one genotypes of papaya germplasm from Spain, Brazil, Ecuador, China, Taiwan, India, and several locations in Bangladesh were genotyped using ten polymorphic simple sequence repeat markers to investigate their molecular diversity as well as their genetic relatedness. The highest numbers of alleles, gene diversity and polymorphic information content were seen in the P3K1024CC and P6K900CC markers. This result confirms the suitability of these markers in the assay of the genetic diversity of papaya genotypes. The model-based population structure and the distance-based assessment categorized the genotypes into six different subcategories. The analysis of molecular variance revealed that 11% of the entire genetic diversity was due to differences among the populations, while 89% was a result of differences within the population. The FST value of 0.136 showed a high level of genetic diversity among the groups alongside a negative FIS (− 0.232) and FIT (− 0.065). The diverse material revealed by our research expands the current papaya genetic resources, which can be used effectively in genomic studies in papaya improvement programs as well as in germplasm conservation studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The beautiful shapes, colors, and sizes of its appealing fruit are some of the reasons why papaya is very popular. Due to the potential benefits of papaya, much attention from growers and researchers has been directed toward the plant. Researchers have chosen papaya as a model plant based on its diploid chromosome number and its relatively small genome size of 372 Mb (Oliveira et al. 2015). Additionally, papaya is the first crop species to be exposed to genome sequencing (Wei and Wing 2008). This plant is the second-most-consumed fruit in Southeast Asia, a point which highlights the importance of papaya. Papaya contains the cysteine protease enzyme papain, which assists the rate of digestion, mainly while acting on protein-rich meals (Adiaha and Adiaha 2017). The fruit also contains significant amounts of calcium, iron, vitamin B complex, potassium, and fiber. The combination of all these benefits makes papaya an important fruit crop within the subtropical and tropical zones.
Apart from the health benefits of papaya, farmers have an interest in planting papaya because of its commercial value. In some countries like Bangladesh, weather conditions are a determining factor in the profitability of any crop. The reason is that these countries frequently experience sudden changes in environmental conditions, which can result in significant production losses for farmers. However, papaya can withstand such challenges as a fruit crop. Papaya plants can survive as long as 20 years, although their commercial life is up to 3 years due to excessive plant height and pathological attack (Jiménez et al. 2014). If papaya is adequately managed, it is a lucrative crop for farmers who use an irrigation method such as the homestead pond for the extended dry summer months. Due to its nutritional value, including being a fundamental source of papain, papaya brings high revenue per unit area.
Carica papaya L. belongs to the Caricaceae family. The Caricaceae family was derived from Africa but spread to Central America over 0.35 billion years ago (Carvalho and Renner 2014). Between 0.27 and 0.19 billion years ago, the species of Caricaceae moved from Central America to South America (Carvalho and Renner 2014). Despite the controversy surrounding the details of its origin, sufficient information has been gathered (Chávez-Pesqueira and Núnez-Farfán 2016) to show that papaya originated from the Caribbean islands of Mesoamerica. Based on its life history, it is believed that C. papaya colonized gradually within the lowlands of rainforests and sustained a genetic diversity with its genetic potential. However, human disturbance in Northern Mesoamerica exposed C. papaya to a high level of threat to its extension and genetic diversity (Chávez-Pesqueira and Núnez-Farfán 2016).
An important precursor to systematic plant breeding is the knowledge of genetic variation, particularly for the establishment of new varieties. The initiation of plant breeding programs requires a sound understanding of the genetic diversity of existing plant material. Moreover, the same knowledge is crucial for the conservation of genetic diversity within a specific gene pool. When morphological and biochemical characterization are combined with molecular characterization, the latter can be a useful device for the recognition of genetic differentiation, population structure, and heterozygosity level. Hence, the use of DNA markers is an option which alleviates time, effort, and cost. Estimation of genotypic differentiation via DNA markers can increase the reliability compared to physiological or morphological characteristics. (Feng et al. 2016; Liu et al. 2018). Researchers have applied several molecular markers for the genetic diversity evaluation of papaya, such as SSR (De Oliveira et al. 2010; Matos et al. 2013; Vidal et al. 2014; Luciano-Rosario et al. 2018), AFLP (Ratchadaporn et al. 2007; Calderón et al. 2016; Chan-León et al. 2017), and RAPD (Madarbokus and Ranghoo-Sanmukhiya 2012; Sudha et al. 2013; Sabara and Vakharia 2018).
In the past, SSR markers or microsatellites have been effectively applied to the identification of molecular variation among the papaya cultivar. Some SSRs have been established to assess the papaya germplasm (Chen et al. 2007; Yu et al. 2009; Blas et al. 2012). Some of the useful properties of SSR markers include DNA profiling, kinship evaluation, and genetic linkage analysis. By considering all these points, we aim to determine the genetic diversity and population structure of papaya germplasm sampled from different locations across the world. This can be used effectively in genomic studies in papaya improvement programs as well as in germplasm conservation studies.
2 Materials and methods
Plant materials –
Thirty-one samples of papaya were taken from India, China, Spain, Ecuador, Brazil, Bangladesh, and Taiwan for this research (Table S1).
DNA extraction of the collected germplasm –
An adjusted CTAB approach (Allen et al. 2006) was used for DNA isolation of the sampled germplasm. In this study, two grams of fresh leaf samples collected from 15-day-old seedlings of each genotype were used. A spectrophotometer was used to quantify the extracted DNA, while the DNAs were run on 0.8% agarose gel to determine their quality. The dilution of the DNA samples was performed using Tris–EDTA, and the working concentration of the DNA was set as 50 ng μL−1.
PCR condition and gel electrophoresis –
The genotypic analysis of the papaya was performed with ten polymorphic SSR markers. PCR amplification was done within a 25 μL reaction mixture which was made up of 25 mM MgCl2, 10× PCR buffer, 10 pmol each of forward and reverse primer, 50 ng template DNA, 0.3 units of Taq DNA polymerase, and 2.5 mM dNTPs. The PCR reactions, which were performed in a Bio-Rad Thermal Cycler, include: initial denaturation at 95 °C followed by 35 cycles of 95 °C for 45 s, annealing at 54–59 °C (Table S2) for 45 s, and extension at 72 °C for 1 min. The last extension was made to operate at 72 °C for 5 min. The PCR products were separated using a 3.5% metaphor agarose gel (Huda et al. 2019).
Statistical analyses –
The data computation for the allele frequency and allele number, gene diversity, and polymorphic information content (PIC) was performed by the use of Power Marker 3.23 software (Liu and Muse 2005). The estimation of genotypic distance was done for all the samples, while the genotypic relation and clustering pattern were shown in an unweighted neighbor-joining (NJ) tree by DARwin 5.0.158 software (Perrier and Jacquemoud-Collet 2010). The analysis of the molecular variance (AMOVA) was conducted with the use of GenAlEx 6.5 (Peakall and Smouse 2006). The analysis of Wright’s F statistics was performed with the same software. STRUCTURE 2.3.3 software (Pritchard et al. 2000) was used for the assessment of the population structure of all sampled genotypes. A triple replication run was done for each K in which the value of K fell between 1 and 10. By using the Monte Carlo chain replicates of 100,000, the burn-in period for every run was 100,000 steps. A web-based program called Structure Harvester (http://taylor0.biology.ucla.edu/structureHarvester/) was used for the determination of the final population. MeV 4.9 software (Howe et al. 2011) was used for the production of the graphic representation of the dissimilarity matrix, while the dissimilarity matrix and the 3D dispersion analysis were conducted with the use of Genes software (Cruz 2013). Sigma Plot 12.0 software (Chaturvedi et al. 2015) was used for the construction of the 3D scatter plot.
3 Results
Polymorphism of SSR markers –
To determine the polymorphism among the 31 papaya genotypes, ten SSR markers were utilized (Supplementary file S3). The ten markers amplified 54 alleles in total (Table 1). Generally, the number of alleles ranged between 4 and 8, while on average, the number of alleles stood at 6. The highest allele number was amplified by the marker P3K1024CC (8) and preceded by the markers CPM1842A5 and P3K168CC, both of which produced seven alleles each. P6K883CC produced the lowest number of alleles (4), followed by the marker P8K395CC, which produced five alleles. The smallest major allele frequency (0.29) was revealed by the marker P6K900CC, while the highest major allele frequency (0.52) was produced by the marker CPM1580CC.
Ten SSR loci were used to evaluate the allelic diversity, and the level of polymorphism among the 31 papaya genotypes, as a result, showed variability among the markers. A PIC range from 0.62 to 0.77 was shown by the studied markers; the marker P3K1024CC showed the highest PIC value at 0.77, followed by the marker P6K900CC, which revealed a PIC value of 0.768. On the other hand, the lowest PIC value (0.62) was shown by the marker CPM1580CC, followed by a 0.64 PIC value found by the marker P8K395CC. The gene diversity ranged between 0.66 and 0.80, with an average value of 0.74. The marker P6K900CC found the highest gene diversity value at 0.80, followed by P3K1024CC (0.79). The lowest gene diversity value was 0.66, which was discovered by CPM1580CC, preceded by the 0.71 estimated by the marker P8K395CC.
Dissimilarity matrix and dispersion analysis –
The genotype G22 was the most genetically dissimilar sample compared to other genotypes in the dissimilarity matrix except for G18, G23, and G24 (Fig. 1). Also, a considerable dissimilarity was shown by G19, G23, G29, and G31 to the other genotypes. The most similar genotypes were G1, G2, G4, G5, G6, G7, G8, G8, G9, and G10, but G3 showed a distance relation with G11, and G4 showed a distance relation with G9. The 3D dispersion graph (Fig. 2) also supports the result given by the dissimilarity matrix.

Dissimilarity matrix showing the genetic distances among the genotypes. The distance between genotypes as indicated by the gradient of color; the red color denotes the highest dissimilarity, and the green color means the lowest genetic distance. Also, the red color represents the diagonal

Genetic distances being indicated by 3D dispersion analysis. The larger dot dispersion shows the prominent genetic divergence of the population. The number 1 to 31 indicated the accessions
Clustering of genotypes based on SSR marker data –
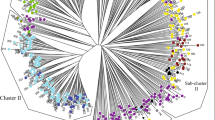
The unweighted neighbor-joining tree shown in Fig. 3 was created using DARwin 5.0.158 software. This figure disclosed the genetic relatedness among the 31 papaya genotypes with the use of ten SSR markers. The unweighted neighbor-joining tree formed six clusters of genotypes; cluster I is made up of G1, G2, G5, G10, G11, G12, G13, G14, G15, and G16, while cluster II includes G3, G4, G6, G7, and G8. Cluster III is made up of only G9; cluster IV contains G17, G18, G19, G21, G24, G25, G26, G27, and G30. Cluster V contains G20, G28, G29, and G31, while cluster VI is made up of G22 and G23.

Unweighted neighbor-joining tree showing genetic relationship among the papaya genotypes using ten SSR markers. The roman numbers represent the clusters, while the numeric numbers stand for the genotypes
Population structure of the studied papaya genotypes –
Evanno’s ΔK (Evanno et al. 2005) was used to find the K value. The highest K value determines the optimum number of populations or groups for the studied genotypes. Among the 31 studied papaya genotypes, the highest K value was 6 (Fig. 4). In evaluating the population structure, the pure and admixtures are the two groups of individuals which can be considered. The pure individual group consists of the individuals beyond 0.8 membership, while those below 0.8 were categorized as admixtures.

K value for the recognition of the probable number of subpopulations among the total population. K values ranged from 1 to 10
The overall population of the 31 studied papaya genotypes was grouped into six subgroups based on the K value (Fig. 5).

A bar plot of papaya genotypes showing the distribution pattern of population structure. Every upright colored bar represents a distinct genotype. The x-axis represents the papaya genotypes, while the y-axis corresponds to the subgroups
Analysis of Molecular Variance –
Significant genetic variations (p > 0.001) among the germplasm were shown by the analysis of molecular variance (AMOVA), which was acquired using the SSR data of the papaya genotypes. The analysis showed that 11% of the total genetic differences were a result of differences between the populations, while 89% of the genetic differences were due to differences within the population (Supplementary file S4).
Fixation Indices –
A − 0.065 value for FIS and a − 0.232 value for FIT were shown by Wright’s F statistics on all ten studied SSR loci (Table 2). The FST value for the polymorphic loci within all lines was shown to be 0.136. The implication of this result is a high level of genetic dissimilarity among individuals. The pairwise FST estimation among the sub-clusters revealed that the groups are considerably diverse from one another.
4 Discussion
Polymorphism of SSR markers –
Compared with previous reports, the average number of alleles per locus (6) of the 31 papaya genotypes collected from different countries of the world was higher (Oliveira et al. 2015; Warnakula et al. 2017). The allele numbers recognized by the studied markers fall between 4 and 8 (Table 1). The results obtained were considerably higher than the outcomes of Warnakula et al. (2017), in which 33 alleles from 13 SSRs with an average of 2.5 alleles were obtained. Oliveira et al. (2015) discovered an average allele number of 4.5 with the use of 59 polymorphic SSR markers for papaya germplasm collected across the world. De Oliveira et al. (2010) conducted research on 30 papaya samples and 18 landraces with 59 SSR markers and discovered a lower average number of alleles per SSR locus (4.02). Ocampo Pérez et al. (2005) studied the papaya germplasm collected from the Caribbean region with 15 SSR primers and found 99 allele numbers, ranging from 3 to 14 with an average value of 6.6. Lesser genetic diversity among the studied papaya samples was shown by the relatively low number of alleles which were amplified by the markers. This occurrence is possible since papaya has a very narrow genetic base (Kim et al. 2002). Nevertheless, it is possible to conclude from this study that the markers P3K1024CC, CPM1842A5, and P3K168CC are valuable markers for further genetic studies on papaya. Also, the three markers can be handy in the establishment of genetic maps and the estimation of linkage disequilibria among alleles.
In this study, the mean PIC value (0.70) obtained is significantly lower than that of Asudi et al. (2010), who found an average PIC value of 0.81 by using seven SSR markers in 42 Kenyan papaya germplasm. A PIC value beyond 0.5 in a given molecular marker denotes that the marker is highly polymorphic and applicable for genetic study (Eltaher et al. 2018). During the characterization of the studied papaya genotypes, all ten SSR markers used revealed a PIC value beyond 0.5. The results of our study suggest that the marker P3K1024CC is the most potent marker in the study of these papaya genotypes.
To determine the power of any SSR marker in a genetic differentiation study, gene diversity is a vital measure (Salem and Sallam 2016). Gene diversity is also a measure of expected heterozygosity (Harris and DeGiorgio 2017; Luo et al. 2019). In this situation, a higher value shows a higher ability of a specific marker to detect polymorphism. The gene diversity found in this study (mean 0.74) is higher than reported by Kaluram et al. (2018), who reported an average gene diversity of 0.59 with the use of ten SSR markers in 23 papaya genotypes.
Dissimilarity matrix –
A crucial factor in the selection of parents for breeding programs is the genetic distance among the germplasm. The reason is that hybridization between distantly related parents will end in more useful progenies in the segregation of generations. Measures such as the distance matrix, dispersion analysis, and cluster analysis revealed that many genotypes show distant relation to other genotypes. Hence, these genotypes have less significance for breeding programs, but their conservation and maintenance can still be useful, as they provide a few beneficial attributes. The distantly related other genotypes could be noted as a useful material to establish a breeding program.
Population structure of the studied papaya germplasm –
The analysis of the population structure revealed the homogeneous groups of individuals and the admixtures among the papaya samples. In all subpopulations, a significant number of admixtures were seen. Compared to the frequently used methodologies such as growth habit and geographic location, this means of classification is more dependable (Kilian et al. 2006; Malysheva-Otto et al. 2006; Yahiaoui et al. 2008) in finding out the population structure. Among the studied germplasm, the relatively high number of admixtures may be a result of the sourcing of the samples from different locations across the globe. For example, the genotypes G17, G18, G19, G22, and G23, which were obtained from exotic sources, displayed more admixtures. However, the local genotypes that show relatively lower admixtures may denote the domestication process. The reproductive biology of papaya is another possible reason for this type of population distribution. Papaya is predominantly a cross-pollinated species, which can result in enhanced variation and more admixture types. Moreover, the population is made up of only hermaphrodite and female fruits, which can lead to the declination of variation. In light of this, morphological and horticultural trait information of the studied germplasm, combined with the outcomes of the structure software, can be more conclusive.
AMOVA for genetic diversity of studied papaya genotypes –
Results of AMOVA indicated that 11% of the total genetic diversity was due to differences between populations, while 89% of the overall genetic diversity was a result of differences within the population. A few possible reasons for the relatively higher variation differences might include the reproductive system (Luo et al. 2007), dispersion of the species among farmers (Ov et al. 2011), and evolutionary changes within the species. The outcome of this study shows that the samples within the subpopulation are genetically diverse and are useful as a potential material of parental line for a breeding program.
Wright’s F statistics –
A moderate genetic diversity (FST = 0.136) within the studied papaya germplasm was observed. A prominent genetic diversity can be seen in a larger population with a higher number of individuals (Chaluvadi et al. 2014). The negative FIS and FIT imply a low level of inbreeding, which can be related to the outcrossing nature of the papaya fruit (Brown et al. 2012).
An analysis of the diversity pattern of papaya genotypes obtained from different nations across the world was performed in this study. Several statistical methods used in this study showed that a series of variations are present among the studied 31 genotypes. Additionally, this study found that the variable materials used can serve as a fundamental source for the establishment of a mapping population and the choice of parental lines within any hybrid breeding program.
References
Adiaha MS, Adiaha MS (2017) Effect of nutritional, medicinal and pharmacological properties of papaya (Carica papaya L.) to human development: a review. World Sci News 2:238–249
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF (2006) A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc 1:2320
Asudi GO, Ombwara FK, Rimberia FK, Nyende AB, Ateka EM, Wamocho LS, Onyango A (2010) Morphological diversity of Kenyan papaya germplasm. Afr J Biotech 9:8754–8762
Blas AL, Yu Q, Veatch OJ, Paull RE, Moore PH, Ming R (2012) Genetic mapping of quantitative trait loci controlling fruit size and shape in papaya. Mol Breeding 29:457–466
Brown JE, Bauman JM, Lawrie JF, Rocha OJ, Moore RC (2012) The structure of morphological and genetic diversity in natural populations of Carica papaya (Caricaceae) in Costa Rica. Biotropica 44:179–188
Calderón MV, Mijangos-Cortés JO, Zavala MJ, Teyer LFS, Quiroz AM, Ortiz MMG, Fernando AC, Francisco EG, Ortiz GF, Santamaría JM (2016) Genetic characterization by amplified fragment length polymorphism (AFLP) markers and morphochemical traits of Carica papaya L. genotypes. Afr J Biotechnol 15:948–959
Carvalho FA, Renner SS (2014) The phylogeny of the Caricaceae. In: Ming R, Moore PH (eds) Genetics and genomics of papaya. Springer, New York, pp 81–92
Chaluvadi SR, Khanam S, Aly MA, Bennetzen JL (2014) Genetic diversity and population structure of native and introduced date palm (Phoenix dactylifera) germplasm in the United Arab Emirates. Trop Plant Biol 7:30–41
Chan-León AC, Quiroz Moreno A, Fuentes Ortíz G, Espadas-Gil FL, Talavera-May CR, Montalvo Fernández G, Ramírez-Prado J, Zapata-Pérez O, Santamaría JM (2017) Genetic profiling of wild accessions of papaya (Carica papaya L.) collected in Yucatan state by using amplified fragment length polymorphism (AFLP) markers. Acta Hortic 1250:69–76
Chaturvedi SK, Zaidi N, Alam P, Khan JM, Qadeer A, Siddique IA, Asmat S, Zaidi Y, Khan RH (2015) Unraveling comparative anti-amyloidogenic behavior of pyrazinamide and D-cycloserine: a mechanistic biophysical insight. PLoS ONE 10:e0136528
Chávez-Pesqueira M, Núnez-Farfán J (2016) Genetic diversity and structure of wild populations of Carica papaya in Northern Mesoamerica inferred by nuclear microsatellites and chloroplast markers. Ann Bot 118:1293–1306
Chen C, Yu Q, Hou S, Li Y, Eustice M, Skelton RL, Veatch O, Herdes RE, Diebold L, Saw J, Feng Y (2007) Construction of a sequence-tagged high-density genetic map of papaya for comparative structural and evolutionary genomics in Brassicales. Genetics 177:2481–2491
Cruz CD (2013) Genes: a software package for analysis in experimental statistics and quantitative genetics. Acta Sci Agron 35:271–276
De Oliveira EJ, Amorim VBO, Matos ELS, Costa JL, da Silva Castellen M, Pádua JG, Dantas JLL (2010) Polymorphism of microsatellite markers in papaya (Carica papaya L.). Plant Mol Biol Rep 28:519–530
Eltaher S, Sallam A, Belamkar V, Emara HA, Nower AA, Salem KF, Poland J, Baenziger PS (2018) Genetic diversity and population structure of F3: 6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front Genet 9:76
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Feng S, He R, Lu J, Jiang M, Shen X, Jiang Y, Wang ZA, Wang H (2016) Development of SSR markers and assessment of genetic diversity in medicinal Chrysanthemum morifolium cultivars. Front Genet 7:113
Harris AM, DeGiorgio M (2017) An unbiased estimator of gene diversity with improved variance for samples containing related and inbred individuals of any ploidy. G3: Genes Genomes Genet 7:671–691
Howe EA, Sinha R, Schlauch D, Quackenbush J (2011) RNA-seq analysis in MeV. Bioinformatics 27:3209–3210
Huda MN, Hasan M, Abdullah HM, Sarker U (2019) Spatial distribution and genetic diversity of wild date palm (Phoenix sylvestris) growing in coastal Bangladesh. Tree Genet Genomes 15:3
Kaluram JP, Singh SK, Goswami AK, Singh P, Hussain Z, Singh AK (2018) Genetic diversity in qualitative and quantitative traits of papaya. Hortic Soc India (Regd.) 75:165–171
Kilian B, Özkan H, Kohl J, von Haeseler A, Barale F, Deusch O, Brandolini A, Yucel C, Martin W, Salamini F (2006) Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol Genet Genomics 276:230–241
Kim MS, Moore PH, Zee F, Fitch MM, Steiger DL, Manshardt RM, Paull RE, Drew RA, Sekioka T, Ming R (2002) Genetic diversity of Carica papaya as revealed by AFLP markers. Genome 45:503–512
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu J, Yang Y, Zhou X, Bao S, Zhuang Y (2018) Genetic diversity and population structure of worldwide eggplant (Solanum melongena L.) germplasm using SSR markers. Genet Resour Crop Evol 65:1663–1670
Luciano-Rosario D, Cruz-Saavedra LA, Siritunga D (2018) Genetic diversity of puerto rican farmer-held papaya (Carica papaya) using SSR markers. HortScience 53:1109–1114
Luo CW, Li K, Chen Y, Sun YY (2007) Floral display and breeding system of Jatropha curcas L. For Stud China 9:114–119
Luo Z, Brock J, Dyer JM, Kutchan TM, Augustin M, Schachtman DP, Ge Y, Fahlgren N, Abdel-Haleem H (2019) Genetic diversity and population structure of a Camelina sativa spring panel. Front Plant Sci 10:184
Madarbokus S, Ranghoo-Sanmukhiya V (2012) Identification of genetic diversity among papaya cultivars in Mauritius using morphological and molecular markers. Int J Life Sci Biotech Pharm Res 1:153–163
Malysheva-Otto LV, Ganal MW, Röder MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:6
Matos ELS, Oliveira EJ, Jesus ON, Dantas JLL (2013) Microsatellite markers of genetic diversity and population structure of Carica papaya. Ann Appl Biol 163:298–310
Ocampo Pérez J, Coppens d’Eeckenbrugge G, Risterucci AM, Dambier D, Ollitrault P (2005) Papaya genetic diversity assessed with microsatellite markers in germplasm from the Caribbean region. In: I international symposium on papaya 740, pp 93–101
Oliveira GAF, Dantas JLL, Oliveira EJ (2015) Development and validation of minisatellite markers for Carica papaya. Biol Plant 59:686–694
Ov I, Espinosa-García FJ, Nuntilde JS, Salvador-Figueroa M (2011) State of the art of genetic diversity research in Jatropha curcas. Sci Res Essays 6:1709–1719
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Molecu Ecol Resour 6:288–295
Perrier X, Jacquemoud-Collet JP (2010) DARwin software. http://darwin.cirad.fr/. Accessed 15 Oct 2019
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ratchadaporn J, Sureeporn K, Khumcha U (2007) An analysis on DNA fingerprints of thirty papaya cultivars (Carica papaya L.) grown in Thailand with the use of amplified fragment length polymorphisms technique. Pak J Biol Sci 10:3072–3078
Sabara P, Vakharia D (2018) Genetic diversity study in papaya (Carica papaya L.) cultivars using RAPD and ISSR markers. NISCAIR-CSIR, Delhi
Salem KF, Sallam A (2016) Analysis of population structure and genetic diversity of Egyptian and exotic rice (Oryza sativa L.) genotypes. Comptes Rendus Biol 339:1–9
Sudha R, Singh DR, Sankaran M, Singh S, Damodaran V, Simachalam P (2013) Genetic diversity analysis of papaya (Carica papaya L.) genotypes in Andaman Islands using morphological and molecular markers. Afr J Agri Res 8:5187–5192
Vidal NM, Grazziotin AL, Ramos HCC, Pereira MG, Venancio TM (2014) Development of a gene-centered SSR atlas as a resource for papaya (Carica papaya) marker-assisted selection and population genetic studies. PloS ONE 9:e112654
Warnakula WADLR, Kottearachchi NS, Yakandawala K (2017) Morphological, SSR and ISSR marker based genetic diversity assessment of mountain papaya germplasm in comparison with Carica papaya. J Natl Sci Found Sri Lanka 45:255–264
Wei F, Wing RA (2008) A fruitful outcome to the papaya genome project. Genome Biol 9:227
Yahiaoui S, Igartua E, Moralejo M, Ramsay L, Molina-Cano JL, Lasa JM, Gracia MP, Casas AM (2008) Patterns of genetic and eco-geographical diversity in Spanish barleys. Theor Appl Genet 116:271–282
Yu Q, Tong E, Skelton RL, Bowers JE, Jones MR, Murray JE, Hou S, Guan P, Acob RA, Luo MC, Moore PH (2009) A physical map of the papaya genome with integrated genetic map and genome sequence. BMC Genom 10:371
Author information
Authors and Affiliations
Contributions
Initial manuscript was written by ASMH as well as the conduction of the experiment; AKMAI examined the study, contributed to experiment structure, and reviewed the manuscript; MGM aided experiment structure, while MH examined the experiment, verified the data analysis, and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hasibuzzaman, A.S.M., Islam, A.K.M.A., Miah, M.G. et al. Phylogeographic diversity and population structure of Carica papaya L. revealed through nuclear microsatellites. Braz. J. Bot 43, 147–154 (2020). https://doi.org/10.1007/s40415-020-00594-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40415-020-00594-8