
Abstract
Neonates and immunosuppressed/immunocompromised pediatric patients are at high risk of invasive fungal diseases. Appropriate antifungal selection and optimized dosing are imperative to the successful prevention and treatment of these life-threatening infections. Conventional amphotericin B was the mainstay of antifungal therapy for many decades, but dose-limiting nephrotoxicity and infusion-related adverse events impeded its use. Despite the development of several new antifungal classes and agents in the past 20 years, and their now routine use in at-risk pediatric populations, data to guide the optimal dosing of antifungals in children are limited. This paper reviews the spectra of activity for approved antifungal agents and summarizes the current literature specific to pediatric patients regarding pharmacokinetic/pharmacodynamic data, dosing, and therapeutic drug monitoring.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
While individualized dosing regimens are optimal, targeted therapy of antifungal agents in children is challenging because of the lack of known pharmacodynamic endpoints for many fungal infections and the unavailability of clinical assays. |
Prescribers should be attuned to the data informing dosing recommendations for antifungal agents and the gaps in the current literature for children. |
This review summarizes the available data on the pharmacokinetics/pharmacodynamics, dosing, and therapeutic drug monitoring of available systemic antifungal agents for treatment and prevention of invasive fungal diseases in children. |
1 Introduction
With the remarkable advances in life-saving and life-prolonging treatments and technologies for premature, immunocompromised, and critically ill infants and children, the number of pediatric patients at risk for invasive fungal disease (IFD) has increased over time. As a result, more and more children are receiving antifungal agents, for either the treatment or the prevention of IFD [1, 2]. Over the past few decades, therapeutic options have expanded, and there has been a shift away from conventional antifungal drugs (e.g., amphotericin products) toward the use of newer agents, such as triazoles and echinocandins [1]. However, pediatric-specific studies are still needed to confirm the therapeutic targets associated with optimal effectiveness and safety for many of these agents, particularly the newer triazole drugs.
Successful treatment of any infection requires the provision of an antimicrobial agent at a dose that achieves therapeutic concentrations at the site of infection. In cases of IFD, substantial interindividual variability in pharmacokinetics (triazoles), narrow therapeutic windows (amphotericin products), and limited oral bioavailability (amphotericin products, echinocandins) complicate antifungal selection and dosing decisions. The maturation of hepatic and renal clearance mechanisms, which can significantly affect the pharmacokinetics of drugs in infants and younger children, further challenges dose optimization in pediatrics [3]. Ultimately, clinicians need to be cognizant of the myriad patient- and drug-related factors influencing antifungal pharmacokinetics in pediatric patients.
The goals of this practical review are to describe the spectrum of activity and pharmacokinetics/pharmacodynamics (PK/PD) of systemic antifungal agents currently available in children. We detail dosing recommendations from infancy to adolescence for drugs currently in use and evaluate the role of therapeutic drug monitoring (TDM) for each. We focus on pediatric data but highlight information that can be extrapolated from adults, as needed. Ultimately, we hope this review will provide clinicians and pharmacists with useful information regarding the current state of antifungal clinical pharmacology in pediatrics.
2 Polyenes
2.1 Spectrum of Activity and Clinical Indications
Amphotericin B (AmB) is the oldest of the systemic antifungal drugs and has long been considered a first-line treatment of IFD because of its potent and broad fungicidal activity. AmB is a polyene macrolide that binds to ergosterol, the principle sterol present in fungal cell membranes, causing membrane disruption, loss of cell contents, and fungal cell death [4]. It is active against most pathogenic yeasts and molds. However, among Candida species, activity against C. lusitaniae [4] and C. auris is variable [5, 6]. Furthermore, while AmB provides the most comprehensive coverage of pathogens from the Mucorales order, increased resistance has been reported with some of the species in this order, such as those in the genera of Cunninghamella and Rhizopus [7].
Four AmB products have been produced for clinical use, all of which have identical spectra of activity: AmB deoxycholate (D-AmB), also known as conventional AmB, and three lipid-based formulations: AmB colloidal dispersion (ABCD), AmB lipid complex (ABLC), and liposomal AmB (L-AmB). Made available in the 1950s, D-AmB was the first formulation for clinical use and served as the cornerstone of antifungal therapy for several decades. Dose-limiting side effects of D-AmB, namely nephrotoxicity and electrolyte disturbances, as well as infusion-related reactions (phlebitis, rigors), were major limitations of D-AmB use and led to the development of lipid formulations in the 1990s. Each of the lipid-based formulations are complexed to lipids in different ways, which protects tissues from the direct toxicity of free AmB.
Nephrotoxicity is the major adverse event of all AmB products and a significant deterrent to their use. The efficacy of the lipid-based formulations of AmB is comparable to that of D-AmB, but safety profiles are better than that of D-AmB [8,9,10]. In a Cochrane review involving four trials and 395 participants, lipid formulations were associated with a significant decreased risk of nephrotoxicity: relative risk 0.47 (95% confidence interval [CI] 0.21–0.90) [11]. Because of their improved safety, lipid preparations are preferred over D-AmB for prevention and treatment of most IFD in children. However, D-AmB remains the product of choice for treatment of neonatal candidiasis [12] because data from observational studies have shown decreased mortality with D-AmB compared with lipid formulations [13], similarly for cryptococcal meningoencephalitis [14]. Lipid preparations, particularly L-AmB, remain the first-line treatment for central nervous system (CNS) candidiasis outside of the neonatal period [12]; mucormycosis [15]; severe endemic mycoses, including pulmonary, disseminated, or CNS blastomycosis [16]; osseous coccidioidomycosis [17]; and acute pulmonary histoplasmosis [18]. AmB is also an alternative therapy for treatment of invasive aspergillosis (IA) in patients who cannot receive voriconazole [19].
2.2 Pharmacokinetics/Pharmacodynamics
AmB exhibits concentration-dependent fungicidal activity and prolonged suppression of fungal growth after the concentration has fallen below the minimum inhibitory concentration (MIC) of the infecting organism [20]. The PK/PD parameter best associated with killing of Candida and Aspergillus species in preclinical studies has been the peak plasma concentration (Cmax)/MIC ratio [20, 21]. As a result, fungicidal activity is promoted through the administration of large dosages that achieve optimal peak concentrations at the site of infection. Unfortunately, dose- and infusion-related toxicities preclude the use of overly large AmB dosages in the clinical setting, and recommended dosages for all AmB formulations are driven based on tolerability.
Each of the AmB products has unique pharmacokinetic properties (Table 1). Conventional AmB is complexed with deoxycholate, a detergent, to make the drug soluble in water. It quickly disassociates from its carrier after infusion and becomes highly (> 95%) protein bound [22]. The pharmacokinetics of D-AmB vary widely among children, with an inverse relationship between age and clearance [23,24,25]. As a result, serum concentrations of AmB in infants are lower than in older children and adults given comparable D-AmB doses [23, 24]. Peak serum concentrations tend to be around 1.5–3.0 mg/L following administration of a 1 mg/kg dose [26], although sizable differences in serum concentrations are seen across pediatric patients [23, 24]. D-AmB has a biphasic plasma concentration profile with an initial half-life of 9–26 h [23, 24, 27] and a terminal half-life as long as 15 days [22]. The plasma half-life has been reported to increase over the course of therapy, particularly in premature infants [24], suggesting that tissue accumulation may occur with prolonged treatment. AmB is not metabolized to any clinically relevant extent, and two-thirds of D-AmB doses are excreted unchanged in the urine and feces [28].
Lipid formulations of AmB were developed to mitigate toxicities related to D-AmB and facilitate the administration of larger dosages. A detailed summary of the different formulations and their properties is beyond the scope of the current review but can be found elsewhere [9, 29]. Compared with D-AmB, both ABCD and ABLC attain lower peak plasma concentrations, smaller area under the plasma concentration–time curve (AUC), larger volumes of distribution (Vd), and shorter terminal half-lives in animal models, likely because of rapid distribution of the drug into tissues [30]. However, these lipid formulations are not well-studied in children. Among three children with hepatosplenic candidiasis treated with ABLC 2.5 mg/kg [31], steady-state plasma concentrations were low at a mean Cmax 1.7–2.0 mg/L on days 7–42 [31]. In a separate population pharmacokinetic study of 28 neonates treated with ABLC [32], clearance was 0.399 L/h/kg, resulting in plasma concentrations similar to those in older children and adults. Meanwhile, in a study involving five children aged < 13 years treated with ABCD 7–7.5 mg/kg [33], pharmacokinetic parameter estimates were comparable to those in children aged > 13 years and adults receiving the same dosages [33].
Compared with D-AmB, L-AmB has a lower Vd [28] and achieves higher Cmax and larger AUC [30]. At a dosage of 5 mg/kg, mean day-1 AUC0–24 was 351 ± 445 µg/mL × h among 13 immunocompromised pediatric patients; this contrasts with a mean AUC0–24 of 24.1 µg/mL × h in children treated with D-AmB 1 mg/kg [26]. Unlike D-AmB, which circulates predominantly as protein-bound (e.g., biologically inactive) drug, L-AmB circulates in three forms: unbound, protein-bound, and liposome-associated drug. While total plasma concentrations are high with L-AmB, the majority of the drug is sequestered within liposomes [22], resulting in a very low unbound fraction (0.005) in plasma [34]. The high fraction of liposome-associated AmB leads to a prolonged circulating half-life and protects individuals from direct toxic effects of free AmB while providing a depot for delivery of AmB to tissues and fungal targets over an extended period [22, 34]. Hence, L-AmB activity is believed to persist long after cessation of therapy.
Complexing AmB into lipids has significant effects on drug distribution to tissues. All formulations of AmB distribute well into the liver and spleen because of uptake by circulating macrophages [35, 36] but have distinct intrapulmonary disposition patterns [37]. Compared with other formulations, ABLC distributes best to lung tissue in animal models [37], achieving concentrations in lung tissue several-fold that of plasma. The lung tissue:plasma ratio for all other AmB formulations is < 1 [37]. However, epithelial lung fluid (ELF) concentrations in critically ill adults are comparable among lipid-based AmB products [38]. The impact of the differential distribution of AmB products in lung tissue and ELF on therapeutic outcomes is unknown.
Lipid preparations of AmB were specifically designed to be renoprotective, raising concerns about their effectiveness in the treatment of fungal urinary tract infections. In a study of 30 neonates with invasive candidiasis (IC) [32], AmB concentrations in the urine following ABLC 2.5–5.0 mg/kg were higher than the MIC for most Candida isolates [32]. Despite these findings, clinical failures with lipid AmB formulations have led to continued recommendations against the use of these products in the treatment of fungal urinary tract infections [39].
Penetration of AmB products into the CNS is of particular clinical importance. However, recommendations regarding the preferred AmB agent for treatment of various CNS infections are conflicting, which is largely driven by the paucity of comparative effectiveness studies rather than demonstration of clinical superiority of one agent over another. In the USA, D-AmB is the preferred initial drug for treatment of CNS candidiasis in infants [12], but D-AmB and L-AmB are given equivalent B-II recommendations in Europe [40]. Meanwhile, D-AmB remains the drug of choice for treatment of cryptococcal meningitis in all ages [14]. However, L-AmB is the preferred agent for treatment of CNS infections in children outside of the neonatal period, including CNS candidiasis [12, 40], mucormycosis [15], and histoplasmosis [18]. In a preclinical rabbit model of Candida meningoencephalitis, L-AmB achieved significantly higher brain tissue concentrations than the other AmB products, whereas cerebrospinal fluid (CSF) concentrations were comparable across all of the products [30]. In this study, D-AmB and L-AmB were equally effective at treating Candida meningoencephalitis and more effective than ABCD or ABLC [30].
2.3 Pediatric Dosing
Each of the four AmB products have unique pharmacological characteristics, and the specific dosage differs by agent (Table 1). Despite these differences, dosing is weight based according to actual body weight for each agent, without a maximum recommended dose [41]. However, a recently published population pharmacokinetics study of L-AmB in morbidly obese adult patients suggested that a fixed dose of 300 or 500 mg may be more appropriate than 3–5 mg/kg for individuals > 100 kg [42], as clearance is not affected by body weight. All of the AmB products are administered once daily regardless of age and, since only small amounts of AmB are excreted in urine and bile, dose adjustments are not required in the setting of renal or hepatic dysfunction. AmB is also not dialyzed, so doses of AmB products do not need to be adjusted in patients receiving renal-replacement therapy. If possible, D-AmB should be avoided in the setting of known kidney disease/injury since it is the most nephrotoxic of the formulations.
Standard dosing of D-AmB in neonates and children is 1 mg/kg/dose, but dosages as high as 1.5 mg/kg could be considered in serious or resistant infections. To reduce the likelihood of infusion reactions, D-AmB should always be infused as a slow infusion (over at least 2–6 h). Some studies have also reported a decreased risk of nephrotoxicity with administration of D-AmB as a continuous infusion [43], although this finding is not universal [44]. While AmB demonstrates concentration-dependent killing, the use of continuous infusions has not been associated with inferior microbiologic or clinical outcomes [44].
Similar to D-AmB, serum concentrations of L-AmB were lower in infants and children than in adults given comparable doses in one report [45]. However, data are conflicting, as a more recent study found that L-AmB pharmacokinetics were similar in adult and pediatric patients [46]. Despite the significant interpatient variability in drug concentrations of L-AmB in children, evidence is insufficient to support different dosing in pediatric and adult patients. Higher L-AmB dosages should be considered when treating resistant or more serious infections in children, such as CNS infections. A dosage of L-AmB 6 mg/kg is recommended for treatment of cryptococcal meningitis to ensure adequate CNS penetration [47]. Meanwhile, dosages of 5–10 mg/kg are recommended by European guidelines for treatment of CNS mucormycosis in children [15]. Dosages > 5 mg/kg demonstrate nonlinear pharmacokinetics in children, and significantly higher drug exposures are attained with these dosages than at dosages < 5 mg/kg [48, 49]. Thus, whether other indications exist for which dosages > 5 mg/kg should be used is unclear. Pediatric data are insufficient to identify specific clinical scenarios in which individualized (i.e., higher or lower) dosages of ABCD and ABLC are warranted.
2.4 Therapeutic Drug Monitoring (TDM): Adverse Events
TDM is not generally available for AmB for several reasons. First, no well-established PK/PD targets have been associated with improved clinical outcomes for any of the AmB products. Although AmB is concentration dependent, and higher Cmax/MIC ratios have been reported in children successfully treated with L-AmB [50], specific targets have not been established to inform dose adjustments for AmB products. Second, while AmB products are associated with nephrotoxicity, toxicodynamic thresholds have also not been specified. Lastly, because the AmB product being used will dictate what type of drug measurement assay should be performed—total drug, protein-bound drug, liposome-associated drug, unbound drug—AmB concentrations are not easily interpretable. For TDM, it is important to be able to accurately identify the active fraction of total drug concentrations (i.e., with L-AmB, both liposome-associated and unbound drug are biologically active). Therefore, assays need to be able to specify different forms of AmB to inform dose adjustments.
Nephrotoxicity and electrolyte wasting are the principal adverse events associated with AmB administration. Nephrotoxicity occurs in 15 to > 50% of children treated with D-AmB [51], although nephrotoxicity is less frequent in children than in adults [51]. AmB-associated nephrotoxicity clinically manifests as increased blood urea nitrogen and serum creatinine, as well as electrolyte wasting [52, 53], primarily in the form of potassium wasting. Hypokalemia requiring potassium supplementation occurs in up to 40% of children treated with high-dose L-AmB (> 3 mg/kg) therapy [46, 49, 54]. Therefore, close laboratory monitoring and avoidance of other nephrotoxic medications, when possible, is advised in all patients treated with AmB products.
Infusion-related reactions are also encountered with administration of AmB products, particularly when administered as a rapid infusion. Fever, rigors, chills, myalgias, arthralgias, and nausea are common and believed to be due to histamine or cytokine release in response to therapy [55]. Hypotension, hypoxia, and cardiac arrhythmias are much rarer. Among the AmB products, infusion-related toxicities are particularly problematic for ABCD. In one trial [56], infusion-related events occurred in more than half of recipients of ABCD, leading some guidelines to discourage its use [19].
3 Azoles
3.1 Spectrum of Activity and Clinical Indications
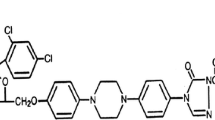
The azole antifungals are classified into two distinct groups: imidazole and triazole antifungals. Structurally, the main difference between the two groups is the number of nitrogens in the 5-membered ring (Fig. 1), where imidazoles have two nonadjacent nitrogens and triazoles have three nitrogens. However, the mechanism of action for both classes of azole antifungals is to inhibit the cytochrome P450 (CYP)-dependent 14-α-sterol demethylase, which interrupts ergosterol biosynthesis of fungal cell membranes and inhibits cell growth [57].

Core structure of azole agents
The clinical indications of imidazoles are mostly limited to topical uses because of their spectrum of activity, adverse effect profile when systemically administered, potency, or solubility [58]. Therefore, imidazoles are frequently administered as topical formulations for the treatment of dermatophytes and vaginal or oral candidiasis (Table 2). Ketoconazole is the only imidazole administered both topically and systemically. However, because of its drug–drug interaction profile, it has fallen out of favor compared with triazole antifungals and is no longer administered systemically in developed countries because safer alternatives are available.
Triazole antifungals have an improved spectrum of activity compared with imidazoles. Fluconazole was the first triazole developed and has activity against most yeasts and thermally dimorphic fungi (those that present as yeasts in temperatures > 37 °C), such as Histoplasma spp. and Blastomyces spp. [59]. It is used extensively in neonates for the prevention and treatment of IC [60]. Newer triazoles, such as itraconazole, posaconazole, and voriconazole, have extended spectra of activity against invasive filamentous fungi, such as Aspergillus spp., but resistance has begun to emerge [61, 62]. The most recently developed second-generation triazole, isavuconazole, was developed to overcome the resistance that limits the efficacy of triazole treatment. However, studies to establish the clinical role of isavuconazole in children are limited. Clinical indications for triazole antifungals differ by agent (Table 3).
3.2 Pharmacokinetics/Pharmacodynamics
The efficacy of azoles is concentration independent [63], with the primary pharmacodynamic endpoint associated with clinical outcomes after azole administration being the exposure to MIC ratio, or AUC0–24/MIC. Azoles also exhibit significant post-antifungal effects [63]. For IC, clinical success is achieved when the AUC0–24/MIC of the unbound azole is > 25 [64]. This averages out to an azole unbound concentration close to the MIC of 1 over 24 h [64]. In contrast, for Aspergillus infections, the proposed AUC/MIC endpoint should be between 2 and 11 [64].
Pharmacokinetic profiles for the triazole antifungals vary. Fluconazole is a hydrophilic compound with low protein binding compared with other agents. It is well-absorbed, and its hydrophilicity limits its Vd to a volume similar to that of total body water. It readily passes through the blood–brain barrier and has a concentration in CSF up to 80% of that observed in the plasma in adults [65]. Fluconazole is bound primarily to α1-acid glycoprotein [66] and undergoes only minimal metabolism (~ 11%) by UGT2B7 [66, 67]. Overall, fluconazole is eliminated primarily unchanged, with 80–90% of parent eliminated in the urine [66, 68].
Itraconazole is a weak base that is highly lipophilic with poor water solubility. Bioavailability varies widely according to formulation. Capsule formulations require a low gastric pH for dissolution, so absorption is appreciably affected by gastric acidity [69, 70]. Coadministration with an H2-receptor antagonist, such as famotidine or ranitidine, decreases both the Cmax and the AUC0–24 by approximately half [71]. A lower gastric pH, such as after a meal, along with longer gastric emptying time and a higher fat content, doubles the bioavailability compared with the fasted state and increases the exposure by > 160% [72]. Itraconazole has a highly variable pharmacokinetic profile, making it difficult to achieve target concentrations. After oral administration, it has lower accumulation in children aged < 12 years than in adults, with the youngest children exhibiting the lowest plasma concentrations, which could be due to maturation in intestinal metabolism or absorption [73, 74]. Another study of itraconazole in children also demonstrated high pharmacokinetic variability and demonstrated a correlation between itraconazole pharmacokinetics and ethnicity and sex [75]. Itraconazole is highly protein bound, mostly to albumin and also to red blood cells [76]. Despite this high protein binding, it distributes extensively into tissues because of its lipophilic nature, which is shown by its high Vd and concentrations two to three times higher in tissues than in plasma [77]. However, it poorly distributes into the CSF, eye fluid, and saliva [77]. It undergoes metabolism by CYP3A4 to 30 different metabolites, with hydroxyl-itraconazole being the primary metabolite that also displays antifungal activity. It has negligible renal elimination of either parent or metabolites, with most elimination into the feces [76].
Voriconazole is a structural analog to fluconazole but has a wider spectrum of activity. It was the first triazole to demonstrate superior efficacy and safety to D-AmB in the treatment of IA [78] and is now the first-line treatment for IA in both children and adults [19]. In adults, it is well-absorbed, with a bioavailability of approximately 96% [79]. Absorption is decreased when administered with food, with a reduction in Cmax and AUC0–24 of up to 60 and 80%, respectively [80]. It is extensively metabolized in the liver by CYP3A4, CYP2C19, CYP2C9, and flavin-containing monooxygenase 3 [79, 81, 82]. Almost all of its metabolites, including the main circulating metabolite, voriconazole N-oxide, are renally eliminated [79]. Studies have suggested that polymorphisms in CYP2C19, including poor and ultrarapid metabolizers, contribute in part to this high variability [83, 84]. The pharmacokinetics of voriconazole in children differs significantly from that in adults. Overall, variability of AUC, Cmax, and clearance ranges from 32 to 175% in adults and children [79, 85]. While bioavailability is high in adults, it is significantly reduced to 44–65% in children [86, 87]. One physiologically based pharmacokinetic model suggested that first-pass intestinal metabolism could be responsible for the lower bioavailability in pediatric patients [88]. In children, voriconazole has linear pharmacokinetics, which has been attributed to the higher abundance and capacity of hepatic CYP2C19 and FMO3 in children than in adults, yielding a clearance threefold higher in children aged 2–12 years compared with that of adults [82]. In preclinical studies [89], autoinduction has been observed, a process by which metabolism of the drug increases over time; this has also been reported in clinical cases with declining concentrations over time [90, 91].
Posaconazole was initially derived from itraconazole and is also highly lipophilic. It is available in a delayed-release tablet, oral suspension, and intravenous formulation. Posaconazole’s lipophilicity allows it to distribute extensively into the tissues, conferring a high Vd and a long terminal half-life. But, as with itraconazole, it is highly protein bound to albumin, and its penetration into CSF fluid is poor. Posaconazole lipophilicity also contributes to large variability in pharmacokinetic parameters, such as clearance and bioavailability, which can vary between subjects by up to 50 and 80%, respectively [92,93,94,95]. Two studies demonstrated that the clearance of posaconazole in children aged 6 months to 13 years was approximately 0.8 L/h/kg [96, 97], an almost fourfold increase compared with adults, and variability between subjects was > 60%. Posaconazole undergoes hepatic metabolism by glucuronidation, but only to a small degree, with approximately 17–34% of the total dose converted to glucuronide metabolites and the rest remaining unchanged as the parent compound is eliminated primarily through the feces [98, 99]. Despite not requiring the CYP450 pathway for metabolism, posaconazole is a potent inhibitor of CYP3A4 [100, 101].
Overall, posaconazole absorption is affected by meals for both the suspension and the tablet formulations, increasing the bioavailability up to 168–290% depending on the fat content of the meal [102]; administration with a high-fat meal increases the gastric residence time and increases solubility. However, bioavailability is saturable such that increasing the dose decreases the percent absorbed [96]. As a result, the bioavailability of posaconazole increases when the total daily dose is divided over multiple doses, with a two- and threefold increase after administration every 12 and 6 h, respectively [93]. There are important differences in the bioavailability of posaconazole between the suspension and delayed-release tablet. In a trial of posaconazole as prophylaxis in hematopoietic cell transplant (HCT) recipients, trough levels were significantly higher in children receiving the tablet than in those receiving the suspension [103]. A recently published nonrandomized trial reported that dosages as high as 18 mg/kg/day divided every 8 h failed to achieve a therapeutic target of Cave of 500–2000 ng/mL in 90% of children treated with the oral suspension [104]. Similarly, simulations performed in a separate study reported that 200 mg in tablet form taken three times daily resulted in 72% probability of target attainment (minimum plasma concentration [Cmin] > 1 mg/L) for children aged 7–12 years, whereas the same dosage in suspension form achieved this target in roughly 40% [96]. Meanwhile, a recently presented abstract reported that over 90% of children aged 2–17 years reached Cave of 500 ng/mL when administered a novel powder for oral suspension at 4.5 mg/kg/day [105], although this formulation is not yet commercially available.
Isavuconazole is the active metabolite of the prodrug isavuconazonium sulfate, a water-soluble prodrug cleaved and almost entirely cleared by plasma esterases [106]. Isavuconazonium is cleared in 98–99% of adult patients within 1–2 h after the start of intravenous administration [107, 108]. After oral administration, the prodrug is hydrolyzed in the intestinal lumen with no quantifiable concentration of the prodrug in the plasma but a high bioavailability of isavuconazole [106]. Isavuconazole, the active moiety, has a long elimination half-life of approximately 56–130 h once absorbed and does not reach steady state until day 14 with once-daily dosing [106, 108]. It is highly protein bound to albumin, with high bioavailability, and undergoes extensive hepatic metabolism [108, 109]. Exposure and half-life increase significantly with mild to moderate hepatic impairment, but dosage adjustments are not recommended because of the morbidity associated with IFDs. At the time of writing, no pharmacokinetic data for children have been published, as pediatric trials are ongoing.
3.3 Pediatric Dosing
3.3.1 Fluconazole
Fluconazole demonstrates a higher clearance in children than in adults, with a half-life of 20 versus 30 h, respectively [110]. Vd is much higher in neonates than in older children or adults, which is reasonable given the hydrophilicity of fluconazole and the relative total body water of neonates compared with older populations [110]. For neonates, it is recommended to administer a loading dose of 25 mg/kg followed by 12 mg/kg/day to achieve target fluconazole plasma concentrations in IC [110, 111]; there is no such loading dose recommendation for children outside of the neonatal period. Oropharyngeal candidiasis is treated with lower dosages: 6 mg/kg on day 1 followed by 3 mg/kg/dose once daily. Dosing is the same for intravenous and enteral formulations.
3.3.2 Itraconazole
The current recommendation for itraconazole dosing in children is 3–5 mg/kg/day to maintain a trough concentration of > 0.5 mg/L [112]. However, studies have demonstrated that even a 5 mg/kg dose does not reliably produce goal trough concentrations in children [113]. In fact, one study suggested that a dose of 8–10 mg/kg divided over two doses reached target trough concentrations better than the recommended dose of 5 mg/kg/day [114]. Given the high variability in absorption, and significant differences in bioavailability of oral formulations, TDM is warranted.
3.3.3 Voriconazole
Differences in clearance mean that recommended dosages of voriconazole are roughly twofold higher in children than in adults. Weight-based oral dosing for children aged > 2 years is 9 mg/kg twice daily (maximum of 350 mg total). Meanwhile, 8 mg/kg twice daily is used as maintenance dosing with the intravenous formulation. Population pharmacokinetic modeling has suggested that higher dosages (9 mg/kg three times daily for 3 days) may more rapidly attain therapeutic concentrations than current twice-daily dosing without notable drug accumulation [115], but this dosage has not been fully evaluated. Similarly, optimal dosing has not been established in children aged < 2 years, although limited studies have suggested that higher doses may be necessary to maintain adequate trough concentrations [116]. Because parenteral voriconazole contains the excipient sulfobutyl ether β-cyclodextrin, which can accumulate in patients with renal impairment, intravenous voriconazole should be avoided in patients with creatinine clearance < 50 mL/min.
3.3.4 Posaconazole
To date, posaconazole is only approved for use in children aged ≥ 13 years. The dosage in this age group is the same as in adults and varies by formulation. Less is known about optimal dosing in younger children. To our knowledge, only a few studies aimed to elucidate the pharmacokinetics and determine the dosing of posaconazole in children aged < 13 years [96, 104, 105]. In a study by Boonsathorn et al. [96] using TDM data collected via routine clinical care, modeling and simulations were performed to evaluate dosing needed to achieve targeted trough concentrations. The authors found low bioavailability of the oral suspension and recommended that children aged 6 months to 6 years receive 200 mg suspension four times daily and children aged 7–12 years receive 300 mg suspension four times daily [96]. The finding of low serum concentrations in young children treated with oral suspension is consistent with a recent study by Arrieta et al. [104], which reported that oral suspension at 12–18 mg/kg/day in two to three divided doses failed to achieve a target of Cave of 500–2500 ng/mL in > 90% of children aged 2–17 years; no specific dosing recommendations were given by these authors. Boonsathorn et al. [96] also included recommendations about dosing of the delayed-release (“gastro-resistant”) tablet, but few pharmacokinetic samples (n = 12) were included from subjects taking this formulation, precluding conclusions about the optimal dosing of delayed-release tablets in children aged < 13 years. Meanwhile, data from an open-label, dose-escalation trial of both an intravenous formulation and a novel powder for oral suspension showed that > 90% of children aged 2–17 years achieved target Cave > 500 ng/mL at dosing of 4.5–6 mg/kg/day with both formulations [105].
As with adults and older children, significant differences in drug concentrations are achieved with the various posaconazole formulations. As such, dosing will likely differ by formulation when this drug is approved in children aged < 13 years. Until dosing is better determined, TDM and concentration-dependent dose adjustments may be beneficial if this drug is used in younger children. Similar to voriconazole, the intravenous formulation of posaconazole contains cyclodextrin, which can accumulate in patients with renal function impairment. Use of the intravenous formulation should be based on a careful risk/benefit assessment in patients with creatinine clearance < 50 mL/min.
3.3.5 Isavuconazole
The optimal dosage of isavuconazole in children has not been established. However, a recent conference abstract reported that an intravenous dose of 10 mg/kg (maximum 372 mg) administered to children aged 1–18 years produced exposures similar to those in adults [117]. In adults, dosing of the intravenous and enteral formulations are the same. Because its intravenous formulation does not contain cyclodextrin, dosages do not need to be adjusted in patients with renal impairment [118], unlike with voriconazole.
3.4 TDM: Adverse Events
TDM is used for triazole agents to optimize clinical outcomes and limit adverse effects. Although the AUC/MIC ratio is the best determinant for efficacy, AUC correlates well with trough concentrations for azoles, as determined by linear regression [119], so trough concentrations are most often used for TDM. Low variability in fluconazole pharmacokinetic parameters decreases the utility of TDM for this agent and, therefore, is not routine. However, van der Elst et al. [120] reported that 40% of critically ill pediatric patients with cancer exhibited subtherapeutic fluconazole Cmin concentrations (< 11 mg/L) and, therefore, TDM should be considered in this population because of the higher mortality risk for invasive fungal infections [120].
Voriconazole trough concentrations between 1 and 6 mg/L have demonstrated improved clinical outcomes while minimizing adverse effects [121]. Dose adjustments after TDM improves target attainment in adult patients [122], but frequent TDM may be required in children because of the higher variability in pharmacokinetics observed in this population [123]. Voriconazole levels should be measured every 3–5 days until appropriate concentrations are attained. Additionally, if voriconazole is administered for a prolonged period (i.e., > 2 months), repeat drug concentrations should be obtained because autoinduction can lead to subtherapeutic concentrations over time [90, 91].
A similar practice is occurring with posaconazole because of the high variability of absorption and clearance in children. Goal trough concentrations of ≥ 0.7 mg/L for prophylaxis and ≥ 1 mg/L for treatment are recommended [124, 125]. Because of the improved bioavailability of posaconazole tablets, TDM can be performed after 3 days, as with the intravenous formulation, whereas steady state may not be achieved until > 7 days with the oral suspension. The TDM targets for itraconazole for both prophylaxis and treatment are > 0.5 mg/L [126], and monitoring should occur 5–7 days after initiation of therapy or with dose adjustments. The exposure–response profile has not been fully elucidated for isavuconazole, so TDM targets have not been established.
It is noteworthy that all of the triazoles demonstrate clinically significant interactions with hepatic CYP enzymes to varying degrees, mostly as inhibitors [127]. This can result in increases in other hepatically metabolized drugs, such as immunosuppressive drugs [128]. Triazole dosages may need to be adjusted when coadministered with other CYP-inducing or -inhibiting medications, and close monitoring of serum levels is important. Isavuconazole has fewer drug–drug interactions than other azoles: in a study of adult HCT recipients, isavuconazole only modestly affected levels of tacrolimus and sirolimus [129].
Hepatotoxicity is the most notable side effect of triazole agents, although the incidence of hepatotoxicity with azoles is similar to that seen with AmB products [130]. Visual disturbance and rash/photosensitivity are unique side effects of voriconazole compared with other azoles, occurring in as many as 45 and 8% of adults, respectively [78, 131]. Furthermore, voriconazole is a known photosensitizing agent, and multiple studies have demonstrated that voriconazole exposure produces a higher risk for developing cutaneous squamous cell carcinomas, even after adjusting for sun exposure [132,133,134]. Cancer risk has correlated with duration of voriconazole exposure and fairer skin [135]. A more complete list of toxicities can be found in Table 4.
4 Echinocandins
4.1 Spectrum of Activity and Clinical Indications
Two echinocandin agents are currently approved for use in children in the USA and Europe: caspofungin and micafungin. Clinical use of these agents has increased substantially in recent years among hospitalized children in the USA [1]. Anidulafungin, the most recently licensed agent in this class, does not yet have a labeled pediatric indication. All three commercially available echinocandin agents exert activity by inhibiting β(1-3)-glucan synthase activity and preventing synthesis of the fungal cell wall [136, 137]. They demonstrate similar spectra and degree of activity with potent fungicidal activity against yeasts, most notably Candida species [138, 139], as well as fungistatic activity against Aspergillus species [140, 141]. They have little to no activity against Cryptococcus neoformans, Trichosporon species, and Saccharomyces cerevisiae [142], nor against species in the Mucorales order [143]. Echinocandin resistance in Candida spp. results from amino acid substitutions in the FKS gene, which confers decreased affinity of glucan synthase to the drugs [144]. Fortunately, echinocandin resistance is uncommon among Candida species [144,145,146,147]: C. albicans (0.0–0.1%), C. parapsilosis (0.0–0.1%), C. tropicalis (0.5–0.7%), C. krusei (0.0–1.7%), and C. glabrata (1.7–3.5%), as reported by the SENTRY Antimicrobial Surveillance Program from 1997 to 2006 [146].
Extensive clinical experience and durable antifungal activity has led to the adoption of echinocandins as first-line therapy for IC in neonates, children, and adults by the European Society for Clinical Microbiology and Infectious Diseases [40] and the Infectious Diseases Society of America [12]. Although pediatric trials are few, echinocandins have demonstrated effectiveness and safety comparable to that of amphotericin products for the treatment of IC in infants and children [148,149,150], as well as empiric treatment of febrile neutropenia in pediatric patients [151]. Most isolates of C. auris (> 95%) are susceptible to echinocandins [152], so these drugs are also considered first-line therapy for this emerging, multidrug-resistant pathogen [153]. Although echinocandins are active against many Aspergillus species in vitro [142], they are reserved for treatment of refractory cases or as salvage therapy [19].
4.2 Pharmacokinetics/Pharmacodynamics
Preclinical studies have determined that echinocandins exhibit time- and concentration-dependent fungal killing of Candida spp. with significant post-antifungal effects (PAFE) [154,155,156,157], meaning that fungicidal activity persists even after concentrations have declined. The pharmacodynamic parameter best associated with effectiveness against Candida species is the ratio between the AUC24/MIC ratio [154,155,156]. The specific pharmacodynamic targets are generally similar for the three agents but vary by Candida species [156]. In an in vivo study by Andes et al. [156], the pharmacodynamic target for C. albicans (mean free drug 24-hour AUC (fAUC24)/MIC of 20.6 ± 32) was significantly higher than for C. glabrata (mean 7.0 ± 8.3) and C. parapsilosis (mean 7.6 ± 7.1) for each agent. Because echinocandins are fungistatic against Aspergillus species, it is difficult to define MICs so instead a minimum effective concentration (MEC) ratio is used to define activity [158], which is the concentration at which hyphae transition to abnormal forms. However, no specific AUC/MEC ratio has been established as a pharmacodynamic target for clinical care Table 5.
Echinocandins are large molecules with poor bioavailability [136] and, thus far, are only available for parenteral administration. The pharmacologic properties of the three agents are similar, demonstrating linear pharmacokinetics over a range of clinically relevant dosages [159,160,161] and distributing well into most tissues [162]. However, they do not penetrate well into the eye [161, 163, 164] or CSF [161, 165] and distribute slowly into urine [166]. There is debate regarding the clinical significance of echinocandins’ poor urine penetration, which differs from their parenchymal penetration into kidney: preclinical studies have found that drug concentrations in the kidneys are comparable to those in other organs [161, 162, 167] and that concentrations persist in the kidneys well after serum concentrations decline [154]. To that end, there have been numerous reports of successful treatment of Candida urinary tract infections with echinocandins [168,169,170,171]. Despite this, data are insufficient to support recommendations for their use in the treatment of urinary tract infections [12, 19], at least as first-line therapy.
Similarly, despite poor CSF penetration, echinocandin concentrations in brain tissue exceed those in CSF [161, 172], and case reports of successful treatment of Candida meningitis [173, 174] and CNS aspergillosis [175] have been published. Preclinical studies and population pharmacokinetic analyses support the use of higher dosages of micafungin for the treatment of Candida meningoencephalitis in neonates [172, 176, 177]. Based on a dose-dependent penetration into the CNS and dose-microbiological responses demonstrated in preclinical studies [172], a dosage of 10 mg/kg is recommended by European guidelines for treatment of hematogenous Candida meningoencephalitis in neonates [40]. However, despite the pharmacokinetic data, the Infectious Diseases Society of America continues to recommend echinocandins only for salvage therapy or in cases of toxicity to other agents [12]. Data are also insufficient to guide the optimal dosing of caspofungin for neonatal meningitis or for any of the echinocandins for treatment of CNS infections outside of the neonatal period.
Significant differences exist in the metabolism and elimination of the three agents. Anidulafungin undergoes nonenzymatic chemical degradation [178], whereas micafungin and caspofungin are subject to hepatic metabolism [179, 180], albeit via different mechanisms. As a result, dosing of micafungin and caspofungin should be adjusted in patients with moderate or severe hepatic dysfunction, whereas this is not necessary for anidulafungin. None of the echinocandins undergo significant renal elimination, so dosage adjustments are not needed in patients with renal impairment, including those receiving continuous venovenous hemofiltration or hemodialysis [181,182,183].
All three agents are highly protein bound (92–99%), predominantly to albumin [166], and have long half-lives in plasma of up to 24–72 h, with steady state attained after several days [136]. Critically ill adult patients with hypoalbuminemia have higher caspofungin clearance and a resultant lower AUC0–24 [184]. This has been hypothesized to be due to the presence of extensive protein binding, in which small reductions in serum albumin lead to a larger free fraction of drug available for elimination. However, decreased protein binding may also result in increased distribution of unbound drug to tissues, improving echinocandins’ effectiveness against tissue-based infections. The impact of serum albumin on echinocandins’ distribution and clearance, and thus dosing, in infants and children is unclear.
4.3 Pediatric Dosing
4.3.1 Micafungin
Children exhibit a nonlinear, inverse relationship between weight and clearance of micafungin [185, 186]. As weight decreases, relatively larger dosages are needed to attain similar exposures to those in heavier patients. As a result, larger weight-based doses of micafungin (per kg) are needed for treatment in infants and smaller children [185]. Data support dosing of micafungin at 2–4 mg/kg once daily for treatment of candidemia in children aged ≥ 4 months [185]. Because children weighing > 50 kg achieve exposures similar to those in adults when receiving a fixed dosage of 100 mg per day, adult dosing is recommended in heavier children [185]. The use of higher dosages (3–5 mg/kg) less often (every 2–3 days or twice weekly) has been evaluated as an approach to prophylaxis in children at risk for IFD [187,188,189]. These regimens attained pharmacodynamic targets against susceptible isolates in most children [187,188,189] but have not been adopted into clinical practice. Lehrnbecher et al. [190] conducted an in-depth review of intermittent dosing strategies [190].
On the other hand, neonates require substantially larger dosages (10–15 mg/kg) to adequately treat disseminated candidiasis [176] because this disease often involves the CNS (i.e., meningoencephalitis) in this age group [172]. Several small observational studies of preterm and term neonates and infants have demonstrated that dosages up to 15 mg/kg/day are well-tolerated in infants [176, 191, 192]. As a result, higher dosages (4–10 mg/kg/day) are endorsed by European guidelines for treatment of IC in neonates, with specific recommendations for the use of 10 mg/kg/day when meningoencephalitis is suspected [40]. Despite these reports, the Infectious Diseases Society of America recommends that echinocandins only be used in neonates as salvage therapy or in settings in which other agents are not tolerated [12].
4.3.2 Caspofungin
The clearance and Vd of caspofungin are more closely related to body surface area (BSA) than weight alone [193,194,195,196]. Dosing scaled to BSA better approximates adult dosing than use of mg/kg dosing for this agent [194]. As a result, BSA-informed dosing of 70 mg/m2 as a loading dose followed by 50 mg/m2 for maintenance is recommended for children aged ≥ 3 months [193]. BSA dosing is also recommended for neonates and infants aged < 3 months: dosages of 25 mg/m2 achieved plasma exposure similar to that in adults receiving standard 50-mg doses in a study of 18 neonates and young infants [197], forming the basis for dosing recommendations in this age group. Caspofungin is the only echinocandin for which dosing adjustments are recommended in patients with hepatic dysfunction. Clearance of caspofungin is not affected by mild liver dysfunction [184, 198, 199], but it is decreased in patients with moderate hepatic impairment, leading to recommendations for use of lower doses in such patients [200].
4.3.3 Anidulafungin
Although anidulafungin is not yet approved in children, pharmacokinetic studies have been performed in pediatric patients across a range of ages [201, 202]. Dosages of 0.75 and 1.5 mg/kg/day achieved AUCs comparable to those achieved with 50-mg and 100-mg doses in adults, respectively [201, 202]. A loading dose of twice the maintenance dose is recommended for adults on day 1 and would presumably also be advised for children.
4.4 TDM: Adverse Events
Echinocandins are generally well-tolerated. The most common adverse events include infusion reactions and elevation of hepatic transaminases [195, 196, 201,202,203], which is most often mild. In general, TDM is not performed for echinocandins. Because of the extent of protein binding (> 95%), clinical assays that reliably measure free drug concentrations would be necessary to determine the amount of active drug in plasma. TDM may be beneficial when using echinocandins for treatment of organisms with decreased susceptibility to ensure that total plasma concentrations are in line with published studies.
5 Other Agents
5.1 Flucytosine (5-FC)
Flucytosine, also known as 5-fluorocytosine (5-FC), is one of the oldest antifungal drugs. It inhibits protein and DNA synthesis following conversion from 5-FC to 5-fluorouracil (5-FU) within fungal cells [204]. Human cells lack the enzyme to convert 5-FC to 5-FU, although intestinal microbes can convert the drug [205], which can lead to systemic 5-FU levels and possible toxicity. Flucytosine is active in vitro against many yeasts and some molds, but its clinical utility is largely limited to adjunctive therapy for cryptococcal meningitis [14]. Because of the rapid emergence of resistance when used as monotherapy, 5-FC is almost always administered in combination with an AmB product. Flucytosine and AmB provide additive activity against C. neoformans and C. albicans [47, 206]. As a result, coadministration can facilitate the use of lower dosages in the treatment of these organisms than are required when either agent is used alone.
Flucytosine is available in both enteral and parenteral formulations. Because it is highly bioavailable, intravenous administration is generally restricted to critically ill patients who cannot take enteral medications. The standard dosage of 5-FC is 100 mg/kg divided every 6 h, which is recommended for both children and adults, although higher dosages are sometimes used. Neonates achieve higher serum concentrations than older children [207, 208], therefore 75 mg/kg/day is the typical dose for infants aged < 30 days. Dose adjustments are also needed in patients with impaired renal function. The pharmacokinetics of 5-FC demonstrate significant interindividual variability [209] and, because 5-FC exhibits concentration-dependent toxicity, which manifests most frequently as hepatotoxicity (elevated transaminases) and bone marrow suppression (leukopenia, thrombocytopenia) [210], TDM is paramount. Peak (1–3 h post-dose) serum concentrations > 100 mg/L are associated with toxicity [211], thus TDM should be used routinely in children treated with 5-FC, with peak concentrations 50–100 mg/L and trough levels 25–50 mg/L considered acceptable [204].
5.2 Terbinafine
Terbinafine is an allylamine drug with broad antifungal activity. It exerts its action by inhibiting the fungal enzyme squalene epoxidase and, ultimately, ergosterol formation [212]. Clinically, terbinafine is most often used to treat tinea capitis or onychomycosis because of excellent penetration into nail, skin, and hair follicles [213]. In clinical trials, terbinafine was noninferior to griseofulvin for treatment of tinea capitis [214]. Terbinafine is highly protein bound (> 99%) and accumulates in skin and adipose tissue, leading to a terminal half-life > 150 h in plasma [215]. In addition, the penetration of terbinafine into other pertinent tissues, such as the brain, is unknown. As a result, its role as monotherapy for treatment of noncutaneous infections is questionable. Terbinafine has also shown in vitro synergistic activity with azoles against several clinically relevant molds, including Aspergillus species, Fusarium species, Rhizopus species, Scedosporium species, and organisms from the Mucorales order [216]. Therefore, terbinafine may have an adjunctive clinical role in the treatment of refractory or resistant mold infections in immunocompromised children, although data documenting the clinical utility of this agent for these pathogens are limited.
Terbinafine is approved by the US FDA for children aged ≥ 4 years. It is administered orally as granules (125 or 187.5 mg) or as a 250-mg tablet once daily. Children require larger dosages of terbinafine per kg of body weight than adults to achieve similar systemic exposures [217]. For tinea capitis, a 6-week course of therapy with 125 mg (< 25 kg), 187.5 mg (25–35 kg), or 250 mg (> 35 kg) once daily is advised [217]. At dosages used for tinea capitis, terbinafine is well-tolerated, with anorexia and gastrointestinal disturbance the most often reported adverse events [212, 217].
High-dose regimens (> 250 mg) have been used in the treatment of refractory mold infections [218]. In a physiologically based pharmacokinetic model [219], plasma terbinafine concentrations significantly accumulated over the first 7 days of therapy with high-dose regimens. Of the dosing regimens studied, 500 mg twice daily achieved the highest drug concentrations and pharmacodynamic target attainment (Cmax/MIC, AUC/MIC). However, without knowledge of the pharmacodynamic target associated with improved clinical outcomes in the treatment of molds, the optimal dosage for this indication is unknown.
5.3 Griseofulvin
Griseofulvin is a fungistatic antifungal with good activity against organisms that cause dermatophyte infections, such as Microsporum and Trichophyton species [220]. The drug is made soluble through its preparation as microsize and ultramicrosize particles, which increases the surface area of the drug and enhances its absorption. Its bioavailability is further enhanced by ingestion of the drug with a high-fat meal or food [221]. Griseofulvin distributes well into skin, nails, hair, liver, and muscle [220], but its clinical utility is limited by its narrow spectrum of activity to the treatment of tinea infections. In a Cochrane review of therapies for tinea capitis [222], griseofulvin was superior to terbinafine in the treatment of M. canis infections but inferior in the treatment of T. tonsurans. Although griseofulvin is carcinogenic in small animals [223], these same toxic effects have not been found in human studies, and the drug is generally well-tolerated. It has fewer hepatotoxic effects than other agents used to treat dermatophyte infections, including ketoconazole, itraconazole, fluconazole, and terbinafine [224].
6 Future Directions
Despite the development of newer and safer antifungal agents, the management of IFD in children remains challenging. Population pharmacokinetic studies have been performed in infants and children, but many of these studies involved a small number of diverse pediatric subjects. Full characterization of the effects of clinical factors (i.e., critical illness, obesity, organ dysfunction, age, drug interactions) on the pharmacokinetics of available antifungal agents in all children continues to be elucidated. Ongoing research in this area will be beneficial as the number of children at risk for IFD expands with time. However, the low overall incidence of IFD makes performance of adequately powered trials challenging. Therefore, well-designed observational studies will continue to be needed to provide comparative effectiveness data on antifungals in children.
IFD seems like the type of infectious process for which personalized medicine would be beneficial: high mortality, limited therapeutic options, variability in drug dosage–exposure relationships. Unfortunately, TDM is neither available nor feasible for most antifungal agents and, when performed, delayed turnaround in drug levels often affects the clinical applicability of results. With the availability of Bayesian dose adaptation software programs and continued investigations into dose–concentration–outcome relationships, the potential exists for implementation of individualized antifungal dosing to improve outcomes in IFD. However, advances in antifungal TDM are necessary to make results clinically actionable and bring the expanding amount of population pharmacokinetic data to the bedside.
An area not discussed in this review is the role of combination therapy in the treatment of IFD. As described elsewhere [178], certain antifungal combinations provide synergistic fungicidal activity in vitro. How well this translates to humans and improves outcomes of IFD is unknown. Dual therapy may be advantageous for some pathogens (i.e., more resistant organisms), infections of sites where drug delivery is impeded, such as the CNS, or in immunocompromised patients, who lack adequate immunity to clear infections once established. Translating research from the laboratory to the patient is particularly challenging in this area but is an important avenue for continued investigation.
Finally, since the 1990s, there has been a welcome expansion in the number of systemically available antifungal agents. This has included three new triazoles, three agents in the novel echinocandin class, and evolution of less toxic lipid formulations of amphotericin. Unfortunately, the immediate availability of these newer agents is often limited to adult patients as pediatric-specific PK/PD data are never available at the time of initial drug approval. Clinicians caring for children at risk for or diagnosed with an IFD are placed in the precarious position of relying on older agents with known pediatric pharmacokinetic parameters but potentially conferring greater toxicity versus the option of extrapolating adult pharmacokinetic data of newer agents to off-label use in children. Fortunately, physician advocates and legislators in both the USA and in Europe recognized this delay in or absence of pediatric-specific PK/PD data. In the past two decades, a series of legislative acts have helped to resolve this knowledge gap; this is described in detail elsewhere [225]. A collaborative infrastructure between pharmaceutical agencies and the FDA and European Medicines Agency has improved the number of pediatric-specific indications for antifungal agents. However, the time from adult approval to pediatric approval still ranges from 7 to 8 years. Furthermore, pediatric indications for certain antifungal agents, such as posaconazole, remain elusive up to 13 years after the initial adult approval. Additional legislation is needed to shorten this time between adult approval and completion of pediatric-specific studies.
References
Downes KJ, Ellis D, Lavigne S, Bryan M, Zaoutis TE, Fisher BT. The use of echinocandins in hospitalized children in the United States. Med Mycol. 2018. https://doi.org/10.1093/mmy/myy084.
Prasad PA, Coffin SE, Leckerman KH, Walsh TJ, Zaoutis TE. Pediatric antifungal utilization: new drugs, new trends. Pediatr Infect Dis J. 2008;27(12):1083–8.
Lestner JM, Smith PB, Cohen-Wolkowiez M, Benjamin DK Jr, Hope WW. Antifungal agents and therapy for infants and children with invasive fungal infections: a pharmacological perspective. Br J Clin Pharmacol. 2013;75(6):1381–95.
Andes D. Antifungal agents pharmacokinetics and pharmacodynamics of amphotericin B. In: Nightingale CH, Ambrose PG, Drusano GL, Murakawa T, editors. Antimicrobial pharmacodynamics in theory and clinical practice. 2nd ed. New York: Informa Healthcare USA, Inc.; 2007.
Chowdhary A, Prakash A, Sharma C, Kordalewska M, Kumar A, Sarma S, et al. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009-17) in India: role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J Antimicrob Chemother. 2018;73(4):891–9.
Prakash A, Sharma C, Singh A, Kumar Singh P, Kumar A, Hagen F, et al. Evidence of genotypic diversity among Candida auris isolates by multilocus sequence typing, matrix-assisted laser desorption ionization time-of-flight mass spectrometry and amplified fragment length polymorphism. Clin Microbiol Infect. 2016;22(3):277 (e1–9).
Vitale RG, de Hoog GS, Schwarz P, Dannaoui E, Deng S, Machouart M, et al. Antifungal susceptibility and phylogeny of opportunistic members of the order mucorales. J Clin Microbiol. 2012;50(1):66–75.
Goldman RD, Koren G. Amphotericin B nephrotoxicity in children. J Pediatr Hematol Oncol. 2004;26(7):421–6.
Hamill RJ. Amphotericin B formulations: a comparative review of efficacy and toxicity. Drugs. 2013;73(9):919–34.
Botero Aguirre JP, Restrepo Hamid AM. Amphotericin B deoxycholate versus liposomal amphotericin B: effects on kidney function. Cochrane Database Syst Rev. 2015;23(11):CD010481.
Blyth CC, Hale K, Palasanthiran P, O’Brien T, Bennett MH. Antifungal therapy in infants and children with proven, probable or suspected invasive fungal infections. Cochrane Database Syst Rev. 2010;17(2):CD006343.
Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62(4):e1–50.
Ascher SB, Smith PB, Watt K, Benjamin DK, Cohen-Wolkowiez M, Clark RH, et al. Antifungal therapy and outcomes in infants with invasive Candida infections. Pediatr Infect Dis J. 2012;31(5):439–43.
Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of america. Clin Infect Dis. 2010;50(3):291–322.
Cornely OA, Arikan-Akdagli S, Dannaoui E, Groll AH, Lagrou K, Chakrabarti A, et al. ESCMID and ECMM joint clinical guidelines for the diagnosis and management of mucormycosis 2013. Clin Microbiol Infect. 2014;20(Suppl 3):5–26.
Chapman SW, Dismukes WE, Proia LA, Bradsher RW, Pappas PG, Threlkeld MG, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46(12):1801–12.
Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112–46.
Wheat LJ, Freifeld AG, Kleiman MB, Baddley JW, McKinsey DS, Loyd JE, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45(7):807–25.
Patterson TF, Thompson GR III, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63(4):e1–60.
Andes D, Stamsted T, Conklin R. Pharmacodynamics of amphotericin B in a neutropenic-mouse disseminated-candidiasis model. Antimicrob Agents Chemother. 2001;45(3):922–6.
Wiederhold NP, Tam VH, Chi J, Prince RA, Kontoyiannis DP, Lewis RE. Pharmacodynamic activity of amphotericin B deoxycholate is associated with peak plasma concentrations in a neutropenic murine model of invasive pulmonary aspergillosis. Antimicrob Agents Chemother. 2006;50(2):469–73.
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob Agents Chemother. 2002;46(3):834–40.
Benson JM, Nahata MC. Pharmacokinetics of amphotericin B in children. Antimicrob Agents Chemother. 1989;33(11):1989–93.
Starke JR, Mason EO Jr, Kramer WG, Kaplan SL. Pharmacokinetics of amphotericin B in infants and children. J Infect Dis. 1987;155(4):766–74.
Koren G, Lau A, Klein J, Golas C, Bologa-Campeanu M, Soldin S, et al. Pharmacokinetics and adverse effects of amphotericin B in infants and children. J Pediatr. 1988;113(3):559–63.
Nath CE, McLachlan AJ, Shaw PJ, Coakley JC, Earl JW. Amphotericin B dose optimization in children with malignant diseases. Chemotherapy. 2007;53(2):142–7.
Nath CE, McLachlan AJ, Shaw PJ, Gunning R, Earl JW. Population pharmacokinetics of amphotericin B in children with malignant diseases. Br J Clin Pharmacol. 2001;52(6):671–80.
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother. 2002;46(3):828–33.
Adler-Moore JP, Proffitt RT. Amphotericin B lipid preparations: what are the differences? Clin Microbiol Infect. 2008;14(Suppl 4):25–36.
Groll AH, Giri N, Petraitis V, Petraitiene R, Candelario M, Bacher JS, et al. Comparative efficacy and distribution of lipid formulations of amphotericin B in experimental Candida albicans infection of the central nervous system. J Infect Dis. 2000;182(1):274–82.
Walsh TJ, Whitcomb P, Piscitelli S, Figg WD, Hill S, Chanock SJ, et al. Safety, tolerance, and pharmacokinetics of amphotericin B lipid complex in children with hepatosplenic candidiasis. Antimicrob Agents Chemother. 1997;41(9):1944–8.
Wurthwein G, Groll AH, Hempel G, Adler-Shohet FC, Lieberman JM, Walsh TJ. Population pharmacokinetics of amphotericin B lipid complex in neonates. Antimicrob Agents Chemother. 2005;49(12):5092–8.
Amantea MA, Bowden RA, Forrest A, Working PK, Newman MS, Mamelok RD. Population pharmacokinetics and renal function-sparing effects of amphotericin B colloidal dispersion in patients receiving bone marrow transplants. Antimicrob Agents Chemother. 1995;39(9):2042–7.
Hong Y, Shaw PJ, Tattam BN, Nath CE, Earl JW, Stephen KR, et al. Plasma protein distribution and its impact on pharmacokinetics of liposomal amphotericin B in paediatric patients with malignant diseases. Eur J Clin Pharmacol. 2007;63(2):165–72.
Collette N, van der Auwera P, Lopez AP, Heymans C, Meunier F. Tissue concentrations and bioactivity of amphotericin B in cancer patients treated with amphotericin B-deoxycholate. Antimicrob Agents Chemother. 1989;33(3):362–8.
Vogelsinger H, Weiler S, Djanani A, Kountchev J, Bellmann-Weiler R, Wiedermann CJ, et al. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J Antimicrob Chemother. 2006;57(6):1153–60.
Groll AH, Lyman CA, Petraitis V, Petraitiene R, Armstrong D, Mickiene D, et al. Compartmentalized intrapulmonary pharmacokinetics of amphotericin B and its lipid formulations. Antimicrob Agents Chemother. 2006;50(10):3418–23.
Weiler S, Falkensammer G, Hammerer-Lercher A, Anliker M, Vogelsinger H, Joannidis M, et al. Pulmonary epithelial lining fluid concentrations after use of systemic amphotericin B lipid formulations. Antimicrob Agents Chemother. 2009;53(11):4934–7.
Fisher JF, Sobel JD, Kauffman CA, Newman CA. Candida urinary tract infections—treatment. Clin Infect Dis. 2011;52(Suppl 6):S457–66.
Hope WW, Castagnola E, Groll AH, Roilides E, Akova M, Arendrup MC, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18(Suppl 7):38–52.
Payne KD, Hall RG. Dosing of antifungal agents in obese people. Expert Rev Anti-infect Ther. 2016;14(2):257–67.
Wasmann RE, Smit C, van Dongen EPH, Wiezer RMJ, Adler-Moore J, de Beer YM, et al. Fixed dosing of liposomal amphotericin b in morbidly obese individuals. Clin Infect Dis. 2019. https://doi.org/10.1093/cid/ciz885.
Eriksson U, Seifert B, Schaffner A. Comparison of effects of amphotericin B deoxycholate infused over 4 or 24 hours: randomised controlled trial. BMJ. 2001;322(7286):579–82.
Falagas ME, Karageorgopoulos DE, Tansarli GS. continuous versus conventional infusion of amphotericin B deoxycholate: a meta-analysis. PloS One. 2013;8(10):e77075.
Kotwani RN, Gokhale PC, Bodhe PV, Kirodian BG, Kshirsagar NA, Pandya SK. A comparative study of plasma concentrations of liposomal amphotericin B (L-AMP-LRC-1) in adults, children and neonates. Int J Pharm. 2002;238(1–2):11–5.
Seibel NL, Shad AT, Bekersky I, Groll AH, Gonzalez C, Wood LV, et al. Safety, Tolerability, and pharmacokinetics of liposomal amphotericin B in immunocompromised pediatric patients. Antimicrob Agents Chemother. 2017;61:e01477–16.
O’Connor L, Livermore J, Sharp AD, Goodwin J, Gregson L, Howard SJ, et al. Pharmacodynamics of liposomal amphotericin B and flucytosine for cryptococcal meningoencephalitis: safe and effective regimens for immunocompromised patients. J Infect Dis. 2013;208(2):351–61.
Lestner JM, Groll AH, Aljayyoussi G, Seibel NL, Shad A, Gonzalez C, et al. Population pharmacokinetics of liposomal amphotericin B in immunocompromised children. Antimicrob Agents Chemother. 2016;60(12):7340–6.
Walsh TJ, Goodman JL, Pappas P, Bekersky I, Buell DN, Roden M, et al. Safety, tolerance, and pharmacokinetics of high-dose liposomal amphotericin B (AmBisome) in patients infected with Aspergillus species and other filamentous fungi: maximum tolerated dose study. Antimicrob Agents Chemother. 2001;45(12):3487–96.
Hong Y, Shaw PJ, Nath CE, Yadav SP, Stephen KR, Earl JW, et al. Population pharmacokinetics of liposomal amphotericin B in pediatric patients with malignant diseases. Antimicrob Agents Chemother. 2006;50(3):935–42.
Bes DF, Rosanova MT, Sberna N, Arrizurieta E. Deoxycholate amphotericin B and nephrotoxicity in the pediatric setting. Pediatr Infect Dis J. 2014;33(8):e198–206.
Branch RA. Prevention of amphotericin B-induced renal impairment. A review on the use of sodium supplementation. Arch Intern Med. 1988;148(11):2389–94.
Medoff G, Kobayashi GS. Strategies in the treatment of systemic fungal infections. N Engl J Med. 1980;302(3):145–55.
Sunakawa K, Tsukimoto I, Tsunematsu Y, Honda M, Iwai N, Maniwa T, et al. Evaluation of the safety and efficacy of liposomal amphotericin B (L-AMB) in children. J Infect Chemother. 2012;18(4):456–65.
Arning M, Kliche KO, Heer-Sonderhoff AH, Wehmeier A. Infusion-related toxicity of three different amphotericin B formulations and its relation to cytokine plasma levels. Mycoses. 1995;38(11–12):459–65.
Bowden R, Chandrasekar P, White MH, Li X, Pietrelli L, Gurwith M, et al. A double-blind, randomized, controlled trial of amphotericin B colloidal dispersion versus amphotericin B for treatment of invasive aspergillosis in immunocompromised patients. Clin Infect Dis. 2002;35(4):359–66.
Bellmann R, Smuszkiewicz P. Pharmacokinetics of antifungal drugs: practical implications for optimized treatment of patients. Infection. 2017;45(6):737–79.
Fromtling RA. Overview of medically important antifungal azole derivatives. Clin Microbiol Rev. 1988;1(2):187–217.
Powers-Fletcher MV, Kendall BA, Griffin AT, Hanson KE. Filamentous fungi. Microbiol Spectr. 2016. https://doi.org/10.1128/microbiolspec.DMIH2-0002-2015.
Ramos-Martin V, O’Connor O, Hope W. Clinical pharmacology of antifungal agents in pediatrics: children are not small adults. Curr Opin Pharmacol. 2015;24:128–34.
Resendiz Sharpe A, Lagrou K, Meis JF, Chowdhary A, Lockhart SR, Verweij PE, et al. Triazole resistance surveillance in Aspergillus fumigatus. Med Mycol. 2018;56(suppl_1):83–92.
Verweij PE, Zhang J, Debets AJM, Meis JF, van de Veerdonk FL, Schoustra SE, et al. In-host adaptation and acquired triazole resistance in Aspergillus fumigatus: a dilemma for clinical management. Lancet Infect Dis. 2016;16(11):e251–60.
Lepak AJ, Andes DR. Antifungal pharmacokinetics and pharmacodynamics. Cold Spring Harb Perspect Med. 2014;5(5):a019653.
Andes D. Pharmacokinetics and pharmacodynamics of antifungals. Infect Dis Clin N Am. 2006;20(3):679–97.
Gerhart JG, Watt KM, Edginton A, Wade KC, Salerno SN, Benjamin DK Jr, et al. Physiologically-based pharmacokinetic modeling of fluconazole using plasma and cerebrospinal fluid samples from preterm and term infants. CPT Pharmacometr Syst Pharmacol. 2019;8(7):500–10.
Debruyne D. Clinical pharmacokinetics of fluconazole in superficial and systemic mycoses. Clin Pharmacokinet. 1997;33(1):52–77.
Bourcier K, Hyland R, Kempshall S, Jones R, Maximilien J, Irvine N, et al. Investigation into UDP-glucuronosyltransferase (UGT) enzyme kinetics of imidazole- and triazole-containing antifungal drugs in human liver microsomes and recombinant UGT enzymes. Drug Metab Dispos. 2010;38(6):923–9.
Brammer KW, Coakley AJ, Jezequel SG, Tarbit MH. The disposition and metabolism of [14C]fluconazole in humans. Drug Metab Dispos Biol Fate Chem. 1991;19(4):764–7.
Bae SK, Park SJ, Shim EJ, Mun JH, Kim EY, Shin JG, et al. Increased oral bioavailability of itraconazole and its active metabolite, 7-hydroxyitraconazole, when coadministered with a vitamin C beverage in healthy participants. J Clin Pharmacol. 2011;51(3):444–51.
Mouton JW, van Peer A, de Beule K, Van Vliet A, Donnelly JP, Soons PA. Pharmacokinetics of itraconazole and hydroxyitraconazole in healthy subjects after single and multiple doses of a novel formulation. Antimicrob Agents Chemother. 2006;50(12):4096–102.
Lim SG, Sawyerr AM, Hudson M, Sercombe J, Pounder RE. Short report: the absorption of fluconazole and itraconazole under conditions of low intragastric acidity. Aliment Pharmacol Ther. 1993;7(3):317–21.
Van Peer A, Woestenborghs R, Heykants J, Gasparini R, Gauwenbergh G. The effects of food and dose on the oral systemic availability of itraconazole in healthy subjects. Eur J Clin Pharmacol. 1989;36(4):423–6.
de Repentigny L, Ratelle J, Leclerc JM, Cornu G, Sokal EM, Jacqmin P, et al. Repeated-dose pharmacokinetics of an oral solution of itraconazole in infants and children. Antimicrob Agents Chemother. 1998;42(2):404–8.
Schmitt C, Perel Y, Harousseau JL, Lemerle S, Chwetzoff E, le Moing JP, et al. Pharmacokinetics of itraconazole oral solution in neutropenic children during long-term prophylaxis. Antimicrob Agents Chemother. 2001;45(5):1561–4.
Allegra S, Fatiguso G, De Francia S, Favata F, Pirro E, Carcieri C, et al. Pharmacokinetic evaluation of oral itraconazole for antifungal prophylaxis in children. Clin Exp Pharmacol Physiol. 2017;44(11):1083–8.
De Beule K, Van Gestel J. Pharmacology of itraconazole. Drugs. 2001;61(Suppl 1):27–37.
Heykants J, Van Peer A, Van de Velde V, Van Rooy P, Meuldermans W, Lavrijsen K, et al. The clinical pharmacokinetics of itraconazole: an overview. Mycoses. 1989;32(Suppl 1):67–87.
Herbrecht R, Denning DW, Patterson TF, Bennett JE, Greene RE, Oestmann JW, et al. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408–15.
Leveque D, Nivoix Y, Jehl F, Herbrecht R. Clinical pharmacokinetics of voriconazole. Int J Antimicrob Agents. 2006;27(4):274–84.
Purkins L, Wood N, Kleinermans D, Greenhalgh K, Nichols D. Effect of food on the pharmacokinetics of multiple-dose oral voriconazole. Br J Clin Pharmacol. 2003;56(Suppl 1):17–23.
Yanni SB, Annaert PP, Augustijns P, Bridges A, Gao Y, Benjamin DK Jr, et al. Role of flavin-containing monooxygenase in oxidative metabolism of voriconazole by human liver microsomes. Drug Metab Dispos. 2008;36(6):1119–25.
Yanni SB, Annaert PP, Augustijns P, Ibrahim JG, Benjamin DK Jr, Thakker DR. In vitro hepatic metabolism explains higher clearance of voriconazole in children versus adults: role of CYP2C19 and flavin-containing monooxygenase 3. Drug Metab Dispos. 2010;38(1):25–31.
Wang G, Lei HP, Li Z, Tan ZR, Guo D, Fan L, et al. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol. 2009;65(3):281–5.
Weiss J, Ten Hoevel MM, Burhenne J, Walter-Sack I, Hoffmann MM, Rengelshausen J, et al. CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J Clin Pharmacol. 2009;49(2):196–204.
Friberg LE, Ravva P, Karlsson MO, Liu P. Integrated population pharmacokinetic analysis of voriconazole in children, adolescents, and adults. Antimicrob Agents Chemother. 2012;56(6):3032–42.
Karlsson MO, Lutsar I, Milligan PA. Population pharmacokinetic analysis of voriconazole plasma concentration data from pediatric studies. Antimicrob Agents Chemother. 2009;53(3):935–44.
Walsh TJ, Driscoll T, Milligan PA, Wood ND, Schlamm H, Groll AH, et al. Pharmacokinetics, safety, and tolerability of voriconazole in immunocompromised children. Antimicrob Agents Chemother. 2010;54(10):4116–23.
Zane NR, Thakker DR. A physiologically based pharmacokinetic model for voriconazole disposition predicts intestinal first-pass metabolism in children. Clin Pharmacokinet. 2014;53(12):1171–82.
Roffey SJ, Cole S, Comby P, Gibson D, Jezequel SG, Nedderman AN, et al. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab Dispos. 2003;31(6):731–41.
Hsu AJ, Dabb A, Arav-Boger R. Autoinduction of voriconazole metabolism in a child with invasive pulmonary aspergillosis. Pharmacotherapy. 2015;35(4):e20–6.
Mulanovich V, Lewis RE, Raad II, Kontoyiannis DP. Random plasma concentrations of voriconazole decline over time. J Infect. 2007;55(5):e129–30.
Torres HA, Hachem RY, Chemaly RF, Kontoyiannis DP, Raad II. Posaconazole: a broad-spectrum triazole antifungal. Lancet Infect Dis. 2005;5(12):775–85.
Ezzet F, Wexler D, Courtney R, Krishna G, Lim J, Laughlin M. Oral bioavailability of posaconazole in fasted healthy subjects: comparison between three regimens and basis for clinical dosage recommendations. Clin Pharmacokinet. 2005;44(2):211–20.
Kersemaekers WM, van Iersel T, Nassander U, O’Mara E, Waskin H, Caceres M, et al. Pharmacokinetics and safety study of posaconazole intravenous solution administered peripherally to healthy subjects. Antimicrob Agents Chemother. 2015;59(2):1246–51.
Sansone-Parsons A, Krishna G, Calzetta A, Wexler D, Kantesaria B, Rosenberg MA, et al. Effect of a nutritional supplement on posaconazole pharmacokinetics following oral administration to healthy volunteers. Antimicrob Agents Chemother. 2006;50(5):1881–3.
Boonsathorn S, Cheng I, Kloprogge F, Alonso C, Lee C, Doncheva B, et al. Clinical pharmacokinetics and dose recommendations for posaconazole in infants and children. Clin Pharmacokinet. 2019;58(1):53–61.
Vanstraelen K, Colita A, Bica AM, Mols R, Augustijns P, Peersman N, et al. Pharmacokinetics of posaconazole oral suspension in children dosed according to body surface area. Pediatr Infect Dis J. 2016;35(2):183–8.
Ghosal A, Hapangama N, Yuan Y, Achanfuo-Yeboah J, Iannucci R, Chowdhury S, et al. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of posaconazole (Noxafil). Drug Metab Dispos Biol Fate Chem. 2004;32(2):267–71.
Krieter P, Flannery B, Musick T, Gohdes M, Martinho M, Courtney R. Disposition of posaconazole following single-dose oral administration in healthy subjects. Antimicrob Agents Chemother. 2004;48(9):3543–51.
Krishna G, Moton A, Ma L, Savant I, Martinho M, Seiberling M, et al. Effects of oral posaconazole on the pharmacokinetic properties of oral and intravenous midazolam: a phase I, randomized, open-label, crossover study in healthy volunteers. Clin Ther. 2009;31(2):286–98.
Wexler D, Courtney R, Richards W, Banfield C, Lim J, Laughlin M. Effect of posaconazole on cytochrome P450 enzymes: a randomized, open-label, two-way crossover study. Eur J Pharm Sci. 2004;21(5):645–53.
Courtney R, Wexler D, Radwanski E, Lim J, Laughlin M. Effect of food on the relative bioavailability of two oral formulations of posaconazole in healthy adults. Br J Clin Pharmacol. 2004;57(2):218–22.
Doring M, Cabanillas Stanchi KM, Queudeville M, Feucht J, Blaeschke F, Schlegel P, et al. Efficacy, safety and feasibility of antifungal prophylaxis with posaconazole tablet in paediatric patients after haematopoietic stem cell transplantation. J Cancer Res Clin Oncol. 2017;143(7):1281–92.
Arrieta AC, Sung L, Bradley JS, Zwaan CM, Gates D, Waskin H, et al. A non-randomized trial to assess the safety, tolerability, and pharmacokinetics of posaconazole oral suspension in immunocompromised children with neutropenia. PLoS One. 2019;14(3):e0212837.
Groll A, Abdel-Azim H, Lehrnbecher T, Steinbach W, Paschke A, Mangin E, et al. Safety, tolerability and pharmacokinetics of posaconazole intravenous solution and oral powder for suspension in children with neutropenia. Amsterdam: European Congress of Clinical Microbiology & Infectious Diseases; 2019.
Rybak JM, Marx KR, Nishimoto AT, Rogers PD. Isavuconazole: pharmacology, pharmacodynamics, and current clinical experience with a new triazole antifungal agent. Pharmacotherapy. 2015;35(11):1037–51.
Pettit NN, Carver PL. Isavuconazole: a new option for the management of invasive fungal infections. Ann Pharmacother. 2015;49(7):825–42.
Schmitt-Hoffmann A, Roos B, Heep M, Schleimer M, Weidekamm E, Brown T, et al. Single-ascending-dose pharmacokinetics and safety of the novel broad-spectrum antifungal triazole BAL4815 after intravenous infusions (50, 100, and 200 milligrams) and oral administrations (100, 200, and 400 milligrams) of its prodrug, BAL8557, in healthy volunteers. Antimicrob Agents Chemother. 2006;50(1):279–85.
Kovanda LL, Desai AV, Lu Q, Townsend RW, Akhtar S, Bonate P, et al. Isavuconazole population pharmacokinetic analysis using nonparametric estimation in patients with invasive fungal disease (results from the VITAL study). Antimicrob Agents Chemother. 2016;60(8):4568–76.
Watt K, Manzoni P, Cohen-Wolkowiez M, Rizzollo S, Boano E, Jacqz-Aigrain E, et al. Triazole use in the nursery: fluconazole, voriconazole, posaconazole, and ravuconazole. Curr Drug Metab. 2013;14(2):193–202.
Piper L, Smith PB, Hornik CP, Cheifetz IM, Barrett JS, Moorthy G, et al. Fluconazole loading dose pharmacokinetics and safety in infants. Pediatr Infect Dis J. 2011;30(5):375–8.
Stockmann C, Constance JE, Roberts JK, Olson J, Doby EH, Ampofo K, et al. Pharmacokinetics and pharmacodynamics of antifungals in children and their clinical implications. Clin Pharmacokinet. 2014;53(5):429–54.
Leong YH, Boast A, Cranswick N, Curtis N, Gwee A. Itraconazole dosing and drug monitoring at a tertiary children’s hospital. Pediatr Infect Dis J. 2019;38(1):60–4.
Simon A, Besuden M, Vezmar S, Hasan C, Lampe D, Kreutzberg S, et al. Itraconazole prophylaxis in pediatric cancer patients receiving conventional chemotherapy or autologous stem cell transplants. Support Care Cancer. 2007;15(2):213–20.
Gastine S, Lehrnbecher T, Muller C, Farowski F, Bader P, Ullmann-Moskovits J, et al. Pharmacokinetic modeling of voriconazole to develop an alternative dosing regimen in children. Antimicrob Agents Chemother. 2018;62:e01194–17. https://doi.org/10.1128/AAC.01194-17.
Gerin M, Mahlaoui N, Elie C, Lanternier F, Bougnoux ME, Blanche S, et al. Therapeutic drug monitoring of voriconazole after intravenous administration in infants and children with primary immunodeficiency. Ther Drug Monit. 2011;33(4):464–6.
Arrieta AF, Steinbach W, Muller W, Sue P, Yin D, Danziger-Isakov L, et al. An open-label, phase I, multi-centre study to evaluate the pharmacokinetic, safety and tolerability profile of intravenous isavuconazonium sulfate in paediatric patients. Amsterdam: European Congress of Clinical Microbiology & Infectious Diseases; 2019.
McCarthy MW, Moriyama B, Petraitiene R, Walsh TJ, Petraitis V. Clinical pharmacokinetics and pharmacodynamics of isavuconazole. Clin Pharmacokinet. 2018;57(12):1483–91.
Seyedmousavi S, Mouton JW, Melchers WJ, Bruggemann RJ, Verweij PE. The role of azoles in the management of azole-resistant aspergillosis: from the bench to the bedside. Drug Resist Updates. 2014;17(3):37–50.
van der Elst KC, Pereboom M, van den Heuvel ER, Kosterink JG, Scholvinck EH, Alffenaar JW. Insufficient fluconazole exposure in pediatric cancer patients and the need for therapeutic drug monitoring in critically ill children. Clin Infect Dis. 2014;59(11):1527–33.
Bruggemann RJ, Donnelly JP, Aarnoutse RE, Warris A, Blijlevens NM, Mouton JW, et al. Therapeutic drug monitoring of voriconazole. Ther Drug Monit. 2008;30(4):403–11.
Luong ML, Al-Dabbagh M, Groll AH, Racil Z, Nannya Y, Mitsani D, et al. Utility of voriconazole therapeutic drug monitoring: a meta-analysis. J Antimicrob Chemother. 2016;71(7):1786–99.
Lempers VJ, Meuwese E, Mavinkurve-Groothuis AM, Henriet S, van der Sluis IM, Hanff LM, et al. Impact of dose adaptations following voriconazole therapeutic drug monitoring in pediatric patients. Med Mycol. 2019;57(8):937–43. https://doi.org/10.1093/mmy/myz006.
Dekkers BGJ, Bakker M, van der Elst KCM, Sturkenboom MGG, Veringa A, Span LFR, et al. Therapeutic drug monitoring of posaconazole: an update. Curr Fungal Infect Rep. 2016;10:51–61.
Jancel T, Shaw PA, Hallahan CW, Kim T, Freeman AF, Holland SM, et al. Therapeutic drug monitoring of posaconazole oral suspension in paediatric patients younger than 13 years of age: a retrospective analysis and literature review. J Clin Pharm Ther. 2017;42(1):75–9.
Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother. 2014;69(5):1162–76.
Groll AH, Townsend R, Desai A, Azie N, Jones M, Engelhardt M, et al. Drug–drug interactions between triazole antifungal agents used to treat invasive aspergillosis and immunosuppressants metabolized by cytochrome P450 3A4. Transpl Infect Dis. 2017. https://doi.org/10.1111/tid.12751.
Lempers VJ, Martial LC, Schreuder MF, Blijlevens NM, Burger DM, Aarnoutse RE, et al. Drug-interactions of azole antifungals with selected immunosuppressants in transplant patients: strategies for optimal management in clinical practice. Curr Opin Pharmacol. 2015;24:38–44.
Kieu V, Jhangiani K, Dadwal S, Nakamura R, Pon D. Effect of isavuconazole on tacrolimus and sirolimus serum concentrations in allogeneic hematopoietic stem cell transplant patients: A drug-drug interaction study. Transpl Infect Dis. 2019;21(1):e13007.
Blyth CC, Palasanthiran P, O’Brien TA. Antifungal therapy in children with invasive fungal infections: a systematic review. Pediatrics. 2007;119(4):772–84.
Walsh TJ, Pappas P, Winston DJ, Lazarus HM, Petersen F, Raffalli J, et al. Voriconazole compared with liposomal amphotericin B for empirical antifungal therapy in patients with neutropenia and persistent fever. N Engl J Med. 2002;346(4):225–34.
Tang H, Shi W, Song Y, Han J. Voriconazole exposure and risk of cutaneous squamous cell carcinoma among lung or hematopoietic cell transplant patients: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80(2):500–7 (e10).
Mansh M, Binstock M, Williams K, Hafeez F, Kim J, Glidden D, et al. Voriconazole exposure and risk of cutaneous squamous cell carcinoma, aspergillus colonization, invasive aspergillosis and death in lung transplant recipients. Am J Transplant. 2016;16(1):262–70.
Wojenski DJ, Bartoo GT, Merten JA, Dierkhising RA, Barajas MR, El-Azhary RA, et al. Voriconazole exposure and the risk of cutaneous squamous cell carcinoma in allogeneic hematopoietic stem cell transplant patients. Transpl Infect Dis. 2015;17(2):250–8.
Cohen BE, Krivitskiy I, Bui S, Forrester K, Kahn J, Barbers R, et al. Comparison of skin cancer incidence in Caucasian and non-Caucasian liver vs. lung transplant recipients: a tale of two regimens. Clin Drug Investig. 2019;39(2):197–203.
Patil A, Majumdar S. Echinocandins in antifungal pharmacotherapy. J Pharm Pharmacol. 2017;69(12):1635–60.
Chang CC, Slavin MA, Chen SC. New developments and directions in the clinical application of the echinocandins. Arch Toxicol. 2017;91(4):1613–21.
Gil-Alonso S, Quindos G, Canton E, Eraso E, Jauregizar N. Killing kinetics of anidulafungin, caspofungin and micafungin against Candida parapsilosis species complex: evaluation of the fungicidal activity. Revista Iberoamericana de Micologia. 2019;36(1):24–9.
Bartizal K, Gill CJ, Abruzzo GK, Flattery AM, Kong L, Scott PM, et al. In vitro preclinical evaluation studies with the echinocandin antifungal MK-0991 (L-743,872). Antimicrob Agents Chemother. 1997;41(11):2326–32.
Aruanno M, Glampedakis E, Lamoth F. Echinocandins for the treatment of invasive aspergillosis: from laboratory to bedside. Antimicrob Agents Chemother. 2019;63:e00399–19. https://doi.org/10.1128/AAC.00399-19.
Bowman JC, Hicks PS, Kurtz MB, Rosen H, Schmatz DM, Liberator PA, et al. The antifungal echinocandin caspofungin acetate kills growing cells of Aspergillus fumigatus in vitro. Antimicrob Agents Chemother. 2002;46(9):3001–12.
Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M. Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J Clin Microbiol. 2013;51(8):2571–81.
Dannaoui E. Antifungal resistance in mucorales. Int J Antimicrob Agents. 2017;50(5):617–21.
Perlin DS. Echinocandin resistance in candida. Clin Infect Dis. 2015;1(61 Suppl 6):S612–7.
Pfaller MA, Castanheira M, Messer SA, Moet GJ, Jones RN. Echinocandin and triazole antifungal susceptibility profiles for Candida spp., Cryptococcus neoformans, and Aspergillus fumigatus: application of new CLSI clinical breakpoints and epidemiologic cutoff values to characterize resistance in the SENTRY Antimicrobial Surveillance Program (2009). Diagn Microbiol Infect Dis. 2011;69(1):45–50.
Pfaller MA, Diekema DJ, Turnidge JD, Castanheira M, Jones RN. Twenty years of the SENTRY Antifungal Surveillance Program: results for candida species from 1997–2016. Open Forum Infect Dis. 2019;6(Suppl 1):S79–s94.
Fraser M, Borman AM, Thorn R, Lawrance LM. Resistance to echinocandin antifungal agents in the United Kingdom in clinical isolates of Candida glabrata: fifteen years of interpretation and assessment. Med Mycol. 2019. https://doi.org/10.1093/mmy/myz053.
Cornely OA, Vazquez J, De Waele J, Betts R, Rotstein C, Nucci M, et al. Efficacy of micafungin in invasive candidiasis caused by common Candida species with special emphasis on non-albicans Candida species. Mycoses. 2014;57(2):79–89.
Mohamed WA, Ismail M. A randomized, double-blind, prospective study of caspofungin vs. amphotericin B for the treatment of invasive candidiasis in newborn infants. J Trop Pediatr. 2012;58(1):25–30.
Benjamin DK Jr, Kaufman DA, Hope WW, Smith PB, Arrieta A, Manzoni P, et al. A phase 3 study of micafungin versus amphotericin B deoxycholate in infants with invasive candidiasis. Pediatr Infect Dis J. 2018;37(10):992–8.
Maertens JA, Madero L, Reilly AF, Lehrnbecher T, Groll AH, Jafri HS, et al. A randomized, double-blind, multicenter study of caspofungin versus liposomal amphotericin B for empiric antifungal therapy in pediatric patients with persistent fever and neutropenia. Pediatr Infect Dis J. 2010;29(5):415–20.
Kordalewska M, Lee A, Park S, Berrio I, Chowdhary A, Zhao Y, et al. Understanding echinocandin resistance in the emerging pathogen Candida auris. Antimicrob Agents Chemother. 2018;62:e00238–18. https://doi.org/10.1128/AAC.00238-18.
U.S. Centers for Disease Control and Prevention. Recommendations for treatment of Candida auris infections. 2018. https://www.cdc.gov/fungal/candida-auris/c-auris-treatment.html#treatment. Cited 17 July 2019.
Louie A, Deziel M, Liu W, Drusano MF, Gumbo T, Drusano GL. Pharmacodynamics of caspofungin in a murine model of systemic candidiasis: importance of persistence of caspofungin in tissues to understanding drug activity. Antimicrob Agents Chemother. 2005;49(12):5058–68.
Andes D, Diekema DJ, Pfaller MA, Prince RA, Marchillo K, Ashbeck J, et al. In vivo pharmacodynamic characterization of anidulafungin in a neutropenic murine candidiasis model. Antimicrob Agents Chemother. 2008;52(2):539–50.
Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, Lepak A. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother. 2010;54(6):2497–506.
Nguyen KT, Taa P, Hoang BT, Cheng S, Hao B, Nguyen MH, et al. Anidulafungin is fungicidal and exerts a variety of postantifungal effects against Candida albicans, C. glabrata, C. parapsilosis, and C. krusei isolates. Antimicrob Agents Chemother. 2009;53(8):3347–52.
Kurtz MB, Heath IB, Marrinan J, Dreikorn S, Onishi J, Douglas C. Morphological effects of lipopeptides against Aspergillus fumigatus correlate with activities against (1,3)-beta-d-glucan synthase. Antimicrob Agents Chemother. 1994;38(7):1480–9.
Tabata K, Katashima M, Kawamura A, Tanigawara Y, Sunagawa K. Linear pharmacokinetics of micafungin and its active metabolites in Japanese pediatric patients with fungal infections. Biol Pharm Bull. 2006;29(8):1706–11.
Wasmann RE, Muilwijk EW, Burger DM, Verweij PE, Knibbe CA, Bruggemann RJ. Clinical pharmacokinetics and pharmacodynamics of micafungin. Clin Pharmacokinet. 2018;57(3):267–86.
Groll AH, Gullick BM, Petraitiene R, Petraitis V, Candelario M, Piscitelli SC, et al. Compartmental pharmacokinetics of the antifungal echinocandin caspofungin (MK-0991) in rabbits. Antimicrob Agents Chemother. 2001;45(2):596–600.
Niwa T, Yokota Y, Tokunaga A, Yamato Y, Kagayama A, Fujiwara T, et al. Tissue distribution after intravenous dosing of micafungin, an antifungal drug, to rats. Biol Pharm Bull. 2004;27(7):1154–6.
Goldblum D, Fausch K, Frueh BE, Theurillat R, Thormann W, Zimmerli S. Ocular penetration of caspofungin in a rabbit uveitis model. Gr Arch Clin Exp Ophthalmol. 2007;245(6):825–33.
Mochizuki K, Sawada A, Suemori S, Kawakami H, Niwa Y, Kondo Y, et al. Intraocular penetration of intravenous micafungin in inflamed human eyes. Antimicrob Agents Chemother. 2013;57(8):4027–30.
Strenger V, Farowski F, Muller C, Hofer N, Dornbusch HJ, Sperl D, et al. Low penetration of caspofungin into cerebrospinal fluid following intravenous administration of standard doses. Int J Antimicrob Agents. 2017;50(2):272–5.
Stone JA, Xu X, Winchell GA, Deutsch PJ, Pearson PG, Migoya EM, et al. Disposition of caspofungin: role of distribution in determining pharmacokinetics in plasma. Antimicrob Agents Chemother. 2004;48(3):815–23.
Damle B, Stogniew M, Dowell J. Pharmacokinetics and tissue distribution of anidulafungin in rats. Antimicrob Agents Chemother. 2008;52(7):2673–6.
Gabardi S, Martin S, Sura M, Mohammed A, Golan Y. Micafungin treatment and eradication of candiduria among hospitalized patients. Int Urol Nephrol. 2016;48(11):1881–5.
Grau S, Luque S, Echeverria-Esnal D, Sorli L, Campillo N, Montero M, et al. Urinary micafungin levels are sufficient to treat urinary tract infections caused by Candida spp. Int J Antimicrob Agents. 2016;48(2):212–4.
Rezai MS, Vaezi A, Fakhim H, Soleimani A, Mohammad Jafari H, Mohseni S, et al. Successful treatment with caspofungin of candiduria in a child with Wilms tumor; review of literature. Journal de Mycologie Medicale. 2017;27(2):261–5.
Sobel JD, Bradshaw SK, Lipka CJ, Kartsonis NA. Caspofungin in the treatment of symptomatic candiduria. Clin Infect Dis. 2007;44(5):e46–9.
Hope WW, Mickiene D, Petraitis V, Petraitiene R, Kelaher AM, Hughes JE, et al. The pharmacokinetics and pharmacodynamics of micafungin in experimental hematogenous Candida meningoencephalitis: implications for echinocandin therapy in neonates. J Infect Dis. 2008;197(1):163–71.
Jans J, Bruggemann RJ, Christmann V, Verweij PE, Warris A. Favorable outcome of neonatal cerebrospinal fluid shunt-associated Candida meningitis with caspofungin. Antimicrob Agents Chemother. 2013;57(5):2391–3.
Liu KH, Wu CJ, Chou CH, Lee HC, Lee NY, Hung ST, et al. Refractory candidal meningitis in an immunocompromised patient cured by caspofungin. J Clin Microbiol. 2004;42(12):5950–3.
Okugawa S, Ota Y, Tatsuno K, Tsukada K, Kishino S, Koike K. A case of invasive central nervous system aspergillosis treated with micafungin with monitoring of micafungin concentrations in the cerebrospinal fluid. Scand J Infect Dis. 2007;39(4):344–6.
Auriti C, Falcone M, Ronchetti MP, Goffredo BM, Cairoli S, Crisafulli R, et al. High-dose micafungin for preterm neonates and infants with invasive and central nervous system candidiasis. Antimicrob Agents Chemother. 2016;60(12):7333–9.
Hope WW, Smith PB, Arrieta A, Buell DN, Roy M, Kaibara A, et al. Population pharmacokinetics of micafungin in neonates and young infants. Antimicrob Agents Chemother. 2010;54(6):2633–7.
Vazquez JA. Anidulafungin: a new echinocandin with a novel profile. Clin Ther. 2005;27(6):657–73.
Chandrasekar PH, Sobel JD. Micafungin: a new echinocandin. Clin Infect Dis. 2006;42(8):1171–8.
Balani SK, Xu X, Arison BH, Silva MV, Gries A, DeLuna FA, et al. Metabolites of caspofungin acetate, a potent antifungal agent, in human plasma and urine. Drug Metab Dispos. 2000;28(11):1274–8.
Weiler S, Seger C, Pfisterer H, Stienecke E, Stippler F, Welte R, et al. Pharmacokinetics of caspofungin in critically ill patients on continuous renal replacement therapy. Antimicrob Agents Chemother. 2013;57(8):4053–7.
Aguilar G, Ferriols R, Lozano A, Ezquer C, Carbonell JA, Jurado A, et al. Optimal doses of caspofungin during continuous venovenous hemodiafiltration in critically ill patients. Crit Care. 2017;21(1):17.
Roger C, Wallis SC, Muller L, Saissi G, Lipman J, Bruggemann RJ, et al. Caspofungin population pharmacokinetics in critically ill patients undergoing continuous veno-venous haemofiltration or haemodiafiltration. Clin Pharmacokinet. 2017;56(9):1057–68.
Kurland S, Furebring M, Lowdin E, Eliasson E, Nielsen EI, Sjolin J. Pharmacokinetics of caspofungin in critically ill patients in relation to liver dysfunction: differential impact of plasma albumin and bilirubin levels. Antimicrob Agents Chemother. 2019;63:e02466–18. https://doi.org/10.1128/AAC.02466-18.
Hope WW, Kaibara A, Roy M, Arrieta A, Azie N, Kovanda LL, et al. Population pharmacokinetics of micafungin and its metabolites M1 and M5 in children and adolescents. Antimicrob Agents Chemother. 2015;59(2):905–13.
Hope WW, Seibel NL, Schwartz CL, Arrieta A, Flynn P, Shad A, et al. Population pharmacokinetics of micafungin in pediatric patients and implications for antifungal dosing. Antimicrob Agents Chemother. 2007;51(10):3714–9.
Mehta PA, Vinks AA, Filipovich A, Bleesing J, Jodele S, Jordan MB, et al. Alternate-day micafungin antifungal prophylaxis in pediatric patients undergoing hematopoietic stem cell transplantation: a pharmacokinetic study. Biol Blood Marrow Transplant. 2010;16(10):1458–62.
Bochennek K, Balan A, Muller-Scholden L, Becker M, Farowski F, Muller C, et al. Micafungin twice weekly as antifungal prophylaxis in paediatric patients at high risk for invasive fungal disease. J Antimicrob Chemother. 2015;70(5):1527–30.
Chandra S, Fukuda T, Mizuno K, Davies SM, Teusink-Cross A, Tarin R, et al. Micafungin antifungal prophylaxis in children undergoing HSCT: can we give higher doses, less frequently? A pharmacokinetic study. J Antimicrob Chemother. 2018;73(6):1651–8.
Lehrnbecher T, Bochennek K, Klingebiel T, Gastine S, Hempel G, Groll AH. Extended dosing regimens for fungal prophylaxis. Clin Microbiol Rev. 2019;32:e00010–19. https://doi.org/10.1128/CMR.00010-19.
Heresi GP, Gerstmann DR, Reed MD, van den Anker JN, Blumer JL, Kovanda L, et al. The pharmacokinetics and safety of micafungin, a novel echinocandin, in premature infants. Pediatr Infect Dis J. 2006;25(12):1110–5.
Smith PB, Walsh TJ, Hope W, Arrieta A, Takada A, Kovanda LL, et al. Pharmacokinetics of an elevated dosage of micafungin in premature neonates. Pediatr Infect Dis J. 2009;28(5):412–5.
Li CC, Sun P, Dong Y, Bi S, Desai R, Dockendorf MF, et al. Population pharmacokinetics and pharmacodynamics of caspofungin in pediatric patients. Antimicrob Agents Chemother. 2011;55(5):2098–105.
Yang XM, Leroux S, Storme T, Zhang DL, de Beaumais TA, Shi HY, et al. Body surface area-based dosing regimen of caspofungin in children: a population pharmacokinetics confirmatory study. Antimicrob Agents Chemother. 2019;63:e00248–19. https://doi.org/10.1128/AAC.00248-19.
Neely M, Jafri HS, Seibel N, Knapp K, Adamson PC, Bradshaw SK, et al. Pharmacokinetics and safety of caspofungin in older infants and toddlers. Antimicrob Agents Chemother. 2009;53(4):1450–6.
Walsh TJ, Adamson PC, Seibel NL, Flynn PM, Neely MN, Schwartz C, et al. Pharmacokinetics, safety, and tolerability of caspofungin in children and adolescents. Antimicrob Agents Chemother. 2005;49(11):4536–45.
Saez-Llorens X, Macias M, Maiya P, Pineros J, Jafri HS, Chatterjee A, et al. Pharmacokinetics and safety of caspofungin in neonates and infants less than 3 months of age. Antimicrob Agents Chemother. 2009;53(3):869–75.
Gustot T, Ter Heine R, Brauns E, Cotton F, Jacobs F, Bruggemann RJ. Caspofungin dosage adjustments are not required for patients with Child-Pugh B or C cirrhosis. J Antimicrob Chemother. 2018;73(9):2493–6.
Muilwijk EW, Schouten JA, van Leeuwen HJ, van Zanten AR, de Lange DW, Colbers A, et al. Pharmacokinetics of caspofungin in ICU patients. J Antimicrob Chemother. 2014;69(12):3294–9.
Yang QT, Zhai YJ, Chen L, Zhang T, Yan Y, Meng T, et al. Whole-body physiology-based pharmacokinetics of caspofungin for general patients, intensive care unit patients and hepatic insufficiency patients. Acta Pharmacol Sin. 2018;39(9):1533–43.
Benjamin DK Jr, Driscoll T, Seibel NL, Gonzalez CE, Roden MM, Kilaru R, et al. Safety and pharmacokinetics of intravenous anidulafungin in children with neutropenia at high risk for invasive fungal infections. Antimicrob Agents Chemother. 2006;50(2):632–8.
Cohen-Wolkowiez M, Benjamin DK Jr, Piper L, Cheifetz IM, Moran C, Liu P, et al. Safety and pharmacokinetics of multiple-dose anidulafungin in infants and neonates. Clin Pharmacol Ther. 2011;89(5):702–7.
Benjamin DK Jr, Deville JG, Azie N, Kovanda L, Roy M, Wu C, et al. Safety and pharmacokinetic profiles of repeated-dose micafungin in children and adolescents treated for invasive candidiasis. Pediatr Infect Dis J. 2013;32(11):e419–25.
Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother. 2000;46(2):171–9.
Harris BE, Manning BW, Federle TW, Diasio RB. Conversion of 5-fluorocytosine to 5-fluorouracil by human intestinal microflora. Antimicrob Agents Chemother. 1986;29(1):44–8.
Hope WW, Warn PA, Sharp A, Reed P, Keevil B, Louie A, et al. Optimization of the dosage of flucytosine in combination with amphotericin B for disseminated candidiasis: a pharmacodynamic rationale for reduced dosing. Antimicrob Agents Chemother. 2007;51(10):3760–2.
Pasqualotto AC, Howard SJ, Moore CB, Denning DW. Flucytosine therapeutic monitoring: 15 years experience from the UK. J Antimicrob Chemother. 2007;59(4):791–3.
Soltani M, Tobin CM, Bowker KE, Sunderland J, MacGowan AP, Lovering AM. Evidence of excessive concentrations of 5-flucytosine in children aged below 12 years: a 12-year review of serum concentrations from a UK clinical assay reference laboratory. Int J Antimicrob Agents. 2006;28(6):574–7.
Baley JE, Meyers C, Kliegman RM, Jacobs MR, Blumer JL. Pharmacokinetics, outcome of treatment, and toxic effects of amphotericin B and 5-fluorocytosine in neonates. J Pediatr. 1990;116(5):791–7.
Stamm AM, Diasio RB, Dismukes WE, Shadomy S, Cloud GA, Bowles CA, et al. Toxicity of amphotericin B plus flucytosine in 194 patients with cryptococcal meningitis. Am J Med. 1987;83(2):236–42.
Vermes A, van Der Sijs H, Guchelaar HJ. Flucytosine: correlation between toxicity and pharmacokinetic parameters. Chemotherapy. 2000;46(2):86–94.
Gupta AK, Adamiak A, Cooper EA. The efficacy and safety of terbinafine in children. J Eur Acad Dermatol Venereol. 2003;17(6):627–40.
Zehender H, Denouel J, Roy M, Le Saux L, Schaub P. Simultaneous determination of terbinafine (Lamisil) and five metabolites in human plasma and urine by high-performance liquid chromatography using on-line solid-phase extraction. J Chromatogr B Biomed Appl. 1995;664(2):347–55.
Jones TC. Overview of the use of terbinafine (Lamisil) in children. Br J Dermatol. 1995;132(5):683–9.
Nejjam F, Zagula M, Cabiac MD, Guessous N, Humbert H, Lakhdar H. Pilot study of terbinafine in children suffering from tinea capitis: evaluation of efficacy, safety and pharmacokinetics. Br J Dermatol. 1995;132(1):98–105.
Vazquez JA. Clinical practice: combination antifungal therapy for mold infections: much ado about nothing? Clin Infect Dis. 2008;46(12):1889–901.
Abdel-Rahman SM, Herron J, Fallon-Friedlander S, Hauffe S, Horowitz A, Riviere GJ. Pharmacokinetics of terbinafine in young children treated for tinea capitis. Pediatr Infect Dis J. 2005;24(10):886–91.
Revankar SG, Nailor MD, Sobel JD. Use of terbinafine in rare and refractory mycoses. Future Microbiol. 2008;3(1):9–17.
Dolton MJ, Perera V, Pont LG, McLachlan AJ. Terbinafine in combination with other antifungal agents for treatment of resistant or refractory mycoses: investigating optimal dosing regimens using a physiologically based pharmacokinetic model. Antimicrob Agents Chemother. 2014;58(1):48–54.
Araujo OE, Flowers FP, King MM. Griseofulvin: a new look at an old drug. DICP. 1990;24(9):851–4.
Ginsburg CM, McCracken GH Jr, Petruska M, Olsen K. Effect of feeding on bioavailability of griseofulvin in children. J Pediatr. 1983;102(2):309–11.
Chen X, Jiang X, Yang M, Bennett C, Gonzalez U, Lin X, et al. Systemic antifungal therapy for tinea capitis in children: an abridged cochrane review. J Am Acad Dermatol. 2017;76(2):368–74.
Knasmuller S, Parzefall W, Helma C, Kassie F, Ecker S, Schulte-Hermann R. Toxic effects of griseofulvin: disease models, mechanisms, and risk assessment. Crit Rev Toxicol. 1997;27(5):495–537.
Khoza S, Moyo I, Ncube D. Comparative hepatotoxicity of fluconazole, ketoconazole, itraconazole, terbinafine, and griseofulvin in rats. J Toxicol. 2017;2017:6746989.
Fisher BT. The changing landscape for paediatric regulation of pharmaceutical agents with a focus on antifungal agents. Curr Fungal Infect Rep. 2016;10(1):1–6.
Seibel NL, Schwartz C, Arrieta A, Flynn P, Shad A, Albano E, et al. Safety, tolerability, and pharmacokinetics of micafungin (FK463) in febrile neutropenic pediatric patients. Antimicrob Agents Chemother. 2005;49(8):3317–24.
Autmizguine J, Guptill JT, Cohen-Wolkowiez M, Benjamin DK Jr, Capparelli EV. Pharmacokinetics and pharmacodynamics of antifungals in children: clinical implications. Drugs. 2014;74(8):891–909.
Debruyne D, Ryckelynck JP. Clinical pharmacokinetics of fluconazole. Clin Pharmacokinet. 1993;24(1):10–27.
Michael C, Bierbach U, Frenzel K, Lange T, Basara N, Niederwieser D, et al. Voriconazole pharmacokinetics and safety in immunocompromised children compared to adult patients. Antimicrob Agents Chemother. 2010;54(8):3225–32.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
KJD is supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under award number K23HD091365 and has received research support from Merck & Co., Inc. and Pfizer, Inc. unrelated to the current work. BTF has received research support from Pfizer, Inc. and Merck Pharmaceuticals unrelated to the current work. BTF also serves as the Chair of a data safety monitoring board for Astellas. NRZ is supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under award number 1K99HD096123. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the above supporting agencies.
Rights and permissions
About this article

Cite this article
Downes, K.J., Fisher, B.T. & Zane, N.R. Administration and Dosing of Systemic Antifungal Agents in Pediatric Patients. Pediatr Drugs 22, 165–188 (2020). https://doi.org/10.1007/s40272-020-00379-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-020-00379-2