
Abstract
Purpose of review
This article provides updates on antifungals, dosing strategies for safe and effective therapy in the critically ill, including special populations, and the understanding of resistance over the last 5 years.
Recent findings
Reports of adverse effects with echinocandins have risen while antifungal resistance to this class has increased, especially in Candida glabrata. New formulations of posaconazole and isuvaconazole have been developed. Alternative dosing strategies including combination therapy are being evaluated for difficult to treat fungal infections. Other highlights include additional data on dosing patients with severe organ dysfunction, including those on continuous renal replacement therapy, and new breakpoints for individual Candida species being established for the echinocandins and triazole classes.
Summary
Increasing resistance in Candida spp. has made susceptibility testing a standard of care for critically ill patients. New formulations of the triazole antifungals have made prevention and treatment of mold infections more of a reality. There are many implications that must be considered when treating critically ill patients due to alterations in pharmacokinetics and pharmacodynamics in order to ensure adequate treatment. This article exposes the need for further clinical research in treating invasive infections in this patient population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is considerable morbidity and mortality associated with invasive fungal infections, especially in critically ill patients. Factors placing this patient population at high risk include the use of broad spectrum antibiotics, parenteral nutrition, immunosuppression (especially transplant recipients and those receiving chemotherapy), intra-abdominal surgery, prolonged intensive care unit (ICU) stay, and renal failure [1]. Of the fungal infections associated with this patient population, Candida spp. are by far the most common accounting for 70 to 90%. Timely and adequate antifungal treatment plays a crucial role in patient outcomes [2]. Critically ill patients often have altered pharmacokinetics and pharmacodynamics, which greatly impact distribution, bioavailability, and clearance of life-saving medications [3]. New agents and dosage forms have recently been released or reformulated which add to an arsenal of antifungals that better combat these diseases. Knowledge of the medications and specific caveats associated with dosing can have significant impact on patients in the ICU.
Echinocandins
The echinocandins (caspofungin, micafungin, and anidulafungin) exert their antifungal effect by inhibiting the synthesis of the polysaccharide 1,3 beta-D-glucan causing degradation of the cell wall leading to cell lysis. Echinocandins possess concentration-dependent fungicidal activity against all Candida species, along with fungistatic activity against Aspergillus species. Candida strains with drug minimum inhibitory concentration (MICs) of <2 mcg/mL were considered susceptible for all three drugs prior to 2012 [4]. Revised breakpoints have been established by the Clinical and Laboratory Standards Institute (CLSI) based on data from clinical studies, pharmacokinetic/pharmacodynamic studies, and epidemiologic cutoff values for each agent and species (Table 1) [5]. From a pharmacodynamic standpoint, AUC/MIC is the parameter associated with outcomes in treatment of Candida species [6], whereas the Cmax/MEC should be optimized when echinocandins are used for invasive aspergillosis [7].
Because of their long half-lives, all echinocandins may be administered once daily. Dosing is summarized in Table 2 [8••]. Of note, US labeling states that the daily dose of caspofungin may be increased to 70 mg daily in patients without adequate response or receiving concomitant inducers of drugs clearance (e.g., rifampin, carbamazepine, dexamethasone, phenytoin, and efavirenz), while the European labeling recommends a daily dose of 70 mg in patients weighing more than 80 kg [9, 10].
Echinocandins possess excellent in vitro activity against Candida species, and standard dosing regimens may be adequate for many ICU patients. Nonetheless, the potential need for higher echinocandin dosing in critically ill patients has been investigated in multiple recent studies. In 2009, high-dose caspofungin (150 mg daily) was compared to standard dosing in non-ICU patients with invasive candidiasis. It was found that both regimens were equally effective, with slightly more adverse events in the high-dose group [11]. However, a recent study of caspofungin pharmacokinetics in 20 ICU patients revealed suboptimal drug exposure in 50% of the patients when standard doses were used. A weight-based dosing strategy of 1 mg/kg per day was proposed to be more suitable in this population [12]. Additionally, a recent study described the pharmacokinetic variability of micafungin 100 mg daily in ICU patients with sepsis and mechanical ventilation. Total micafungin exposure was lower in ICU patients compared to non-ICU patients. The authors suggested that the recommended dose of 100 mg daily will rarely attain target AUC/MIC values in Candida albicans or Candida glabrata infections with MIC >0.015 mg/L and any cases of infection due to Candida parapsilosis [13]. The impact of this effect on clinical outcomes remains to be seen.
As a class, the echinocandins are generally quite safe, but recent publications point to an association between echinocandins and cardiotoxicity. The proposed mechanism of toxicity involves mitochondrial damage due to inhibition of oxidative phosphorylation during loading doses [14, 15•]. A case series of three ICU patients receiving empiric anidulafungin or caspofungin described temporary drops in cardiac index or mean arterial pressure (MAP) during infusion [16]. Of note, all of these patients had a history of left ventricular hypertrophy or dysfunction. The difference in toxicity between agents may be attributable to the higher lipophilicity of anidulafungin and caspofungin compared to micafungin [15•, 17], but polymorphic ventricular tachycardia has been reported in a patient receiving micafungin [18]. A recent small, prospective study evaluating hemodynamic parameters in 15 ICU patients before and after the administration of caspofungin or anidulafungin found no clinically significant changes in heart rate, blood pressure, cardiac index, or required dose of vasopressors [19]. Close hemodynamic monitoring may be prudent in critically ill patients receiving echinocandins, especially during loading doses.
Dose adjustment of echinocandins based on organ function is typically not required. This feature is especially attractive in the critically ill population who are at a high risk of multiple organ failure. In hepatic dysfunction (Child–Pugh scores 7–9, class B), the recommended dose of caspofungin is reduced to 35 mg daily given after the typical loading dose [10]. However, a recent study suggested that caspofungin dose reduction for liver dysfunction may be associated with inadequate drug exposure in ICU patients with a Child–Pugh B score. This may be driven by hypoalbuminemia and the authors suggest avoiding dose reduction in this population [9]. There is insufficient information regarding the use of caspofungin or micafungin in patients with severe liver disease (Child–Pugh class C), however, anidulafungin may be used without dose adjustment in ICU patients with any degree of hepatic dysfunction due to minimal alternations in pharmacokinetics [20]. The echinocandins are not dialyzable, but drug exposure may be altered in patients undergoing continuous renal replacement (CRRT) therapy due to decreased protein binding and drug loss via filter membrane adsorption [21]. Pharmacokinetic parameters of each echinocandin have been investigated in small populations of patients requiring various modalities of CRRT. Minimal echinocandin adsorption by hemofilters is reported across studies, suggesting that regular doses are likely adequate in patients on CRRT [21,22,23].
Echinocandin pharmacokinetics have also been described in the case reports of patients requiring extracorporeal membrane oxygenation (ECMO), which is associated with an increased volume of distribution and the potential for drug loss in the ECMO circuit. Reported plasma concentrations of caspofungin in ECMO patients vary between undetectable [24] and normal [25]. A case report of anidulafungin used during ECMO reported similar Cmax and Cmin levels in critically ill patients not requiring ECMO and suggested that anidulafungin dose adjustments during ECMO are unnecessary [26]. However, a recent ex vivo study evaluating the sequestration of highly protein bound drugs (including caspofungin) in ECMO circuits reported significant drug loss to the circuit, with only 56% recovery of caspofungin at 24 h [27]. The extent of echinocandin drug loss during in vivo ECMO therapy and the possible effect of this interaction on drug dosing and patient outcomes is yet to be determined.
Limited information exists regarding dosing of echinocandins in obesity. Increased body weight has been associated with increased volume of distribution and clearance of all echinocandins [28]. Pharmacokinetics of both caspofungin and micafungin have shown decreased peak concentrations and AUC when standard doses are used, suggesting the need for dose escalation of echinocandins in obesity [29, 30]. A bedside equation for dose optimization of micafungin in obesity based on Monte Carlo simulation has been proposed, but is yet to be evaluated in human subjects [31]. A case report of micafungin therapy for C. glabrata candiduria in a morbidly obese patient (BMI 102 kg/m2) reported successful therapy with the normal dose of 100 mg daily despite significantly decreased serum levels [32]. In a retrospective study evaluating the safety and efficacy of caspofungin in obese patients, the incidence of favorable outcomes in esophageal candidiasis and invasive candidiasis was similar between obese and non-obese patients. Only 2% of the study population was considered morbidly obese (BMI >40), limiting the generalizability of the results to all obese patients [33].
Echinocandins are the recommended initial therapy for most episodes of candidemia and invasive candidiasis in the recently updated Infectious Disease Society of America (IDSA) practice guidelines [8••]. A pooled analysis of nine randomized trials evaluating antifungal therapy in candidemia and invasive candidiasis found that initial treatment with an echinocandin was associated with improved survival rates compared to therapy with a polyene or triazole [34••]. As empiric therapy, no difference could be detected in 30-day survival free from fungal infection when empiric micafungin was administered to patients with ICU-acquired sepsis however [35]. Echinocandins should be avoided as therapy for infections of the CNS and eye because of limited penetration in these sites. Due to minimal urinary excretion (<1–10%), the echinocandins are also generally avoided for the treatment of candiduria. In patients with fluconazole-resistant Candida species or contraindications to azole therapy, micafungin has the highest renal elimination and has been successfully used for treatment of urinary tract infections [36, 37].
With increased use of echinocandins for non-C. albicans species over the last decade, reports of resistance have begun to increase nationwide. Susceptibility rates for C. glabrata have decreased to 93% on average across four sentinel sites monitored by the CDC [38]. The increased MICs are attributed to a mutation in the fks gene encoding for 1,3 beta-D glucan synthase. Prior echinocandin use is the largest established risk factor for this resistance and it correlates to clinically worse clinical outcomes [39•, 40]. C. parapsilosis is associated with higher in vitro MICs for echinocandins compared to other Candida species due to naturally occurring polymorphisms in the fks1 gene [41]. Recent publications however report similar outcomes in patients with C. parapsilosis candidemia treated with either fluconazole or an echinocandin [41, 42]. The 2016 update of the IDSA guidelines no longer clearly supports fluconazole over echinocandin use for C. parapsilosis, but recommends that echinocandin susceptibility testing should be considered in patients with infections due to that species or prior echinocandin use, especially with C. glabrata [8••].
Triazoles
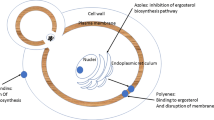
Triazoles inhibit 14-α-demethylase preventing the conversion of lanosterol to ergosterol and ultimately causing a disruption of the fungal cell membrane. Each agent differs pharmacokinetically leading to varying activity between fungal species. Because of the variable differences with in this class if antifungals, great care must be taken when selecting an agent especially in critically ill patients [43].
Fluconazole
Fluconazole is active against most Candida species. The exceptions are Candida krusei which is intrinsically resistant and strains of C. glabrata due to acquired resistance (efflux pumps) [44]. Fluconazole remains the drug of choice for the treatment of oral (100–200 mg/day) and esophageal (200–400 mg) candidiasis [8••]. It is regularly used in treating infections caused by Cryptococcus neoformans and coccidioidomycosis (400–800 mg/day) [45, 46]. In critically ill patients with cryptococcal meningitis, fluconazole may be used as a part of induction therapy (800 mg/day) in combination with amphotericen B when flucytosine is unavailable, but is most commonly use as consolidation (400 mg/day × 8–10 weeks) or suppressive maintenance therapy (200 mg/day × 6–12 months) [47]. When treating invasive candidiasis, a loading dose of 800 mg (12 mg/kg) followed by a maintenance dose of 400 mg (6 mg/kg) daily is recommended [8••]. For infections caused by C. glabrata susceptible to fluconazole, higher doses of a load of 1600 mg (24 mg/kg) followed by 800 mg (12 mg/kg) daily are recommended to overcome the susceptible dose-dependent range seen with C. glabrata. When possible dosing should be based on actual body weight in obese patients [48].
Before deciding on dosing for the critically ill, the pharmacokinetic properties of fluconazole must be considered. Oral formulations have excellent bioavailability (>90%) making for an easy transition from IV to oral therapy. The oral formulation, unlike other agents in this class, is not affected by acid suppression and can be administered via a gastric tube [49, 51]. The volume of distribution and protein binding are low, but fluconazole is still able to reach therapeutic levels in the central nervous system (CNS) and attains good tissue penetration [49, 50]. Fluconazole is predominantly excreted, 60 to 70%, unmetabolized by the kidneys, making it extremely useful in the treatment of candiduria (200 mg/day). If C. glabrata or pyelonephritis is suspected, increasing the dose to 400 mg/day may be considered. Due to the high level of excretion in the urine, dosage adjustments are necessary in renal dysfunction [50, 51]. Fluconazole doses should be reduced by 50% or dosing interval extended to every 48 h for a creatinine clearance (CrCl) < 50 ml/min. Fluconazole is extensively removed by dialysis (25–40% removed in 4 h) due to its low degree of protein binding. Patients undergoing intermittent dialysis should receive unadjusted doses after each dialysis session or 50% of the usual dose given daily [49, 51]. CRRT removes an increased amount of fluconazole and is dependent on the modality of CRRT used. Continuous venovenous hemofiltration (CVVH) requires fluconazole 200–400 mg (3–6 mg/kg) every 24 h; continuous venovenous hemodialysis (CVVHD) requires 400–800 mg (6–12 mg/kg) every 24 h; and continuous venovenous hemodiafiltration (CVVHDF) requires 800 mg (12 mg/kg) every 24 h [52, 53]. Adjustments in dosing are not necessary for hepatic impairment. Fluconazole is an inhibitor of CYP2C9 and CYP3A4, which leads to numerous drug interactions [54, 55].
Voriconazole
Voriconazole is a broad spectrum azole used in the critically ill. It has activity against all Candida spp. (potential cross-resistance does exist with fluconazole; especially C. glabrata), Aspergillus spp., and molds such as Scedosporium spp. and Fusarium spp. Voriconazole is the drug of choice for invasive aspergillosis and is an alternative or step-down therapy in Candida infections [8••, 56•]. It is available in both oral and intravenous formulations. For invasive fungal infections, a loading dose of 6 mg/kg IV every 12 h for two doses on day 1, followed by 4 mg/kg IV twice daily is recommended. Oral bioavailability is excellent (>90% in healthy subjects), which allows for easy conversion of therapy when the patient stabilizes. Recent accounts show more variability in the critically ill with oral bioavailability as low as 63% [49, 57]. Absorption of the oral formulation is decreased by high-fat foods and should be given 1 h before meals or 3 h after meals to optimize absorption. In patients receiving continuous enteral nutrition, ideally, nutrition should be interrupted in order to allow complete absorption of voriconazole [43, 50, 51].
Voriconazole is able to achieve excellent lung, CNS, and vitreous concentrations due to a large volume of distribution and high-tissue penetration [48,49,50]. It is extensively metabolized in the liver via CYP2C19 and is a potent inhibitor of enzymes such as CYP3A4 and CYP2C9, which leads to numerous interactions with other medications. Dosage adjustments are necessary in patients with severe hepatic impairment. The usual loading dose (6 mg/kg IV every 12 h) is followed by a decreased maintenance dose of 2 mg/kg twice daily. Dosage adjustments are not necessary in patients with renal dysfunction or hemodialysis for the oral formulations. Originally, the use of the IV formulation was considered contraindicated in patients with a CrCl < 50 mL/min because of potential concerns with accumulation of the cyclodextrin carrier vehicle. However, this has not been determined to be clinical relevant as kidney damage has only been established with repeated doses of cyclodextrin in animal models [58]. In a human study of 166 patients, neither baseline renal dysfunction, voriconazole route of administration, nor a combination of these factors was seen to impact renal impairment [59].
There are numerous patient factors that may lead to variability with voriconazole levels including pharmacokinetics during critical illness, drug interactions, or genetic factors associated with polymorphisms with CYP2C19. Therapeutic drug monitoring is recommended for patients who are unresponsive to voriconazole therapy or at risk for adverse effects. Trough concentrations should be drawn after 2 to 3 days of therapy and should be in a goal range of 2 to 6 mg/L [60•, 61].
Itraconazole
Itraconazole is a broad spectrum agent with activity against Candida spp., Aspergillus spp., and endemic fungi. Due to its unpredictable pharmacokinetic profile, it is reserved as a step-down, alternative, or salvage therapy [43]. Drug level monitoring is recommended because of its multifaceted pharmacokinetic profile to ensure both efficacy and limit toxicity. Levels are drawn after 5 to 7 days of therapy and the trough should be >1 mcg/mL for treatment doses and >0.5 mcg/mL for prophylaxis dosing [60•, 61]. Itraconazole is available only in oral formulations and, due to its long half-life, should be given as a loading dose of 200 mg three times a day for 3 days, followed by 200 mg once or twice daily [43].
Neither the capsule nor oral solution formulations are readily absorbed. The solution has enhanced absorption compared to the capsule because it is formulated with a cyclodextrin carrier increasing hydrophilicity. Absorption of the capsules is significantly increased in an acidic environment so it should be taken with food or a cola beverage and be separated from acid-suppressive medications. Unlike the capsule, the oral solution is best absorbed on an empty stomach [43]. Itraconazole undergoes liver metabolized primarily through CYP3A4 and it is also a potent CYP340 inhibitor, which leads to several clinically relevant drug interactions [49]. Due to the extensive liver involvement, dosage adjustment is necessary in patients with severe hepatic impairment. Adjustments in dosing are not necessary in the setting of renal dysfunction or hemodialysis [51].
Posaconazole
Posaconazole was originally approved as a suspension for the prophylaxis of invasive candidiasis and aspergillosis in severely immunocompromised patients, but the medication is now available as both delayed-release oral tablets and IV. Posaconazole has an extended spectrum of activity that includes endemic dimgorphic fungi, zygomycetes, and Fusarium spp. The medication possesses a large volume of distribution, but limited CNS penetration. Renal and hepatic dose adjustments are not necessary due its extensive excretion in the feces as unmetabolized drug. Posaconazole is a potent inhibitor of CYP3A4 and has numerous drug interactions [49].
The recommended dose of suspension is 200 mg three times a day for prophylaxis and 800 mg divided two to four times a day for the treatment of invasive fungal infections. Bioavailability of the oral suspension is increased with high fat meals and is highly dependent on both gastric pH and emptying time results in significant clinical variability in posaconazole drug concentrations. Dosing can further be challenging in critically ill patients because absorption is altered when co-administered with acid suppressing agent [62]. In a prospective observational study, 187 posaconazole serum concentrations were analyzed from 31 patients. Of those, 80 (43%) were found to be below the target trough level of 700 μg/L and 68% of patients had at least one concentration below goal [63]. Due to the significant pharmacokinetic challenges seen with posaconazole oral suspension, the IV and delayed-release tablet were introduced in 2014. The delayed-release tablet is designed to disperse the medication at the higher pH environment found in the small intestine, which greatly improves absorption [64]. Dosing for the tablet and IV formulation is the same: two 300 mg doses as a load, separated 12 h apart on the first day, followed by 300 mg once daily starting on the second day of therapy [65]. Like voriconazole, the IV formulation of posaconazole contains cyclodextrin as a carrier vehicle. As previously discussed, the presence of this has not been determined to be clinically relevant [66]. With these changes, the achievement of goal serum concentration should theoretically be easier. A retrospective study evaluating trough levels in oncology patients showed that posaconazole delayed-release tablets were able to attain appropriate trough concentrations in most patients. However, lower mean trough levels were seen in patients experiencing diarrhea (650 ± 80 μg/L vs. 1300 ± 130 μg/L) and in patients with a weight ≥90 kg (740 ± 90 μg/L) as compared to patients with lower weight (1320 ± 140 μg/L). The same trend was seen in patients with a body mass index (BMI) ≥ 30 (890 ± 130 μg/L) vs. patients with a BMI < 30 (1290 ± 140 μg/L) [67]. While no study has specifically evaluated trough concentrations with posaconazole delayed-release tablets or IV formulation in critically ill patients, these findings suggest that therapeutic drug monitoring may need to be considered in this population. Anecdotally, the authors have observed sub-therapeutic concentrations even when the drug is given IV.
Isavuconazole
Isavuconazole is the newest antifungal agent. It was approved by the FDA in 2015 for the treatment of invasive aspergillosis and invasive mucormycosis [68]. Similar to posaconazole, isavuconazole has an extended spectrum with activity against a large range of yeasts, dimorphic fungi, and molds including those in the Zygomycetes family (Mucor spp., Rhizopus spp., Lichtheimia spp., and Rhizomucor spp.) [69]. Isavuconazonium sulfate is a water-soluble prodrug that is rapidly hydrolyzed to isavuconazole, the active component. Isavuconazonium sulfate is available in both oral and intravenous formulations [70•]. For both, the recommended dosing regimen is a loading dose of isavuconazonium sulfate 372 mg (200 mg isavuconazole) every 8 h for six doses, followed by a maintenance dose of 372 mg once daily [69]. Because the prodrug is highly water soluble, the intravenous formulation does not require solubilization by a cyclodextrin vehicle which decreases concern for potential nephrotoxicity.
Dosage adjustments for renal and hepatic impairment are not necessary. Isavuconazole is metabolized in the liver by CYP3A4 and CYP3A5. Although clearance of isavuconazole is impaired in patients with hepatic impairment, dosage adjustments are not recommended for these patients. Since isavuconazole is a substrate of CYP3A4, drug interactions are important considerations [69]. Isavuconazole has 98% oral bioavailabilty, its absorption is not affected by food, and it follows linear pharmacokinetics [71•]. At this time, no correlation between serum concentrations and outcomes has been determined but clinical experience is still lacking. Therefore, therapeutic drug monitoring is not yet routinely recommended for isavuconazole, but the same could be said for other triazoles upon initial approval. Its role in therapy may be that it has the potential for fewer adverse effects than other azoles. In trials for aspergillosis, there were less hepatobiliary and ocular reactions compared to voriconazole. Rather than causing a risk of QTc prolongation, caution is advised when isavuconazole is used in patients with short QTc.
When used for the treatment of invasive aspergillosis, isavuconazole and voriconazole were found to have similar rates of all-cause mortality and success at the end of treatment. An open-label non-comparative trial looked at the use of isavuconazole for the treatment of invasive mucormycosis. Patients had an all-cause mortality rate at day 42 of 38% and an overall success rate of 31%. The mortality rates seen were similar with mortality rates of 35–45% seen in previous studies with amphotericin B for the treatment of invasive mucormycosis.
Amphotericin B
Amphotericin B (AmB) is a member of the polyene antifungal class. It exerts concentration-dependent fungicidal activity by increasing membrane permeability through interactions with ergosterol [72]. Concentrations needed for fungicidal activity are 4–10× the organism’s MIC, and it is important to note only a fraction of AmB is microbiologically active in tissue [73, 74]. Since there is no evidence that distribution takes place in adipose tissue, dosing in the obese can be based on an ideal body weight [48]. The medication produces a post-antifungal effect and pharmacodynamics allow for once daily dosing [75]. Metabolism and route of elimination for AmB are not well understood [48]. Oral absorption is poor, and AmB is most commonly administered intravenously. Intrathecal and inhalation administrations are used in unique circumstances [43, 76].
AmB is a broad spectrum agent and is the drug of choice for cryptococcal meningitis and endemic mycoses in the critically ill. Other uses include salvage therapy or unknown infections when empiric antifungal treatment is desired, especially in cases of neutropenic hosts [43, 56•, 77]. AmB can be used as a prophylactic agent but is most often avoided because of the efficacious aforementioned oral agents with less toxicity. Usual dosing for conventional AmB is 0.7 mg/kg IV daily (0.25–1.5 mg/kg/day), although doses of 0.3 mg/kg/day are suited for the treatment of minor infections [78]. Increased doses, 1 to 1.5 mg/kg/day, can be considered in severe invasive fungal infections or infections caused by organisms with increased resistance to preferred agents [8••, 79]. Treatment of severe invasive infections is often combined with surgery and requires several months of IV therapy [79]. Extended durations and high doses are associated with increased rates of nephrotoxicity [80]. In patients developing nephrotoxicity, the dose may be halved or the dosing interval increased to every other day to prevent further renal compromise. Due to infusion-related reactions like fever, chills, rigors, and cardiac arrhythmias, administration of conventional AmB should be over 4 to 6 h. Those requiring renal replacement therapy do not necessitate a dose adjustment due to AmB being poorly dialyzable. Intrathecal doses of AmB range from 0.1 to 1.5 (typically 0.5) mg and are reserved for severely ill patients with central nervous system infections refractory to IV administration [48].
The use of inhaled amphotericin has been reported since 1959, but there is a limited quality clinical data regarding this method of administration [81]. Nebulization is a strategy for minimizing the toxicities of the medication while providing localized therapy. Though the risk for systemic toxicity is reduced, there is a concern for direct lung toxicity which may be due to alterations in lung surfactant, especially with conventional amphotericin. Selection of a nebulizer is another important consideration due to its vital role in medication aerosolization and subsequent treatment success [76]. Dosing has not been standardized and is dependent on the amphotericin formulation, fungal organism, and nebulizer used [76, 81]. This has led to a variety of reported doses ranging from 3 to 100 mg daily to 50 mg once weekly [75]. As for its place in therapy, nebulized amphotericin may be an option as adjunctive therapy for treatment of tracheobronchial aspergillosis in combination with a systemic triazole, prophylactic therapy for pulmonary aspergillus in lung transplant patients, and as an alternative treatment of allergic bronchopulmonary aspergillosis [56•, 76]. Limited data exists for use in other invasive pulmonary fungal infections.
On account of the toxicities associated with conventional AmB, additional formulations are being investigated. New research on ion complexes, nanoparticles, protein-phospholipid bioparticles, and liquid crystals indicate potential future directions for this drug [82,83,84,85]. Lipid formulations are currently the only alternative designs approved for use. Existing alternative products include amphotericin B colloidal dispersion (ABCD), amphotericin B lipid complex (ABLC), and liposomal amphotericin B (L-AmB). All have shown comparable efficacy to conventional amphotericin B [43]. Dosing differs from conventional AmB and ranges from 3 to 5 mg/kg given as a single daily IV infusion. Infusion times are also decreased when compared to conventional AmB. ABLC and L-AmB can be given over 2 h, while AmB deoxycholate is administered over 4 h. As with conventional AmB, higher doses of L-AmB have been used for cryptococcal meningitis (6 mg/kg/day) and zygomycosis (10–15 mg/kg/day) [79, 86]. Two modern studies have assessed high-dose liposomal amphotericin. The AmBiLoad trial randomized patients to a loading phase of 10 mg/kg/day for 2 weeks compared to the standard regimen (3 mg/kg/day) in treatment of invasive aspergillosis. No difference in efficacy was seen, but nephrotoxicity and hypokalemia were significantly increased with the higher dosing [87, 88••]. The more recent AmbiZygo trial also evaluated doses of 10 mg/kg/day for 4 weeks in patients with mucormycosis. Creatinine levels doubled from baseline in 40% of patients (16/40) with no improvement in mortality (38% at 12 weeks) [89•].
Due to the increased mortality associated with mucormycosis, combination therapy with lipid formulations of amphotericin B paired with echinocandins, deferasirox, or triazoles has been studied [90]. Despite no inherent activity from echinocandins, the combination of capsofungin plus amphotericin B lipid complex has the promising, but limited, data, supporting its use for mucormycosis [91].
Flucytosine
Flucytosine is classified as a pyrimidine analog and exerts activity through inhibition of fungal DNA synthesis [43]. The cost of flucytosine has significantly increased since 2009. A 2-week course of flucytosine increased to roughly $28,000 based on 2015 whole sale cost in the USA [92]. It is primarily used in the treatment of cryptococcal meningitis in combination with another agent due to its increased propensity to develop resistance when used by itself. Most commonly, flucytosine is combined with amphotericin B due to increased rates of CSF clearance compared to amphotericin B with fluconazole [93]. Flucytosine is associated with significant bone marrow suppression and can be seen with traditional dosing of 150 mg/kg/day [78]. Peak serum levels should be measured 3 to 5 days into therapy and be maintained between 30 and 80 mcg/mL in order to decrease adverse effects. Lowering the daily dose to 75–100 mg/kg to minimize toxicity is appropriate when combined with another antifungal agent [46, 91]. Due to renal excretion, dose adjustments are required for patients with renal impairment (creatinine clearance <50 mL/min). Limited data exists regarding dosing in patients requiring renal replacement therapy. Suggested dosing ranges from 20 to 50 mg/kg after dialysis sessions and 37.5 mg/kg every 12–24 h for continuous renal replacement therapy (CVVHD/CVVH) [94].
Conclusion
This update provides pertinent dosing considerations and indications for antifungal agents commonly used in the critically ill. The pharmacokinetic information provided may greatly impact the safety and efficacy of therapy and subsequent patient outcomes when treating invasive fungal infections. Additional data would be useful in establishing dosing for special populations such as those with liver dysfunction, kidney failure, or morbid obesity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ruping MJ, Vehreschild JJ, Cornely OA. Patients at high risk of invasive fungal infections: when and how to treat. Drugs. 2008;68:1941–62.
Calandra T, Roberts JA, Antonelli M, Bassetti M, Vincent JL. Diagnosis and management of invasive candidiasis in the ICU: an updated approach to an old enemy. Crit Care. 2016;20:125. doi:10.1186/s13054-016-1313-6.
Varghese JM, Roberts JA, Lipman J. Antimicrobial pharmacokinetic and pharmacodynamic issues in the critically ill with severe sepsis and septic shock. Crit Care Clin. 2011;27:19–34.
Chen SC, Slavin MA, Sorrell TC. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs. 2011;71:11–41.
Fothergill AW, Sutton DA, McCarthy DI, Wiederhold NP. Impact of new antifungal breakpoints on antifungal resistance in Candida species. J Clin Microbiol. 2014;52:994–7.
Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, Lepak A. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother. 2010;54:2497–506.
Wiederhold NP, Kontoyiannis DP, Chi J, Prince RA, Tam VH, Lewis RE. Pharmacodynamics of caspofungin in a murine model of invasive pulmonary aspergillosis: evidence of concentration-dependent activity. J Infect Dis. 2004;190:1464–71.
•• Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, et al. Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:409–17. This is the latest version of practice guidelines for treatment of candidiasis issued by the Infectious Diseases Society of America.
Martial LC, Bruggemann RJ, Schouten JA, van Leeuwen HJ, van Zanten AR, de Lange DW, et al. Dose reduction of caspofungin in intensive care unit patients with child Pugh B will result in suboptimal exposure. Clin Pharmacokinet. 2016;55:723–33.
Cancidas [Prescribing Information]. Whitehouse Station: Merck & Co., Inc; 2016.
Betts RF, Nucci M, Talwar D, Gareca M, Queiroz-Telles F, Bedimo RJ, et al. A Multicenter, double-blind trial of a high-dose caspofungin treatment regimen versus a standard caspofungin treatment regimen for adult patients with invasive candidiasis. Clin Infect Dis. 2009;48:1676–84.
van der Elst KC, Veringa A, Zijlstra JG, Beishuizen A, Klont R, Brummelhuis-Visser P, Uges DR, Touw DJ, Kosterink JG, van der Werf TS, Alffenaar JC. Low caspofungin exposure in patients in the Intensive Care Unit. Antimicrob Agents Chemother. 2016. doi:10.1128/AAC.01582-16.
Jullien V, Azoulay E, Schwebel C, Le Saux T, Charles PE, Cornet M, et al. Population pharmacokinetics of micafungin in ICU patients with sepsis and mechanical ventilation. J Antimicrob Chemother. 2017;72(1):181–189.
Stover KR, King ST, Cleary JD. Cardiac toxicity of the echinocandins: chance or cause and effect association? J Clin Pharm Ther. 2014;39:1–3.
• Cleary JD, Stover KR. Antifungal-associated drug-induced cardiac disease. Clin Infect Dis. 2015;61 Suppl 6:S662–8. Good review of antifungal-induced cardiac toxicities, especially for echinocandins.
Lichtenstern C, Wolff M, Arens C, Klie F, Majeed RW, Henrich M, et al. Cardiac effects of echinocandin preparations - three case reports. J Clin Pharm Ther. 2013;38:429–31.
Stover KR, Cleary JD. Cardiac response to centrally administered echinocandin antifungals. J Pharm Pharmacol. 2015;67:1279–83.
Shah PJ, Sundareshan V, Miller B, Bergman SJ. Micafungin and a case of polymorphic ventricular tachycardia. J Clin Pharm Ther. 2016;41:362–4.
Lahmer T, Schnappauf C, Messer M, Rasch S, Fekecs L, Beitz A, et al. Influence of echinocandin administration on hemodynamic parameters in medical intensive care unit patients: a single center prospective study. Infection. 2015;43:723–7.
Liu P, Ruhnke M, Meersseman W, Paiva JA, Kantecki M, Damle B. Pharmacokinetics of anidulafungin in critically ill patients with candidemia/invasive candidiasis. Antimicrob Agents Chemother. 2013;57:1672–6.
Gonzalez de Molina F, Martinez-Alberici Mde L, Ferrer R. Treatment with echinocandins during continuous renal replacement therapy. Crit Care. 2014;18:218.
Aguilar G, Azanza JR, Carbonell JA, Ferrando C, Badenes R, Parra MA, et al. Anidulafungin dosing in critically ill patients with continuous venovenous haemodiafiltration. J Antimicrob Chemother. 2014;69:1620–3.
Weiler S, Seger C, Pfisterer H, Stienecke E, Stippler F, Welte R, et al. Pharmacokinetics of caspofungin in critically ill patients on continuous renal replacement therapy. Antimicrob Agents Chemother. 2013;57:4053–7.
Ruiz S, Papy E, Da Silva D, Nataf P, Massias L, Wolff M, et al. Potential voriconazole and caspofungin sequestration during extracorporeal membrane oxygenation. Intensive Care Med. 2009;35:183–4.
Spriet I, Annaert P, Meersseman P, Hermans G, Meersseman W, Verbesselt R, et al. Pharmacokinetics of caspofungin and voriconazole in critically ill patients during extracorporeal membrane oxygenation. J Antimicrob Chemother. 2009;63:767–70.
Aguilar G, Ferriols R, Carbonell JA, Ezquer C, Alonso JM, Villena A, et al. Pharmacokinetics of anidulafungin during venovenous extracorporeal membrane oxygenation. Crit Care. 2016;20:325.
Shekar K, Roberts JA, Mcdonald CI, Ghassabian S, Anstey C, Wallis SC, et al. Protein-bound drugs are prone to sequestration in the extracorporeal membrane oxygenation circuit: results from an ex vivo study. Crit Care. 2015;19(1):164. doi:10.1186/s13054-015-0891-z.
Muilwijk EW, Lempers VJ, Burger DM, Warris A, Pickkers P, Aarnoutse RE, et al. Impact of special patient populations on the pharmacokinetics of echinocandins. Expert Rev Anti-Infect Ther. 2015;13:799–815.
Hall 2nd RG, Swancutt MA, Meek C, Leff R, Gumbo T. Weight drives caspofungin pharmacokinetic variability in overweight and obese people: fractal power signatures beyond two-thirds or three-fourths. Antimicrob Agents Chemother. 2013;57:2259–64.
Hall RG, Swancutt MA, Gumbo T. Fractal geometry and the pharmacometrics of micafungin in overweight, obese, and extremely obese people. Antimicrob Agents Chemother. 2011;55:5107–12.
Pasipanodya JP, Hall 2nd RG, Gumbo T. In silico-derived bedside formula for individualized micafungin dosing for obese patients in the age of deterministic chaos. Clin Pharmacol Ther. 2015;97:292–7.
Zomp A, Bookstaver PB, Ahmed Y, Turner JE, King C. Micafungin therapy in a critically ill, morbidly obese patient. J Antimicrob Chemother. 2011;66:2678–80.
Ryan DM, Lupinacci RJ, Kartsonis NA. Efficacy and safety of caspofungin in obese patients. Med Mycol. 2011;49:748–54.
•• Andes DR, Safdar N, Baddley JW, Playford G, Reboli AC, Rex JH, et al. Impact of treatment strategy on outcomes in patients with candidemia and other forms of invasive candidiasis: a patient-level quantitative review of randomized trials. Clin Infect Dis. 2012;54:1110–22. Meta-analysis of antifungal studies show improved treatment outcomes for invasive candidiasis when echinocandins used.
Timsit JF, Azoulay E, Schwebel C, Charles PE, Cornet M, Souweine B, et al. Empirical micafungin treatment and survival without invasive fungal infection in adults with ICU-acquired sepsis, Candida colonization, and multiple organ failure: the EMPIRICUS randomized clinical trial. JAMA. 2016;316:1555–64.
Gabardi S, Martin S, Sura M, Mohammed A, Golan Y. Micafungin treatment and eradication of candiduria among hospitalized patients. Int Urol Nephrol. 2016;48:1881–5.
Grau S, Luque S, Echeverria-Esnal D, Sorli L, Campillo N, Montero M, et al. Urinary micafungin levels are sufficient to treat urinary tract infections caused by Candida spp. Int J Antimicrob Agents. 2016;48:212–4.
Vallabhaneni S, Cleveland AA, Farley MM, Harrison LH, Schaffner W, Beldavs ZG, et al. Epidemiology and risk factors for echinocandin nonsusceptible Candida glabrata bloodstream infections: data from a large multisite population-based candidemia surveillance program, 2008-2014. Open Forum Infect Dis. 2015;2:ofv163.
• Beyda ND, John J, Kilic A, Alam MJ, Lasco TM, Garey KW. FKS mutant Candida glabrata: risk factors and outcomes in patients with candidemia. Clin Infect Dis. 2014;59:819–25. Risk factor for FKS mutation leading to echinocandin resistance is prior echinocandin use.
Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R, et al. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis. 2013;56:1724–32.
Chiotos K, Vendetti N, Zaoutis TE, Baddley J, Ostrosky-Zeichner L, Pappas P, et al. Comparative effectiveness of echinocandins versus fluconazole therapy for the treatment of adult candidaemia due to Candida parapsilosis: a retrospective observational cohort study of the Mycoses Study Group (MSG-12). J Antimicrob Chemother. 2016;71:3536–9.
Fernandez-Ruiz M, Aguado JM, Almirante B, Lora-Pablos D, Padilla B, Puig-Asensio M, et al. Initial use of echinocandins does not negatively influence outcome in Candida parapsilosis bloodstream infection: a propensity score analysis. Clin Infect Dis. 2014;58:1413–21.
Bergman SJ, Tyagi I, Ronald K. Antifungal dosing in critically ill patients. Curr Fungal Infect Rep. 2010;4:78–86.
Pennisi M, Antonelli M. Clinical aspects of invasive candidiasis in critically ill patients. Drugs. 2009;69 Suppl 1:21–8.
Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63:e112–46.
Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of America. Clin Infect Dis. 2010;50:291–322.
Pappas PG, Chetchotisakd P, Larsen RA, Manosuthi W, Morris MI, Anekthananon T, et al. A phase II randomized trial of amphotericin B alone or combined with fluconazole in the treatment of HIV-associated cryptococcal meningitis. Clin Infect Dis. 2009;48:1775–83.
Bergman SJ, Speil C, Short M, Koirala J. Pharmacokinetic and pharmacodynamic aspects of antibiotic use in high-risk populations. Infect Dis Clin N Am. 2007;21:821-46, x.
Lipp HP. Antifungal agents--clinical pharmacokinetics and drug interactions. Mycoses. 2008;51 Suppl 1:7–18.
Thompson GR, 3rd, Cadena J, Patterson TF. Overview of antifungal agents. Clin Chest Med. 2009;30:203-15, v.
Rex JH SD. Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. Elsevier Health Sciences; 2014.
Trotman RL, Williamson JC, Shoemaker DM, Salzer WL. Antibiotic dosing in critically ill adult patients receiving continuous renal replacement therapy. Clin Infect Dis. 2005;41:1159–66.
Heintz BH, Matzke GR, Dager WE. Antimicrobial dosing concepts and recommendations for critically ill adult patients receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy. 2009;29:562–77.
Nivoix Y, Leveque D, Herbrecht R, Koffel JC, Beretz L, Ubeaud-Sequier G. The enzymatic basis of drug-drug interactions with systemic triazole antifungals. Clin Pharmacokinet. 2008;47:779–92.
Bruggemann RJ, Alffenaar JW, Blijlevens NM, Billaud EM, Kosterink JG, Verweij PE, et al. Clinical relevance of the pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis. 2009;48:1441–58.
• Patterson TF, Thompson 3rd GR, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice guidelines for the diagnosis and management of Aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63:e1–60. Recent update of practice guidelines and summary of literature for the treatment of aspergillosis.
Pascual A, Csajka C, Buclin T, Bolay S, Bille J, Calandra T, et al. Challenging recommended oral and intravenous voriconazole doses for improved efficacy and safety: population pharmacokinetics-based analysis of adult patients with invasive fungal infections. Clin Infect Dis. 2012;55:381–90.
Luke DR, Tomaszewski K, Damle B, Schlamm HT. Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci. 2010;99:3291–301.
Neofytos D, Lombardi LR, Shields RK, Ostrander D, Warren L, Nguyen MH, et al. Administration of voriconazole in patients with renal dysfunction. Clin Infect Dis. 2012;54:913–21.
• Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British society for medical mycology. J Antimicrob Chemother. 2014;69:1162–76. Review of target serum concentrations for efficacy and toxicity when monitoring antifungal agents.
Smith J, Andes D. Therapeutic drug monitoring of antifungals: pharmacokinetic and pharmacodynamic considerations. Ther Drug Monit. 2008;30:167–72.
Percival KM, Bergman SJ. Update on posaconazole pharmacokinetics: comparison of old and new formulations. Curr Fungal Infect Rep. 2014;8:139–45.
Gross BN, Ihorst G, Jung M, Wasch R, Engelhardt M. Posaconazole therapeutic drug monitoring in the real-life setting: a single-center experience and review of the literature. Pharmacotherapy. 2013;33:1117–25.
Krishna G, Ma L, Martinho M, Preston RA, O’Mara E. A new solid oral tablet formulation of posaconazole: a randomized clinical trial to investigate rising single- and multiple-dose pharmacokinetics and safety in healthy volunteers. J Antimicrob Chemother. 2012;67:2725–30.
Noxafil [Prescribing Information]. Whitehouse Station: Merck & Co., Inc; 2016.
Maertens J, Cornely OA, Ullmann AJ, Heinz WJ, Krishna G, Patino H, et al. Phase 1B study of the pharmacokinetics and safety of posaconazole intravenous solution in patients at risk for invasive fungal disease. Antimicrob Agents Chemother. 2014;58:3610–7.
Miceli MH, Perissinotti AJ, Kauffman CA, Couriel DR. Serum posaconazole levels among haematological cancer patients taking extended release tablets is affected by body weight and diarrhoea: single centre retrospective analysis. Mycoses. 2015;58:432–6.
Cresemba [Prescribing Information]. Northbrook: Astellas, Inc.; 2016.
Miceli MH, Kauffman CA. Isavuconazole: a new broad-spectrum triazole antifungal agent. Clin Infect Dis. 2015;61:1558–65.
• Marty FM, Ostrosky-Zeichner L, Cornely OA, Mullane KM, Perfect JR, Thompson 3rd GR, et al. Isavuconazole treatment for mucormycosis: a single-arm open-label trial and case-control analysis. Lancet Infect Dis. 2016;16:828–37. Treatment results leading to orphan drug indication for isavuconazole against mucormycosis.
• Maertens JA, Raad II, Marr KA, Patterson TF, Kontoyiannis DP, Cornely OA, et al. Isavuconazole versus voriconazole for primary treatment of invasive mould disease caused by Aspergillus and other filamentous fungi (SECURE): a phase 3, randomised-controlled, non-inferiority trial. Lancet. 2016;387:760–9. Clinical trial results showing equal efficacy of isavuconazole and fewer adverse effects than voriconazole for invasive asgergillosis.
Lewis 2nd JS, Graybill JR, Fungicidal versus fungistatic: what’s in a word? Expert Opin Pharmacother. 2008;9:927–35.
Andes D, Safdar N, Marchillo K, Conklin R. Pharmacokinetic-pharmacodynamic comparison of amphotericin B (AMB) and two lipid-associated AMB preparations, liposomal AMB and AMB lipid complex, in murine candidiasis models. Antimicrob Agents Chemother. 2006;50:674–84.
Lewis RE, Wiederhold NP. The solubility ceiling: a rationale for continuous infusion amphotericin B therapy? Clin Infect Dis. 2003;37:871–2.
Lewis RE. Pharmacodynamic implications for use of antifungal agents. Curr Opin Pharmacol. 2007;7:491–7.
Kuiper L, Ruijgrok EJ. A review on the clinical use of inhaled amphotericin B. J Aerosol Med Pulm Drug Deliv. 2009;22:213–27.
Alvarez-Lerma F, Mariscal F, Quintana E, Rialp G, Diaz-Reganon J, Perez MJ, et al. Use of liposomal amphotericin B in critically ill patients: a retrospective, multicenter, clinical study. J Chemother. 2009;21:330–7.
Dismukes WE. Antifungal therapy: lessons learned over the past 27 years. Clin Infect Dis. 2006;42:1289–96.
Petrikkos GL. Lipid formulations of amphotericin B as first-line treatment of zygomycosis. Clin Microbiol Infect. 2009;15 Suppl 5:87–92.
Pasqualotto AC. Amphotericin B: the higher the dose, the higher the toxicity. Clin Infect Dis. 2008;47:1110. author reply 1110-1.
Patel TS, Eschenauer GA, Stuckey LJ, Carver PL. Antifungal prophylaxis in lung transplant recipients. Transplantation. 2016;100:1815–26.
Chuealee R, Aramwit P, Noipha K, Srichana T. Bioactivity and toxicity studies of amphotericin B incorporated in liquid crystals. Eur J Pharm Sci. 2011;43:308–17.
Burgess BL, He Y, Baker MM, Luo B, Carroll SF, Forte TM, et al. NanoDisk containing super aggregated amphotericin B: a high therapeutic index antifungal formulation with enhanced potency. Int J Nanomedicine. 2013;8:4733–43.
Jain V, Gupta A, Pawar VK, Asthana S, Jaiswal AK, Dube A, et al. Chitosan-assisted immunotherapy for intervention of experimental leishmaniasis via amphotericin B-loaded solid lipid nanoparticles. Appl Biochem Biotechnol. 2014;174:1309–30.
Gola J, Strzalka-Mrozik B, Kruszniewska-Rajs C, Janiszewski A, Skowronek B, Gagos M, et al. A new form of amphotericin B - the complex with copper (II) ions - downregulates sTNFR1 shedding and changes the activity of genes involved in TNF-induced pathways: AmB-Cu2+ downregulates sTNFR1 shedding and changes the activity of genes involved in TNF-induced pathways. Pharmacol Rep. 2016;69:22–8.
Hamill RJ, Sobel JD, El-Sadr W, Johnson PC, Graybill JR, Javaly K, et al. Comparison of 2 doses of liposomal amphotericin B and conventional amphotericin B deoxycholate for treatment of AIDS-associated acute cryptococcal meningitis: a randomized, double-blind clinical trial of efficacy and safety. Clin Infect Dis. 2010;51:225–32.
Cornely OA, Maertens J, Bresnik M, Ebrahimi R, Ullmann AJ, Bouza E, et al. Liposomal amphotericin B as initial therapy for invasive mold infection: a randomized trial comparing a high-loading dose regimen with standard dosing (AmBiLoad trial). Clin Infect Dis. 2007;44:1289–97.
•• Cornely OA, Arikan-Akdagli S, Dannaoui E, Groll AH, Lagrou K, Chakrabarti A, et al. ESCMID and ECMM joint clinical guidelines for the diagnosis and management of mucormycosis 2013. Clin Microbiol Infect. 2014;20 Suppl 3:5–26. Although no practice guidelines exist from IDSA for treatment of mucormycosis, this European version exhibits the best compilation of literature available.
• Lanternier F, Poiree S, Elie C, Garcia-Hermoso D, Bakouboula P, Sitbon K, et al. Prospective pilot study of high-dose (10 mg/kg/day) liposomal amphotericin B (L-AMB) for the initial treatment of mucormycosis. J Antimicrob Chemother. 2015;70:3116–23. The AmbiZygo trial evaluated 10 mg/kg doses of liposomal amphotericin B over 4 weeks for mucormycosis and showed substantially more nephrotoxicity toxicity without greater efficacy.
Spellberg B, Ibrahim A, Roilides E, Lewis RE, Lortholary O, Petrikkos G, et al. Combination therapy for mucormycosis: why, what, and how? Clin Infect Dis. 2012;54 Suppl 1:S73–8.
Reed C, Bryant R, Ibrahim AS, Edwards Jr J, Filler SG, Goldberg R, et al. Combination polyene-caspofungin treatment of rhino-orbital-cerebral mucormycosis. Clin Infect Dis. 2008;47:364–71.
Merry M, Boulware DR. Cryptococcal meningitis treatment strategies affected by the explosive cost of flucytosine in the United States: a cost-effectiveness analysis. Clin Infect Dis. 2016;62:1564–8.
Brouwer AE, Rajanuwong A, Chierakul W, Griffin GE, Larsen RA, White NJ, et al. Combination antifungal therapies for HIV-associated cryptococcal meningitis: a randomised trial. Lancet. 2004;363:1764–7.
Kunka ME, Cady EA, Woo HC, Thompson Bastin ML. Flucytosine pharmacokinetics in a critically Ill patient receiving continuous renal replacement therapy. Case Rep Crit Care. 2015;2015:927496.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Tyson E. Dietrich, Carolyn J. Pfeifer, Kelsey E. Aker, and Scott J. Bergman declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pharmacology and Pharmacodynamics of Antifungal Agents
Rights and permissions
About this article

Cite this article
Dietrich, T.E., Pfeifer, C.J., Aker, K.E. et al. Antifungal Dosing Strategies for Critically Ill Patients. Curr Fungal Infect Rep 11, 5–15 (2017). https://doi.org/10.1007/s12281-017-0270-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-017-0270-0