
Abstract
The clinical management of glioblastoma (GBM) is still bereft of treatments able to significantly improve the poor prognosis of the disease. Despite the extreme clinical need for novel therapeutic drugs, only a small percentage of patients with GBM benefit from inclusion in a clinical trial. Moreover, often clinical studies do not lead to final interpretable conclusions. From the mistakes and negative results obtained in the last years, we are now able to plan a novel generation of clinical studies for patients with GBM, allowing the testing of multiple anticancer agents at the same time. This assumes critical importance, considering that, thanks to improved knowledge of altered molecular mechanisms related to the disease, we are now able to propose several potential effective compounds in patients with both newly diagnosed and recurrent GBM. Among the novel compounds assessed, the initially great enthusiasm toward trials employing immune checkpoint inhibitors (ICIs) was disappointing due to the negative results that emerged in three randomized phase III trials. However, novel biological insights into the disease suggest that immunotherapy can be a convincing and effective treatment in GBM even if ICIs failed to prolong the survival of these patients. In this regard, the most promising approach consists of engineered immune cells such as chimeric antigen receptor (CAR) T, CAR M, and CAR NK alone or in combination with other treatments. In this review, we discuss several issues related to systemic treatments in GBM patients. First, we assess critical issues toward the planning of clinical trials and the strategies employed to overcome these obstacles. We then move on to the most relevant interventional studies carried out on patients with previously untreated (newly diagnosed) GBM and those with recurrent and pretreated disease. Finally, we investigate novel immunotherapeutic approaches with special emphasis on preclinical and clinical data related to the administration of engineered immune cells in GBM.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
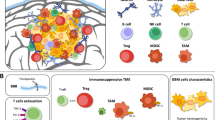
Improved design and planning of clinical studies allows for a more efficient conduct of these trials and allows a larger number of patients to potentially benefit from experimental treatment for glioblastoma (GBM). |
Novel biological insights are leading to the assessment of innovative treatment strategies. Engineered immune cells are cells obtained from the patient and modified to target and attack tumor cells. Although promising, available data about their efficacy in GBM are still limited to date. |
1 Introduction
According to the 2021 World Health Organization (WHO) classification of primary central nervous system (CNS) malignancies, glioblastoma (GBM) is diagnosed as a diffuse astrocytic glioma without IDH/H3R mutations, with microvascular proliferation, necrosis, and/or specific gene alterations such as TERT mutation, epidermal growth factor receptor (EGFR) amplification and chromosome rearrangement (loss of chromosome 10/gain chromosome 7) [1, 2].
The estimated incidence of GBM is 3.22/100.000 cases in the US, making this malignancy the most common primary brain tumor [3, 4]. GBM is associated with a dismal prognosis, with an expected 5-year overall survival (OS) rate of only 6.8% [3, 4]. Since 2005, the standard of care of newly diagnosed GBM is represented by maximal safe resection surgery followed by temozolomide concurrent with and adjuvant to radiotherapy [4,5,6,7]. Nonetheless, the prognosis of newly diagnosed GBM ranges from 12 to 18 months [8,9,10]. Recurrence after surgery is almost certain and life expectancy after tumor relapse decreases to 5–10 months [8,9,10].
Several trials tested novel therapeutic drugs in both newly diagnosed and recurrence settings, however the majority of the trials failed to show significant clinical improvements. An exception is made by tumor-treating fields (TTFs), which were shown to improve the survival of patients with newly diagnosed GBM [11].
The complex biology of the disease, the high mutation heterogeneity observed between cancer cells, the presence of a natural protection represented by the blood–brain barrier (BBB), and an altered tumor-associated microenvironment (TME) are all elements explaining the lack of therapeutic improvements against this disease [12,13,14].
There are few recognized therapeutic options, especially in the recurrence setting. Both locoregional approaches and systemic treatments can be proposed at the time of recurrence [15,16,17]. Considering the lack of effective treatments, the enrollment of GBM patients in clinical trials should be strongly encouraged. Nonetheless, only 10% of GBM patients can access an experimental drug [8, 10, 18]. Immune checkpoint inhibitors (ICIs) have represented a revolution for the management of patients with solid tumor. These same agents failed to show a significant clinical improvement in patients with primary and recurrent GBM [19, 20]. However, novel immune therapeutic approaches such as chimeric antigen receptor (CAR) M and CAR T cells are under investigation [21, 22].
In this review, we summarize ongoing experimental treatments in GBM, including novel trials exploring immunotherapeutic approaches. In the first section of the current manuscript we discuss critical issues and novel methods adopted to improve clinical trial designs. We further proceed with a discussion of more important studies on both newly diagnosed GBM and recurrent disease (progressed on standard radiochemotherapy and adjuvant chemotherapy). In the second and final part of the paper, we focus our attention on immunotherapy trials and, in particular, on the studies employing engineered lymphocytes.
2 Improving Trial Design in Glioblastoma (GBM)
Several biological issues related to GBM could explain the lack of efficacy of several compounds tested in clinical trials, however further improvements in the design of studies might be of utmost importance (Fig. 1).

Summary of novel strategies adopted to improve clinical trial design. Adaptive phase II studies can allow to modify several parameters of the study according to the results of a primary interim analyses. Phase 0 studies can identify drugs more able to cross the brain–blood barrier. R randomization
In a survey published in 2018 [23] exploring clinical trials enrolling patients with GBM registered on ClinicalTrials.gov from 2005 to 2016, the following points emerged:
-
1.
over 60% of clinical studies were phase I/II or II trials and only 5 of 249 trials were randomized;
-
2.
a relevant rate of these studies (1 to 10) stopped due to futility, funding, or lack of accrual;
-
3.
despite the small number of patients required, time to completion of phase II studies was 3–4 years;
-
4.
phase III trials represented only 7% and only 12 of 16 of these trials were supported by a previous phase II study [23].
The lack of an effective and strong surrogate endpoint for OS is a well-known issue in clinical trials carried out on GBM and can partially explain the long time required to complete small phase I/II studies [24,25,26,27,28]. Surrogate endpoints such as progression-free survival (PFS) or overall response rate (ORR) are frequent endpoints adopted within trials on solid malignancies that can significantly reduce the time of study completion. Other endpoints such as post-progression survival could be proposed among clinical trials in the recurrent GBM setting [29].
The increasing number of compounds to test and the long time required for study completion are problems to consider in the trial planning process. Even the development of unpowered early efficacy trials should be discouraged as results of these studies could be difficult to interpret and could lead to the early stop of potentially active compounds.
To address these limitations, patients with GBM should be treated within reference centers and the development of cooperative networks between these institutions should be strongly encouraged. Organizational improvement and increased investments in trial planning and patients on trial tutelage are also important issues [30,31,32]. The inclusion of a comparator arm into phase II studies helped to achieve a more accurate selection of compounds to test in phase III trials [33]. However, randomized phase II trials required a higher number of patients and a longer time to completion compared with the mono-arm phase II trials. The adoption of a Bayesian study design could partially reduce these limitations [34]. In a Bayesian model, the probability is modified in the course of the study according to the results observed. This allows to modify treatment allocation and/or sample size while the study is ongoing, optimizing enrollment and assessment of the most promising compounds [34]. These Bayesian adaptive randomized (AR) trials are assuming increasing interest and trials such as AGILE, INSIGhT, and N2M2 represent excellent examples [35,36,37].
AGILE (The Adaptive Global Innovative Learning Environment) is a multi-arm, platform trial composed of two statistical designs in which Bayesian AR trials constitute the first phase of compound assessment. Patients with IDH wild-type GBM and newly diagnosed/recurrent disease can be enrolled in this trial regardless of MGMT status. The first Bayesian AR phase aims to select more promising compounds and the population in which these are expected to be more active, at the same time reducing the number of patients needed and enlarging the number of potentially active compounds to test [35]. Similar to AGILE, the INSIGhT clinical trial adopted a Bayesian AR trial in the first phase [36]. The difference between AGILE and INSIGhT is mainly represented by the different populations included. In the INSIGhT trial, only patients with full genomic data, unmethylated MGMT, and IDHR132H wild-type could be included. The NCT Neuro Master Match (N2M2) is an umbrella trial for patients with MGMT unmethylated IDH wild-type GBM [37] in which treatments are provided according to the molecular and genomic background of the disease. In this study, there are two distinct phases: a discovery phase and a treatment phase. The treatment phase is carried out through stratification of the population obtained after the discovery phase. The Bayesian model is essential to provide monitoring of toxicity during the two phases [37].
Bayesian AR trials can also be employed to assess the correlation between OS and a surrogate endpoint. Indeed, information provided in the course of a clinical trial can demonstrate a correlation or no-correlation between a surrogate endpoint and OS, allowing to keep or refute the use of the surrogate [38]. The use of a historical control cohort could avoid the presence of a comparator arm but is associated with the risk of several biases. Indeed, there is also a trend toward a progressive OS improvement in patients treated with standard treatment, suggesting that the historical cohort is not associated with a stable survival rate [33, 39]. In addition, it has been well reported that the intertrial variability reflects a variable distribution of the endpoint of interest, which significantly increases the risk to underestimate or overestimate the benchmarks [33].
Other surrogate endpoints such as ORR are difficult to be employed on GBM. Indeed, the assessment of the response requires additional clinical data (type of treatment providers, molecular background of the disease, clinical symptoms, etc.) as well as dimensional and imaging criteria to estimate the response to treatment. The use of functional imaging provided by magnetic resonance imaging (MRI) and positron emission tomography (PET) is increasing and could be a promising tool to achieve integration between molecular, clinical, and imaging data [40,41,42,43,44,45,46]. Novel technologies adopting artificial intelligence algorithms are also working in this direction [47, 48].
Finally, another trial design could be of critical importance in GBM. The ‘phase 0’ studies are trials in which compounds of interest are provided before a planned surgery. Partial or complete removal of the tumor mass can allow assessment of the biological effect of the agent provided and the concentration achieved into the tumor mass or the percentage passing the BBB [49].
Interestingly, the programmed death receptor-1 (PD-1) inhibitor pembrolizumab has been assessed before second surgery in patients with recurrent GBM. Post-surgical assessment of the tumor mass revealed that administration of pembrolizumab resulted in a significant modification of TME [50] and survival data suggested a promising role in this setting.
3 Newly Diagnosed GBM
The standard post-surgical approach for newly diagnosed GBM was established in 2005 and is represented by temozolomide concurrent with and adjuvant to radiotherapy (60 Gy over 6 weeks) [5]. Patients treated with this sequence achieved an OS of 14.6 months [5], with the best clinical improvement observed in patients with MGMT promoter methylation [7]. In the past years, novel therapeutic approaches have been assessed in this setting in an attempt to improve these results.
TTFs consist of low-intensity, 200 kHz alternating electric fields provided to the tumor, resulting in antimitotic effects [11]. In the EF-14 phase III clinical trial, the addition of TTFs to adjuvant temozolomide compared with adjuvant temozolomide alone resulted in PFS and OS improvement (median OS 20 months vs. 16 months, hazard ratio [HR] 0.63, 95% confidence interval [CI] 0.53–0.76; p < 0.0001). The use of TTFs is limited in clinical practice (only 3–12% of patients) due to the refusal of patients to wear the device, the high costs of treatment, and the uncertain mechanism of action associated with doubts about the favorable outcomes observed [11].
The combination of lomustine and temozolomide has been investigated in the CeTeG/NOA-09 trial [51], in which 141 patients with MGMT-methylated GBM were randomized to receive standard adjuvant temozolomide or up to six courses of lomustine (100 mg/m2 on day 1) plus temozolomide (100–200 mg/m2 per day on days 2–6 of the 6-week course) in addition to radiotherapy (59–60 Gy). The OS observed was longer in patients receiving the combination of lomustine and temozolomide (48.1 vs. 31.4 months, HR 0.60, 95% CI 0.35–1.03; p = 0.0492), while no benefit in terms of PFS has been observed. The absence of PFS improvement and the small cohort of patients enrolled exposes the risk of biases and limited the inclusion of this schedule in clinical practice. Furthermore, other trials investigating a dose-dense temozolomide regimen or extensive temozolomide adjuvant treatment failed to show a significant improvement in terms of survival [52, 53], suggesting that a chemotherapy-intensified regimen could not be associated with clinical improvement in newly diagnosed GBM. The ANOCEF group have proposed a randomized phase III trial (NCT03663725), which is still ongoing and is further investigating the use of intensified temozolomide treatment after surgery.
In addition to chemotherapy, other trials assessed targeted agents, including the vascular endothelial growth factor (VEGF) inhibitor bevacizumab. None of these studies identified a survival advantage with the use of bevacizumab, which was instead associated with an increased rate of adverse events [26, 27, 54,55,56].
The proteasome inhibitor marizomib has been assessed in the phase III EORTC 1709 clinical trial. The addition of this agent to standard of care resulted in an increased rate of grade 3/4 treatment adverse events (ataxia, hallucinations, and headache) with no survival advantage, leading the Independent Data Monitoring Committee (IDMC) to recommend enrollment discontinuation [57].
Enzastaurin is a compound able to inhibit the protein kinase C beta, resulting in angiogenesis inhibition and direct cytotoxic activity. Recent studies identified that patients harboring a polymorphism of the Denovo Genomic Marker 1 (DGM1) on chromosome 8 could have enhanced clinical benefit from the administration of enzastaurin [58,59,60,61]. The phase III trial NCT03776071 is currently assessing this agent in newly diagnosed GBM with or without DGM1 polymorphism.
ICIs have been assessed in two different phase III trials in newly diagnosed GBM. Nivolumab (a PD-1 inhibitor) has been assessed in combination with radiotherapy among patients with MGMT unmethylated GBM (Checkmate-498; NCT02617589) and in association with concomitant temozolomide and radiation therapy among patients with MGMT-methylated GBM (CheckMate-548; NCT02667587). No survival advantages emerged from the addition of nivolumab in both clinical trials. Nonetheless, there are some ongoing trials under evaluation assessing ICIs in this setting.
PERGOLA (NCT03899857) is a phase II trial investigating the addition of pembrolizumab to standard treatment in patients with newly diagnosed GBM. The combination between nivolumab and ipilimumab (a cytotoxic T-lymphocyte antigen 4 [CTLA-4] inhibitor) showed a high incidence of adverse events in preliminary studies [62]. A phase II/III study is currently assessing this combination in patients with MGMT unmethylated GBM (NCT04396860).
Depatuxizumab mafodotin (Depatux-m, ABT414) is an antibody drug conjugate targeting EGFR that has been assessed in newly diagnosed and recurrent GBM. Despite encouraging preliminary data [63,64,65,66], the addition of Depatux-m to the standard Stupp protocol did not add a significant survival benefit (press release of the INTELLANCE-1 study).
In conclusion, the prognosis of newly diagnosed GBM remains poor even if a small but non-negligible percentage of patients achieved a long-term survival, being alive at 5 years from diagnosis. These patients are more frequently younger at diagnosis, female, and present with MGMT methylation and enhanced sphingomyelin metabolism [67,68,69]. The EORTC 1419 Eternity trial (NCT03770468) is retrospectively investigating the clinical and molecular features of patients with long-term survival.
4 Recurrent GBM
Treatment options in patients with recurrent GBM are limited and consist of systemic treatments and locoregional approaches [19, 70,71,72,73,74]. Despite these efforts, patients experiencing tumor recurrence have a dismal prognosis [8,9,10]. The absence of an effective standard of care in this setting makes the development of a new therapeutic strategy a clinical priority. Among systemic treatment, nitrosoureas are still considered the treatment of choice. A recent systematic review and meta-analysis suggested that few studies investigated re-operation and re-irradiation in patients with recurrent disease, thus these two approaches should be proposed in selected cases [75].
A large number of compounds have been tested in the recurrent setting without significant improvement. Since about 50% of GBM patients present an amplification of the EGFR, several targeted agents have been assessed without significant efficacy [76,77,78]. The INTELLANCE-2/EORTC_1410 phase II study evaluated Depatux-m alone or in combination with temozolomide versus temozolomide/CCNU (according to the time of the last adjuvant temozolomide cycle). The administration of Depatux-m in combination with temozolomide resulted in a modest OS improvement (9.6 vs. 8.2 months) [79]. The survival benefit was more evident among patients with an EGFR single-nucleotide variation that amplified EGFR sensitiveness to its ligand [80,81,82]. Bevacizumab has been largely investigated in the recurrence setting [74, 83], however the phase III EORTC 26101 trial assessing bevacizumab and lomustine over lomustine alone failed to show a significant OS improvement with the addition of bevacizumab [84]. The multitarget tyrosine kinase inhibitor regorafenib has recently been tested in comparison with lomustine among recurrent GBM patients within the REGOMA phase II trial [70]. The administration of regorafenib resulted in a prolonged survival compared with lomustine (7.4 vs. 5.6 months), leading to further assessment of this targeted agent alone or in combination with nivolumab (NCT04704154). Cyclin-dependent kinase 4/6 (CDK4/6) inhibitors are the treatment of choice in patients with hormone-sensitive and human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer. These agents have also been assessed in GBM, where CDK4/6 are commonly altered. Both palbociclib (tested in patients with RB1 proficiency) [85] and abemaciclib (tested in patients with CDKN2A/B loss and intact RB) [86] failed to show a significant clinical improvement in GBM patients.
Promising preliminary results originate from two different basket trials exploring the BRAF inhibitors among glioma patients with BRAFV600 mutation [87, 88]—the VE-BASKET trial (assessing the BRAF inhibitor vemurafenib) [87] and the ROAR basket trial (assessing the combination between a BRAF and MEK inhibitor) [87, 88]. In particular, among the 45 patients with high-grade gliomas (including 31 GBMs) receiving dabrafenib plus trametinib within the ROAR trial, there were 3 complete responses and 12 partial responses [89].
Finally, the NTRK inhibitor larotrectinib has been assessed in GBM patients harboring the TRK mutation [90] and showed a very impressive result with the achievement of a disease control rate for all enrolled patients (n = 6 patients with GBM) [90].
5 Checkpoint Inhibitors and Chimeric Antigen Receptor (CAR) T Cells for the Treatment of Recurrent GBM
ICIs have failed to show significant clinical efficacy in patients with recurrent GBM. The Check-Mate 143 trial compared nivolumab with bevacizumab in recurrent GBM patients, failing to show a significant survival benefit in patients randomized in the nivolumab arm [20].
The combination between rindopepimunt (a vaccination against EGFRvIII) and bevacizumab showed favorable prolonged PFS of patients with recurrent GBM in a phase II trial. Nevertheless, this agent failed to confirm an OS advantage in a subsequent phase III study [91].
Active immunization with dendritic cells or peptide vaccines is another approach under evaluation [92] and has shown promising results in a phase III trial in newly diagnosed GBM patients [92]. The NCT04277221 phase III trial is currently investigating the dendritic cell vaccine (DCVax®-L) in patients with recurrent GBM.
The repertoire of targets for GBM immunotherapy has been expanded with the introduction of CAR T cells, a novel therapeutic option to overcome the limits of ICIs in the treatment of GBM.
Engineering T cells to express CARs has shown a wide range of anticancer activity (Fig. 2), showing remarkable clinical results in hematological malignancies by targeting the B-cell antigen CD19, with several clinical trials reporting high response rates and durable remissions [93, 94].

A summary of CAR T-cell general mechanism and possible strategies under evaluation to improve engineered cell potential within patients with glioblastoma. CAR chimeric antigen receptor
Although the anticancer activity of CAR T cells in solid tumors, and particularly in GBM, appeared lower than expected, a possible advantage of this novel treatment lies in its great flexibility and adaptability [22, 95]. CAR T cells are T lymphocytes that are modified, through the use of oncoretroviral or lentiviral vectors, to express CARs. These synthetic receptors redirect T cells to cancer cell surface antigens in order to eliminate the targeted cancer cells, bypassing the HLA-restricted nature of T cells [96]. Notably, by combining the specificity of the major histocompatibility complex (MHC)-independent antibody recognition with the anti-tumor potential of T lymphocytes, these cells can virtually target and eliminate any antigenic specificity (Table 1).
Engineered CAR T cells are obtained from the blood of the patient through leukapheresis, then genetically modified to express the chimeric receptor, which selectively binds antigens expressed on the surface of tumor cells. These modified T cells are induced to replicate in the laboratory through a combination of cytokines, including interleukin (IL)‐2, IL‐7, IL‐15, or IL‐21, and then transplanted into the patient [97]. When the CAR construct binds its antigen, there is T-cell activation and proliferation associated with cytokine release and cytolytic degranulation, resulting in an activated immune response against tumor cells [98].
There are three main components of CAR T cells: the extracellular single chain variable domain (scFv), the transmembrane domain, and the intracellular domain for signal transmission. Since the first description of the CAR design, the CAR T structure has continuously evolved and improved. In particular, CARs evolved from the basic first-generation CARs, composed only of the activation domain (CD3ζ chain) for signaling, to second- and third-generation CARs, where the CD3ζ domain has been equipped with one (4-1BB or CD28) or more (4-1BB and CD28) costimulatory domains to enhance CAR T therapeutic efficacy, proliferation, persistence and duration of response [99, 100]. Third-generation CAR T cells have demonstrated improved antitumor activity at the expense of worse toxicity [101, 102].
CAR T-cell targets that have been evaluated preclinically and in early-phase clinical trials for GBM include IL-13Rα2, HER2, EGFRvIII, EphA2, CD 70, B7-H3 and chlorotoxin [103].
5.1 Interleukin (IL)-13Rα2
The IL-13 receptor-α2 (IL-13Rα2) is involved in the activation of the phosphatidylinositol-3 kinase/AKT/mammalian target of rapamycin pathway and is overexpressed in 75% of GBM cells. It has been the first target to be investigated in clinical trials [104]. Several studies have reported the safety and clinical bioactivity of IL-13Rα2-directed CAR T in GBM [105, 106].
Brown et al. described the case of a 50-year-old patient with recurrent multifocal IDH1 wild-type, MGMT- unmethylated GBM who experienced remission of all intracranial and spinal tumors after injection of CAR T cells targeting IL-13Rα2, sustained for 7.5 months after the initiation of therapy [105]. The treatment was administered intracranially six times weekly and then switched to intraventricular administration weekly. All lesions progressively and gradually disappeared until they became unmeasurable. Unfortunately, disease progression was observed after cycle 16, and this recurrence has been attributed to the phenomenon of ‘antigen loss’, i.e. a progressive decreased expression of IL-13Rα2 on GBM cells. However, this report underlines the importance of the route of delivery in CAR T therapy and represents one of the first evidences of how local delivery seems to outperform systemic delivery.
5.2 Human Epidermal Growth Factor Receptor 2 (HER2)
The HER2 is a cell membrane receptor with tyrosine kinase activity and is a member of the EGFR family. Its overexpression in cancer is associated with CNS tumors, including GBM, whereas HER2 expression is absent in both normal neuronal and glial tissue during adulthood. In 2017, a phase I trial was published that enrolled 17 patients with progressive HER 2-positive GBM to receive one or more systemic intravenous infusions of a second-generation HER2-specific CAR T-cell therapy with a CD28 costimulatory domain [107]. No patients experienced dose-limiting toxicity. One patient exhibited a partial response for more than 9 months and three patients achieved stable disease for at least 24 months, with a median OS of 11.1 months and a median PFS of 3.5 months [107]. This study confirmed the safety and feasibility of HER2-directed second-generation CAR T for GBM patients, with encouraging preliminary antitumor activity.
5.3 Epidermal Growth Factor Receptor (EGFR) vIII
EGFRvIII is a constitutively active mutant variant form of the EGFR that is expressed in about 30–40% of GBMs, thus representing a well-established therapeutic target [108]. Recently, a second-generation EGFRvIII CAR T-cell treatment has been tested in a phase I trial in patients with multifocal, MGMT-unmethylated recurrent GBM [109, 110]. Notably, after CAR T infusion, the engineered lymphocytes became detectable in both peripheral blood and tumor samples (obtained from patients who underwent second surgery) [109,110,111]. Moreover, the antigen EGFRvIII density in tumor specimens was substantially reduced, suggesting interesting bioactivity of these CAR T cells. Despite this, only one patient achieved stable disease (18 months post single infusion), while the other two treated patients experienced progressive disease as best response. The toxicity profile was favorable as no dose-limiting toxicities occurred.
In 2019, Goff et al. developed a third-generation EGFRvIII-specific CAR T and launched a phase I clinical trial enrolling patients with recurrent GBM expressing EGFRvIII [109]. Eighteen patients were treated intravenously with an escalating dose of EGFRvIII CAR T cells following lymphodepleting host conditioning. At initial dose levels (107−109 cells), EGFRvIII CAR T cells were well tolerated; however, at the superior dose levels, toxicity was conspicuous, with one treatment‐related death (after intravenous infusion of 6 × 1010 cells), due to respiratory distress and pulmonary edema. One patient developed dyspnea that was successfully managed with continuous positive airway pressure (CPAP). Unfortunately, again, no objective responses were obtained, except one patient who experienced a long PFS of 12.5 months. In summary, despite severe toxicity, the outcomes appeared disappointing, with a median OS of 6.9 months and a median PFS of 1.3 months [109]. The reasons for this failure have been investigated by Sampson et al., who demonstrated that EGFRvIII CAR therapy generates antigenic loss, rendering the tumor resistant to CAR Ts [111]. It is interesting that despite the unsatisfactory results, the researchers were not discouraged and continuous efforts are being made in an attempt to improve and optimize EGFRvIII-targeted CAR Ts [112,113,114].
6 Novel CAR T Approaches to Treat GBM
A novel CAR T equipped with chlorotoxin, a scorpion toxin, has been developed in orthotopic xenograft GBM tumor models, demonstrating safety and long-term remissions [115].
Tang et al. have engineered specific CAR Ts directed against B7-H3, a type I transmembrane protein widely expressed in tumor cells of diverse origin that has demonstrated antitumor activity both in vitro and in vivo in orthotopic GBM models [116]. Starting with the observation that CD70, if overexpressed in GBM cells, is capable of inducing apoptosis on T cells, Jin et al. have developed a CD70 CAR T that demonstrated antitumor activity against CD70+ glioma cells in vitro and in vivo, inducing remission with 100 × 106 CAR T cells [117]. In a follow-up study, the same group further increased the efficiency of CD70 CAR T cells through co-expression of CXCR2, the IL-8 receptor. Since IL-8 expression is known to promote tumor resistance and invasion, using CAR T cells directed towards IL-8, the authors achieved a product able to penetrate tumor tissue more diffusely, obtaining in vivo remission with only 2 × 106 CAR T cells [118].
EphA2 is a protein overexpressed in sarcomas and gliomas [119]. After encouraging results in preclinical studies [118, 120], in the first-in-human NCT03423992 phase I trial, EphA2-redirected CAR T cells were administered with a lymphodepletion regimen consisting of fludarabine and cyclophosphamide, leading to stable disease in only one patient, while the remaining two patients experienced disease progression as best response [121]. Although not exciting, these results deserve further study.
Another interesting strategy is to develop CAR T cells that constitutively secrete IL-18, a cytokine that enhances the immune response through interferon (IFN)-γ secretion and activation of natural killer (NK) and cytotoxic T lymphocytes. Synthetic IL-18 secretion by CAR T cells increases T-cell persistence and anti-tumor activity, thus promoting CAR T cytotoxicity in animal preclinical models [122, 123].
7 The Reasons for CAR T Failure in GBM and Strategies to Overcome Resistance and Obtain Durable Responses
Despite promising early results, several limitations and significant challenges compromise the efficacy of CAR T therapy in GBM, primarily because of the unique immunosuppressive, hostile, and ‘cold’ GBM microenvironment, poor or pro-inflammatory mediators, and also the tumor antigen heterogeneity of this neoplasm [21, 22].
A major basic problem in the manufacturing of CAR T cells is lymphopenia, a limited T-cell bioavailability in patients heavily pretreated with corticosteroids and chemotherapy. The process of CAR T manufacturing implies that patient peripheral T cells have to be isolated, extracted, and expanded. This represents an issue in GBM patients, heavily immunosuppressed and equipped with often anergic and exhausted T lymphocytes [124].
Another obstacle is the presence of the BBB, which is critical for T-cell penetration into the CNS parenchymal tissue. The BBB is composed of endothelial cells, which give rise to a continuous and non-fenestrated endothelium. The endothelial cells are then joined together by occluding cellular junctions (otherwise called tight junctions); this greater compactness prevents the passage of hydrophilic substances or substances with large molecular weight from the blood flow to the interstitium (and therefore to the neurons), with the capacity for much more selective filtering than that carried out by the endothelial cells of the capillaries of other parts of the body. A further factor that contributes to the formation of this anatomical functional unit called BBB is the projection of astrocytic cells, called astrocytic peduncles (also known as ‘glial limiting’), which surround the endothelial cells of the BBB, determining an additional ‘barrier’ [125]. It is a highly selective physiologic boundary that limits the trafficking of activated T cells and the engraftment of the CAR T product into the tumor tissue. Given the challenges of trafficking CAR T cells into parenchymal tissue, many GBM CAR T trials have focused on local intracavitary and intraventricular delivery in favor of intravenous delivery.
Although the BBB limit can be overrun by an intracavitary or intraventricular infusion, the GBM microenvironment is enriched with immunosuppressive myeloid and lymphoid cells in different states of maturation. Furthermore, many soluble factors are secreted by the tumor, such as transforming growth factor (TGF)-β, VEGF, IL-6 and IL-10, involved in the resolutive phases of inflammation and able to inhibit an immune response. These factors suppress T-cell proliferation, decreasing the absolute counts of CD4+ T cells and increasing the proportion of regulatory T cells (T-reg, which exert inhibitory action on CD4+ T cells, cytotoxic CD8+ T cells, dendritic cells, and NK cells), thus hindering immune responses around tumors [126]. Immunosuppressive tumor-associated macrophages (TAMs), in particular, possess immunosuppressive abilities and may be potent inactivators of both CD4+ and CD8+ T cells [127, 128].
The massive infiltration of TAMs and ‘alternatively activated M2 macrophages’ (i.e. immunosuppressive-type macrophages), T-reg, and myeloid-derived suppressor cells is a hallmark feature of GBM: these cells perform an anti-inflammatory action associated with suppression of the immune response, hindering the infiltration of T cells [129,130,131].
Furthermore, the tumor TME is hypoxic and metabolically stressful, and is thus inadequate to provide the sufficient glucose intake required for the high glycolytic activity of T cells. Glucose depletion and the exhaustion of essential metabolic substrates such as tryptophan, arginine, and lysine, which is typical of the dysregulated metabolic state of GBM, result in the accumulation of lactic acid and reduction of pH values, which suppresses T lymphocyte proliferation and effector capacity [132, 133].
Another hurdle to consider is the inadequate trafficking of CAR T cells to the tumor site, resulting in diffuse inability of the T cells to penetrate and infiltrate the TME, depending on the interactions between the chemokine receptors on the CAR (CXCR3 and CCR5) and the chemotactic chemokines secreted by the tumors (often inadequate), thus resulting in unsuccessful trafficking of CAR T cells to the tumor tissue [134].
However, even if the engineered CAR T cells survive and penetrate the ‘cold’ and hostile GBM TME, their cytotoxic action is inhibited and stopped by the immune-suppressive cytokine storm. Furthermore, heterogeneous surface antigen expression can lead to CAR T evasion. Both the spatial and temporal heterogeneity of target expression on GBM cells presents a challenge to CAR T effectiveness. In EGFRvIII- and IL-13Ra2-directed CAR T trials, investigators noted that quantitative target antigen expression tended to shrink over time, a phenomenon known as ‘antigen loss’, responsible for resistance and relapse.
The failure of CAR T therapy in GBM may also be related to a phenomenon named T-cell exhaustion, typical of chronic infections and cancer and consisting in a state of ‘anergy’ and deterioration of T-cell effector functions that are exposed to chronic antigen stimulation or persistent infections/inflammatory signals. Constitutive prolonged activation of the CAR, known as tonic signaling, leads to increased expression of multiple inhibitory receptors of CAR T and to altered transcriptional programs responsible for the relapse and ineffectiveness of CAR T cells [135, 136].
The hypothesized underlying mechanisms at the basis of exhaustion are different and include expression of inhibitory receptor signaling; alterations of key transcriptional factors and epigenetic reprogramming; DNA methylation that promotes T-cell exhaustion and limits the activity of immune checkpoint blockade [137, 138]; and reduction of chromatin accessibility, which is the basis of DNA demethylation processes [139,140,141].
Weber et al. have recently published an interesting study that, through a platform controlling CAR surface expression, prevented tonic signaling on CAR T cells, thus generating a product free from the phenotypic hallmarks of exhaustion, like inhibitory receptor signaling (PD-1, TIM-3, LAG-3), and deprived of exhaustion-associated gene expression profile [142]. The CAR T cells thus modified presented increased antitumor activity and maintained the ability to secrete cytokines over time [142]. The authors concluded that the transient inhibition of CAR surface expression and, consequently, the cessation of CAR T-cell tonic signaling can reverse and prevent the phenomenon of T-cell exhaustion. The same group also demonstrated that the tyrosine kinase inhibitor dasatinib seems to be a valid option to reverse tonic signaling and exhaustion in T cells.
A concern regarding the clinical efficacy of CAR T cells in brain tumors is the impact of antigen density on the potency of CAR Ts and the threshold of antigen expression. In fact, CAR Ts require high antigen density for full T-cell activation. Furthermore, this property has two important implications; first, the risk of relapse with low expression of antigen, and second, the toxicity, considering that even a low level of target antigen expression on normal tissues can prove fatal. The solution is engineering of CAR T cells focusing on antigen density thresholds. A recently published article has reported an interesting strategy aimed at improving CAR T efficacy, and also in the context of a low concentration of antigens [143]. The promising GPC2-CAR, targeting glypican-2 (GPC2), an embryonal antigen with expression restricted to fetal brain tissue, overexpressed on neuroblastoma compared with normal tissues, has been manufactured. GPC2-CARs have been optimized to lower the antigen density threshold and have shown promising antitumor effects in vivo in preclinical models and also in a low antigen density microenvironment [143]. Although fine-tuning CAR T cells to recognize low antigen density potentially increases the risk for on- or off-target toxicity, the toxicity related to this treatment was quite insignificant, likely because GPC2 expression is very limited in postnatal tissues [143].
Another major issue concerning CAR T therapy in GBM is safety. The two main toxicities are cytokine release syndrome (CRS) and a set of neurological sequelae named ‘immune effector cell-associated neurotoxicity syndrome’ (ICANS), responsible for several cases of treatment-related deaths [94, 144,145,146]. CRS is the result of a cytokine storm and involves several symptoms, including fever, nausea, vomiting, diarrhea, and, in the most severe cases, arrythmia, dyspnea, pulmonary oedema and respiratory failure. This syndrome can be managed with the use of the monoclonal antibody tocilizumab or dexamethasone, to reduce the inflammatory reaction [147]. Therefore, it is supposed, even if not definitely demonstrated, that the use of corticosteroids, with their broad spectrum, non-specific anti-inflammatory action, might be associated with detrimental effects on CAR T effectiveness [148,149,150]. The pathogenesis of ICANS is poorly understood. The most likely mechanism involves cytokines spreading into the cerebrospinal fluid, causing inflammation of the CNS. Symptoms related to ICANS are headache, seizures, brain edema, coma, aphasia, delirium. or even rare cases of death [151,152,153,154].
Recently, Tmunity stopped the phase I CAR T-PSMA-TGFβRDN study in patients with prostate cancer after two cases of death as a result of ICANS [146]. Tocilizumab has demonstrated poor efficacy for the management of ICANS and may hypothetically aggravate the severity of neurologic toxicity [155], thus corticosteroids remain the first-line approach.
Innovative sophisticated strategies to overcome the limits of CAR T cells have been developed, including the use of multitargeted CARs, the identification of universal immune receptors targeting multiple antigens (universal CAR T), and the research of novel combination therapies with ICIs.
8 UNIVERSAL CAR T
Conventional CAR T cells are autologous lymphocytes, requiring the collection of patient-specific T cells, which results in interpatient variability. The final cell product has the ability to target only one fixed epitope of a single specific antigen, making the therapy vulnerable to relapse due to tumor heterogeneity and mechanisms of antigen escape, such as antigen loss or low-density antigen. In fact, despite the fact that traditional CAR T therapy may initially achieve tumor regression, relapse often occurs [100, 156].
The traditional CAR T manufacturing protocols are expensive and time-consuming, making an ‘individualized’ product, variable from patient to patient, that is not ‘ready to use’ for critically ill patients and which cannot be simultaneously administered to multiple patients. Moreover, the success of the production depends on the collection of a sufficient number of autologous T cells, which can be inadequate in lymphopenic patients highly pretreated with multiple previous chemotherapies [157].
Over the last few years, significant efforts have been made to further improve the CAR T design. Universal CAR T represents a ‘fourth generation’ of modular CAR T cells, developed to overcome the fixed antigen specificity of traditional CAR T systems, to increase the safety and controllability of CAR manufacturing, and to offer an off-the-shell, ready to use product, obtained from the T cells of a healthy donor. Universal CAR Ts could circumvent the problem of tumor heterogeneity and overcome the issue of quantitatively insufficient or poor-quality CAR T cell products [158, 159].
Modular CAR T technology is obtained by the T cells of a healthy donor and presents a ‘third-party’ intermediate system that splits the antigen-targeting region from T-cell signaling through use of an ‘adaptor’. This is a switch molecule that serves as the targeting element and allows flexibility in tumor targeting, conferring to CAR T cells a broad-range of antigen specificity [160, 161]. Indeed, universal CAR Ts effectively abolish graft-versus-host disease (GVHD) by generating TCR-deficient and HLA class I-deficient T cells that are not capable of recognizing allogenic antigens. The most commonly used methodology for disrupting the TCR gene or HLA class I loci of the allogeneic T cells is genome immune editing, an approach consisting of DNA manipulation through the use of nucleases, which can modify and regulate genomic loci to achieve the required therapeutic effects. Transcription activator-like effector nucleases (TALENs) and the ‘newborn’ clustered regularly interspaced short palindromic repeats (CRISPR) are among the most powerful editing tools available to genetic engineering [161, 162]. Using TALEN editing technology, Poirot et al. have developed T cells disrupted in the αβ TCR gene and CD52 gene expression, and thus deficient of both T-cell receptor and CD52, a protein targeted by alemtuzumab (a chemotherapeutic agent), limiting the risks of alloreactivity GVHD and making T cells resistant to destruction by alemtuzumab [163]. These modified T cells were used for the generation of universal CAR T cells that when administered in combination with the chemotherapeutic agent alemtuzumab demonstrated efficient destruction of CD19 tumor targets. The CRISPR/CRISPR-associated protein 9 system is a gene editing tool able to perform multiplex genome editing, cleaving the DNA at sites of interest. Adopting the CRISPR protocol, it is possible to generate ‘double knockout’ of HLA class I and TCR universal CAR T cells, abolishing the potential for T cells to react to allogeneic antigens and improving CAR T safety [164,165,166].
One relevant challenge regarding the loss of classical HLA class I expression is that HLA-negative allogeneic cells might be recognized by endogenous NKs, becoming vulnerable for NK-mediated lysis and thus rendering the therapy inefficient. A solution to prevent the elimination of administered HLA-negative allogeneic cells by NKs is provided by the enforced expression on donor cells of non-classical HLA class I molecules such as HLA-E or HLA-G, which have been shown to protect cells from NK-mediated cytotoxicity [167,168,169,170,171]
Guo et al. have recently demonstrated that anti-CD19 universal CAR T cells (UCAR T-19) manufactured with the constitutive expression of mutant β2-microglobulin HLA-E and β2-microglobulin HLA-G fusion proteins are protected from NK cell-mediated lysis and show anti-tumor efficacy [167].
9 Multispecific CAR T
The therapeutic successes achieved by CAR T cells on the management of B-cell tumors can be partially explained by the presence of a single antigen (CD19) that is expressed by all tumoral cell clones. Unfortunately, GBM is characterized by high intratumor and intertumoral heterogeneity, which makes the development of a cell clone targeting a single antigen difficult [166, 172]. To overcome this obstacle, the development of CAR T cells simultaneously targeting more than one antigen has been proposed [166, 172,173,174,175]. These lymphocytes can be defined as ‘bivalent’, ‘trivalent’, or ‘multispecific’ according to the number of targets. Tandem CAR T cells are bivalent CAR T cells that have been assessed in GBM [175]. Hegde et al. developed tandem CAR T cells targeting IL-13Rα2 and HER2 [174]. Their tandem CAR was capable of killing GBM cells by binding to either HER2 or IL-13Rα2. However, when these cells simultaneously recognized the two antigens, the immune response was significantly enhanced, resulting in ‘super additive’ T-cell activation. To date, ‘trivalent’ CAR T cells targeting HER2, IL-13Rα2, and EphA2 are being assessed in animal models [175, 176]. The bispecific tumor-targeted T-cell engager (BiTE)-armed CAR T cells is another strategy in which engineered T cells can target both wild-type and EGFRvIII-expressing GBM cells. BiTE-armed CAR T cells successfully eliminated cancer cells and prolonged the survival of mice orthotopically grafted with either GBM cell lines or patient-derived glioma neurospheres [112].
10 Combination Therapies
Evidence in preclinical models of GBM have shown that a promising strategy to overcome the limits of the immunosuppressive TME is the combination of CAR T immunotherapy cells with immune checkpoint blockade.
It has been widely demonstrated that exhausted CAR T cells overexpress inhibitory receptors, including programmed death-1 (PD-1), with corresponding upregulation of PD-1 ligands (PD-L1 and PD-L2) on the tumor cells [176]. Thus, novel combination therapies to limit the PD-L1-mediated immunosuppression and to enhance the efficacy of CAR T-cell therapy are emerging, especially those aimed at genetically engineering CAR T cells to intrinsically express a PD-1-negative receptor on the surface [176]. Chen et al. engineered specific CAR T cells genetically modified to overexpress a PD-1-dominant negative receptor (PD-1 DNR) that demonstrated antitumor activity in both an in vitro and in vivo mouse model [176].
The ongoing single-arm, open-label NCT03726515 trial investigated the combination of high doses of EGFRvIII-directed CAR T cells with the anti-PD1 humanized antibody pembrolizumab after adjuvant radiotherapy. The results from this trial are expected to provide important information regarding the clinical impact of CAR‐T cells combined with checkpoint inhibitors in GBM treatment, as well as the safety and feasibility of these combination strategies.
11 CAR NK and Car M against GBM
To overcome the limits of the application of CAR T cells in solid tumors, novel CAR-based therapeutic strategies are under investigation.
CAR NK therapy is based on the genetic manipulation of NK cells that are modified to express CARs that recognize tumor-associated antigens. NK cells are characterized by broad cytotoxic activity, are the first line of the immune defense, even in the absence of a prior sensitization of class I MHC, and are also active in the presence of low expression of the CAR target antigen or against non-CAR-specific antigens [177, 178]. CAR NK effectors are allogenic off-the-shelf products from donor-derived NKs, exhibiting a low risk of GVHD and limited toxicity compared with CAR T cells, because they do not induce CRS [179]. CAR NK cells targeting EGFRvIII, EGFR/ EGFRVIII, and HER2 have been tested in GBM in preclinical studies only, showing interesting antitumor activity [180,181,182,183,184].
The ongoing CAR2BRAIN study (NCT03383978), which is currently enrolling participants, is a phase I clinical trial evaluating the safety and tolerability of CAR NK cells from the NK-92 cell line (NK-92/5.28.z CAR NK cells) and targeting HER2. The Achilles heel of CAR NKs appears to be their short in vivo survival that on the one hand limits the toxicities and on the other hand reduces their long-term persistence and duration of activity, requiring multiple repeated treatments [185].
A study by the University of Pennsylvania has recently explored an innovative approach based on the transduction of CARs into macrophages (CAR Ms) [186]. CAR M therapy demonstrated multimodal antitumor activity at various levels of the immune response: macrophages infiltrate the TME phagocytizing cancer cells, and indeed they present marked ability in reverting the immunosuppressive TME, repolarizing the cold protumoral M2 phenotype of macrophages towards the hot proinflammatory M1 phenotype, and thus promoting a proinflammatory microenvironment. Finally, CAR M therapy stimulates the cells of the adaptive immune system [186]. Although to date there is no evidence and there are no studies on the use of CAR M in GBM, macrophages seem to be better candidates suited for the application of CAR therapy in GBM, given their ability to penetrate and infiltrate the TME and to directly phagocyte cancer cells, thus overcoming the obstacles associated with the immunosuppressive TME and to T-cell immunotherapy [21].
12 Conclusions
GBM remains one the most aggressive CNS malignancies. Nonetheless, thanks to increasing knowledge regarding molecular mechanisms associated with tumor onset and progression, promising novel treatments will be tested. Previous trials are important despite the negative results that are often observed. Indeed, from the experience acquired by previous studies, we are now able to design more efficient trials capable of testing more compounds at the same time and adopting the minimum required number of patients. In this light, the development of early phase 0 trials assessing the ability of different drugs to penetrate into CNS tissue appears to be of particular interest.
In novel therapeutic approaches, immunotherapy still represents a potential effective treatment. In this regard, the development of genetically engineered immune effector cells equipped with CARs represent a valuable option to strengthen the potential of T-cell immunity.
However, despite the excellent clinical results achieved in hematological malignancies, the effectiveness of CAR T cells in GBM appears lower than expected, with frequent and early antigen-negative relapses and relevant toxicities. The reasons are manifold: the low number of T cells in CNS tissue and the immunosuppressive TME, enriched with M2-like macrophages and T-reg; the use of autologous T cells, which implies a laborious and expensive manufacturing process; and not least, the relevant, sometimes fatal, adverse effects.
Despite all these obstacles, CAR T therapy offers unparalleled advantages, such as the flexibility in tumor targeting, the adaptability to exploit all the cell-killing mechanisms that the immune system offers, and the MHC-independent antigen recognition, to overcome the evasion strategies developed by tumors cells. This justifies the continuous attempts of researchers for improving tumor targeting and developing novel CAR T combinations, and the impetus of the numerous new studies that are underway.
The goal is to optimize the following points: improving safety, protecting CAR T from the immunosuppressive TME, preventing T-cell exhaustion and antigen loss and ameliorating tumor-homing. It is not unexpected that research in the field of non-T-engineered immune cell effectors, NK cells, and macrophages will find solutions to these challenges.
References
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–51. https://doi.org/10.1093/neuonc/noab106.
Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–86. https://doi.org/10.1038/s41571-020-00447-z.
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Suppl 5):v1–100. https://doi.org/10.1093/neuonc/noz150.
Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–113. https://doi.org/10.1093/neuonc/noaa106.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. https://doi.org/10.1056/NEJMoa043330.
Perry JR, Laperriere N, O’Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. 2017;376(11):1027–37. https://doi.org/10.1056/NEJMoa1611977.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. https://doi.org/10.1056/NEJMoa043331.
Wu W, Lamborn KR, Buckner JC, Novotny PJ, Chang SM, O’Fallon JR, et al. Joint NCCTG and NABTC prognostic factors analysis for high-grade recurrent glioma. Neuro Oncol. 2010;12(2):164–72. https://doi.org/10.1093/neuonc/nop019.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66. https://doi.org/10.1016/s1470-2045(09)70025-7.
Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, DeAngelis LM, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. 2008;10(2):162–70. https://doi.org/10.1215/15228517-2007-062.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306–16. https://doi.org/10.1001/jama.2017.18718.
Nunno VD, Franceschi E, Gatto L, Brandes AA. BET inhibitors: the promise of a new generation of immunotherapy in glioblastoma. Immunotherapy. 2021. https://doi.org/10.2217/imt-2021-0296.
Di Nunno V, Franceschi E, Tosoni A, Gatto L, Lodi R, Bartolini S, et al. Glioblastoma: emerging treatments and novel trial designs. Cancers (Basel). 2021;13(15):3750. https://doi.org/10.3390/cancers13153750.
Di Nunno V, Franceschi E, Tosoni A, Di Battista M, Gatto L, Lamperini C, et al. Treatment of recurrent glioblastoma: state-of-the-art and future perspectives. Expert Rev Anticancer Ther. 2020;20(9):785–95. https://doi.org/10.1080/14737140.2020.1807949.
Weller M, Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev. 2020;87: 102029. https://doi.org/10.1016/j.ctrv.2020.102029.
Lombardi G, Idbaih A, Le Rhun E, Preusser M, Zagonel V, French P. A new landscape for systemic pharmacotherapy of recurrent glioblastoma? Cancers (Basel). 2020;12(12):3775. https://doi.org/10.3390/cancers12123775.
Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80: 101896. https://doi.org/10.1016/j.ctrv.2019.101896.
Lim-Fat MJ, Youssef GC, Touat M, Iorgulescu JB, Whorral S, Allen M, et al. Clinical utility of targeted next generation sequencing assay in IDH-wildtype glioblastoma for therapy decision-making. Neuro Oncol. 2021. https://doi.org/10.1093/neuonc/noab282.
Di Nunno V, Franceschi E, Gatto L, Bartolini S, Brandes AA. Predictive markers of immune response in glioblastoma: hopes and facts. Future Oncol. 2020;16(15):1053–63. https://doi.org/10.2217/fon-2020-0047.
Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003–10. https://doi.org/10.1001/jamaoncol.2020.1024.
Gatto L, Nunno VD, Franceschi E, Brandes AA. Chimeric antigen receptor macrophage for glioblastoma immunotherapy: the way forward. Immunotherapy. 2021;13(11):879–83. https://doi.org/10.2217/imt-2021-0054.
Gatto L, Franceschi E, Di Nunno V, Maggio I, Lodi R, Brandes AA. Engineered CAR-T and novel CAR-based therapies to fight the immune evasion of glioblastoma: gutta cavat lapidem. Expert Rev Anticancer Ther. 2021;21(12):1333–53. https://doi.org/10.1080/14737140.2021.1997599.
Vanderbeek AM, Rahman R, Fell G, Ventz S, Chen T, Redd R, et al. The clinical trials landscape for glioblastoma: is it adequate to develop new treatments? Neuro Oncol. 2018;20(8):1034–43. https://doi.org/10.1093/neuonc/noy027.
Alexander BM, Trippa L. Progression-free survival: too much risk, not enough reward? Neuro Oncol. 2014;16(5):615–6. https://doi.org/10.1093/neuonc/nou041.
Brandes AA, Franceschi E (2011) New agents and new end points for recurrent gliomas. J Clin Oncol 29(9):e245–6; author reply e7. https://doi.org/10.1200/jco.2010.33.2809.
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–22. https://doi.org/10.1056/NEJMoa1308345.
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. https://doi.org/10.1056/NEJMoa1308573.
Han K, Ren M, Wick W, Abrey L, Das A, Jin J, et al. Progression-free survival as a surrogate endpoint for overall survival in glioblastoma: a literature-based meta-analysis from 91 trials. Neuro Oncol. 2014;16(5):696–706. https://doi.org/10.1093/neuonc/not236.
Franceschi E, Ermani M, Bartolini S, Bartolotti M, Poggi R, Tallini G, et al. Post progression survival in glioblastoma: where are we? J Neurooncol. 2015;121(2):399–404. https://doi.org/10.1007/s11060-014-1651-7.
Sharrocks K, Spicer J, Camidge DR, Papa S. The impact of socioeconomic status on access to cancer clinical trials. Br J Cancer. 2014;111(9):1684–7. https://doi.org/10.1038/bjc.2014.108.
Tosoni A, Gatto L, Franceschi E, Di Nunno V, Lodi R, Mura A, et al. Association between socioeconomic status and survival in glioblastoma: an Italian single-centre prospective observational study. Eur J Cancer. 2021;145:171–8. https://doi.org/10.1016/j.ejca.2020.12.027.
Winkfield KM. Improving access to cancer clinical trials by reducing the financial burden. Cancer. 2020;126(1):14–6. https://doi.org/10.1002/cncr.32523.
Vanderbeek AM, Ventz S, Rahman R, Fell G, Cloughesy TF, Wen PY, et al. To randomize, or not to randomize, that is the question: using data from prior clinical trials to guide future designs. Neuro Oncol. 2019;21(10):1239–49. https://doi.org/10.1093/neuonc/noz097.
Trippa L, Lee EQ, Wen PY, Batchelor TT, Cloughesy T, Parmigiani G, et al. Bayesian adaptive randomized trial design for patients with recurrent glioblastoma. J Clin Oncol. 2012;30(26):3258–63. https://doi.org/10.1200/jco.2011.39.8420.
Alexander BM, Ba S, Berger MS, Berry DA, Cavenee WK, Chang SM, et al. Adaptive global innovative learning environment for glioblastoma: GBM AGILE. Clin Cancer Res. 2018;24(4):737–43. https://doi.org/10.1158/1078-0432.Ccr-17-0764.
Alexander BM, Trippa L, Gaffey S, Arrillaga-Romany IC, Lee EQ, Rinne ML, et al. Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): a bayesian adaptive platform trial to develop precision medicines for patients with glioblastoma. JCO Precis Oncol. 2019. https://doi.org/10.1200/po.18.00071.
Wick W, Dettmer S, Berberich A, Kessler T, Karapanagiotou-Schenkel I, Wick A, et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol. 2019;21(1):95–105. https://doi.org/10.1093/neuonc/noy161.
Trippa L, Wen PY, Parmigiani G, Berry DA, Alexander BM. Combining progression-free survival and overall survival as a novel composite endpoint for glioblastoma trials. Neuro Oncol. 2015;17(8):1106–13. https://doi.org/10.1093/neuonc/nou345.
Marenco-Hillembrand L, Wijesekera O, Suarez-Meade P, Mampre D, Jackson C, Peterson J, et al. Trends in glioblastoma: outcomes over time and type of intervention: a systematic evidence based analysis. J Neurooncol. 2020;147(2):297–307. https://doi.org/10.1007/s11060-020-03451-6.
Delgado-López PD, Riñones-Mena E, Corrales-García EM. Treatment-related changes in glioblastoma: a review on the controversies in response assessment criteria and the concepts of true progression, pseudoprogression, pseudoresponse and radionecrosis. Clin Transl Oncol. 2018;20(8):939–53. https://doi.org/10.1007/s12094-017-1816-x.
Ellingson BM, Sampson J, Achrol AS, Aghi MK, Bankiewicz K, Wang C, et al. Modified RANO, immunotherapy RANO, and standard RANO response to convection-enhanced delivery of IL4R-targeted immunotoxin MDNA55 in recurrent glioblastoma. Clin Cancer Res. 2021;27(14):3916–25. https://doi.org/10.1158/1078-0432.Ccr-21-0446.
Galldiks N, Niyazi M, Grosu AL, Kocher M, Langen KJ, Law I, et al. Contribution of PET imaging to radiotherapy planning and monitoring in glioma patients—a report of the PET/RANO group. Neuro Oncol. 2021;23(6):881–93. https://doi.org/10.1093/neuonc/noab013.
Holzgreve A, Albert NL, Galldiks N, Suchorska B. Use of PET imaging in neuro-oncological surgery. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13092093.
Nakajo K, Uda T, Kawashima T, Terakawa Y, Ishibashi K, Tsuyuguchi N, et al. Diagnostic performance of [(11)C]methionine positron emission tomography in newly diagnosed and untreated glioma based on the revised World Health Organization 2016 Classification. World Neurosurg. 2021;148:e471–81. https://doi.org/10.1016/j.wneu.2021.01.012.
Nayak L, DeAngelis LM, Brandes AA, Peereboom DM, Galanis E, Lin NU, et al. The Neurologic Assessment in Neuro-Oncology (NANO) scale: a tool to assess neurologic function for integration into the Response Assessment in Neuro-Oncology (RANO) criteria. Neuro Oncol. 2017;19(5):625–35. https://doi.org/10.1093/neuonc/nox029.
Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, Wick W, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16(15):e534–42. https://doi.org/10.1016/s1470-2045(15)00088-1.
Chang K, Beers AL, Bai HX, Brown JM, Ly KI, Li X, et al. Automatic assessment of glioma burden: a deep learning algorithm for fully automated volumetric and bidimensional measurement. Neuro Oncol. 2019;21(11):1412–22. https://doi.org/10.1093/neuonc/noz106.
Kickingereder P, Isensee F, Tursunova I, Petersen J, Neuberger U, Bonekamp D, et al. Automated quantitative tumour response assessment of MRI in neuro-oncology with artificial neural networks: a multicentre, retrospective study. Lancet Oncol. 2019;20(5):728–40. https://doi.org/10.1016/s1470-2045(19)30098-1.
Vogelbaum MA, Krivosheya D, Borghei-Razavi H, Sanai N, Weller M, Wick W, et al. Phase 0 and window of opportunity clinical trial design in neuro-oncology: a RANO review. Neuro Oncol. 2020;22(11):1568–79. https://doi.org/10.1093/neuonc/noaa149.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–86. https://doi.org/10.1038/s41591-018-0337-7.
Herrlinger U, Tzaridis T, Mack F, Steinbach JP, Schlegel U, Sabel M, et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet. 2019;393(10172):678–88. https://doi.org/10.1016/s0140-6736(18)31791-4.
Blumenthal DT, Gorlia T, Gilbert MR, Kim MM, Burt Nabors L, Mason WP, et al. Is more better? The impact of extended adjuvant temozolomide in newly diagnosed glioblastoma: a secondary analysis of EORTC and NRG Oncology/RTOG. Neuro Oncol. 2017;19(8):1119–26. https://doi.org/10.1093/neuonc/nox025.
Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–91. https://doi.org/10.1200/jco.2013.49.6968.
Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(16):2712–8. https://doi.org/10.1200/jco.2009.26.6650.
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1100–8. https://doi.org/10.1016/s1470-2045(14)70379-1.
Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015;17(5):708–17. https://doi.org/10.1093/neuonc/nou356.
Roth P, Reijneveld J, Gorlia T, Dhermain F, De Vos F, Vanlancker M, et al (2019) P14.124 EORTC 1709/CCTG CE.8: a phase III trial of marizomib in combination with standard temozolomide-based radiochemotherapy versus standard temozolomide-based radiochemotherapy alone in patients with newly diagnosed glioblastoma. Neuro-Oncology 21(Suppl 3):iii98–iii. https://doi.org/10.1093/neuonc/noz126.359.
Butowski N, Chang SM, Lamborn KR, Polley MY, Pieper R, Costello JF, et al. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro Oncol. 2011;13(12):1331–8. https://doi.org/10.1093/neuonc/nor130.
Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65(16):7462–9. https://doi.org/10.1158/0008-5472.Can-05-0071.
Kreisl TN, Kotliarova S, Butman JA, Albert PS, Kim L, Musib L, et al. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol. 2010;12(2):181–9. https://doi.org/10.1093/neuonc/nop042.
Wick W, Steinbach JP, Platten M, Hartmann C, Wenz F, von Deimling A, et al. Enzastaurin before and concomitant with radiation therapy, followed by enzastaurin maintenance therapy, in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation. Neuro Oncol. 2013;15(10):1405–12. https://doi.org/10.1093/neuonc/not100.
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–86. https://doi.org/10.1093/neuonc/nox208.
Gan HK, Reardon DA, Lassman AB, Merrell R, van den Bent M, Butowski N, et al. Safety, pharmacokinetics, and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro Oncol. 2018;20(6):838–47. https://doi.org/10.1093/neuonc/nox202.
Lassman AB, van den Bent MJ, Gan HK, Reardon DA, Kumthekar P, Butowski N, et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: results from an international phase I multicenter trial. Neuro Oncol. 2019;21(1):106–14. https://doi.org/10.1093/neuonc/noy091.
Padovan M, Eoli M, Pellerino A, Rizzato S, Caserta C, Simonelli M, et al. Depatuxizumab mafodotin (Depatux-M) plus temozolomide in recurrent glioblastoma patients: real-world experience from a multicenter study of Italian Asso. Cancers (Basel). 2021. https://doi.org/10.3390/cancers13112773.
van den Bent M, Gan HK, Lassman AB, Kumthekar P, Merrell R, Butowski N, et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemother Pharmacol. 2017;80(6):1209–17. https://doi.org/10.1007/s00280-017-3451-1.
Tykocki T, Eltayeb M. Ten-year survival in glioblastoma. A systematic review. J Clin Neurosci. 2018;54:7–13. https://doi.org/10.1016/j.jocn.2018.05.002.
Richardson TE, Kumar A, Xing C, Hatanpaa KJ, Walker JM. Overcoming the odds: toward a molecular profile of long-term survival in glioblastoma. J Neuropathol Exp Neurol. 2020;79(10):1031–7. https://doi.org/10.1093/jnen/nlaa102.
Burgenske DM, Yang J, Decker PA, Kollmeyer TM, Kosel ML, Mladek AC, et al. Molecular profiling of long-term IDH-wildtype glioblastoma survivors. Neuro Oncol. 2019;21(11):1458–69. https://doi.org/10.1093/neuonc/noz129.
Lombardi G, De Salvo GL, Brandes AA, Eoli M, Rudà R, Faedi M, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019;20(1):110–9. https://doi.org/10.1016/s1470-2045(18)30675-2.
Franceschi E, Stupp R, van den Bent MJ, van Herpen C, Laigle Donadey F, Gorlia T, et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol. 2012;14(12):1503–10. https://doi.org/10.1093/neuonc/nos256.
Franceschi E, Bartolotti M, Tosoni A, Bartolini S, Sturiale C, Fioravanti A, et al. The effect of re-operation on survival in patients with recurrent glioblastoma. Anticancer Res. 2015;35(3):1743–8.
Brandes AA, Tosoni A, Franceschi E, Blatt V, Santoro A, Faedi M, et al. Fotemustine as second-line treatment for recurrent or progressive glioblastoma after concomitant and/or adjuvant temozolomide: a phase II trial of Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Cancer Chemother Pharmacol. 2009;64(4):769–75. https://doi.org/10.1007/s00280-009-0926-8.
Brandes AA, Finocchiaro G, Zagonel V, Reni M, Caserta C, Fabi A, et al. AVAREG: a phase II, randomized, noncomparative study of fotemustine or bevacizumab for patients with recurrent glioblastoma. Neuro Oncol. 2016;18(9):1304–12. https://doi.org/10.1093/neuonc/now035.
McBain C, Lawrie TA, Rogozińska E, Kernohan A, Robinson T, Jefferies S. Treatment options for progression or recurrence of glioblastoma: a network meta-analysis. Cochrane Database Syst Rev. 2021;5(1):Cd013579. https://doi.org/10.1002/14651858.CD013579.pub2.
Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer. 2007;96(7):1047–51. https://doi.org/10.1038/sj.bjc.6603669.
Sepúlveda-Sánchez JM, Vaz M, Balañá C, Gil-Gil M, Reynés G, Gallego Ó, et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017;19(11):1522–31. https://doi.org/10.1093/neuonc/nox105.
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27(8):1268–74. https://doi.org/10.1200/jco.2008.17.5984.
Van Den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel JS, et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020;22(5):684–93. https://doi.org/10.1093/neuonc/noz222.
Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18(8):1146–56. https://doi.org/10.1093/neuonc/now009.
Capper D, von Deimling A, Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, et al. Biomarker and histopathology evaluation of patients with recurrent glioblastoma treated with galunisertib, lomustine, or the combination of galunisertib and lomustine. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18050995.
Hoogstrate Y, Vallentgoed W, Kros JM, de Heer I, de Wit M, Eoli M, et al. EGFR mutations are associated with response to depatux-m in combination with temozolomide and result in a receptor that is hypersensitive to ligand. Neurooncol Adv. 2020;2(1):vdz051. https://doi.org/10.1093/noajnl/vdz051.
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. https://doi.org/10.1200/jco.2008.19.8721.
Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377(20):1954–63. https://doi.org/10.1056/NEJMoa1707358.
Taylor JW, Parikh M, Phillips JJ, James CD, Molinaro AM, Butowski NA, et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J Neurooncol. 2018;140(2):477–83. https://doi.org/10.1007/s11060-018-2977-3.
Lee EQ, Trippa L, Fell G, Rahman R, Arrillaga-Romany I, Touat M, et al. Preliminary results of the abemaciclib arm in the Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): A phase II platform trial using Bayesian adaptive randomization. J Clin Oncol. 2021;39(15_suppl):2014. https://doi.org/10.1200/JCO.2021.39.15_suppl.2014.
Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. 2018;36(35):3477–84. https://doi.org/10.1200/jco.2018.78.9990.
Wen P, Stein A, van den Bent M, De Greve J, Dietrich S, De Vos F, et al. ACTR-30. Updated efficacy and safety of dabrafenib plus trametinib in patients with recurrent/refractory braf V600E–mutated high-grade glioma (HGG) and low-grade glioma (LGG). Neuro-Oncology. 2019;21(Suppl 6):vi19–vi20. https://doi.org/10.1093/neuonc/noz175.073.
Wen PY, Stein A, van den Bent M, De Greve J, Wick A, de Vos F, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022;23(1):53–64. https://doi.org/10.1016/s1470-2045(21)00578-7.
Drilon AE, DuBois SG, Farago AF, Geoerger B, Grilley-Olson JE, Hong DS, et al. Activity of larotrectinib in TRK fusion cancer patients with brain metastases or primary central nervous system tumors. J Clin Oncol. 2019; 37(15_suppl):2006. https://doi.org/10.1200/JCO.2019.37.15_suppl.2006.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85. https://doi.org/10.1016/s1470-2045(17)30517-x.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. https://doi.org/10.1186/s12967-018-1507-6.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48. https://doi.org/10.1056/NEJMoa1709866.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44. https://doi.org/10.1056/NEJMoa1707447.
Soler DC, Kerstetter-Fogle A, McCormick TS, Sloan AE. Using chimeric antigen receptor T-cell therapy to fight glioblastoma multiforme: past, present and future developments. J Neurooncol. 2021. https://doi.org/10.1007/s11060-021-03902-8.
Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–40. https://doi.org/10.1016/j.cell.2017.01.016.
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–9. https://doi.org/10.1182/blood-2014-01-552174.
Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol. 2001;167(11):6123–31. https://doi.org/10.4049/jimmunol.167.11.6123.
Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9(9):1183–97. https://doi.org/10.15252/emmm.201607485.
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. https://doi.org/10.1056/NEJMra1706169.
Upreti D, Bakhshinyan D, Bloemberg D, Vora P, Venugopal C, Singh SK. Strategies to enhance the efficacy of T-cell therapy for central nervous system tumors. Front Immunol. 2020;11: 599253. https://doi.org/10.3389/fimmu.2020.599253.
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–28. https://doi.org/10.1016/j.ccell.2015.09.004.
Land CA, Musich PR, Haydar D, Krenciute G, Xie Q. Chimeric antigen receptor T-cell therapy in glioblastoma: charging the T cells to fight. J Transl Med. 2020;18(1):428. https://doi.org/10.1186/s12967-020-02598-0.
Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60(5):1168–72.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–9. https://doi.org/10.1056/NEJMoa1610497.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–72. https://doi.org/10.1158/1078-0432.Ccr-15-0428.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–101. https://doi.org/10.1001/jamaoncol.2017.0184.
Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med. 2005;3:38. https://doi.org/10.1186/1479-5876-3-38.
Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–35. https://doi.org/10.1097/cji.0000000000000260.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aaa0984.
Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20(4):972–84. https://doi.org/10.1158/1078-0432.Ccr-13-0709.
Abbott RC, Verdon DJ, Gracey FM, Hughes-Parry HE, Iliopoulos M, Watson KA, et al. Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin Transl Immunol. 2021;10(5): e1283. https://doi.org/10.1002/cti2.1283.
Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–58. https://doi.org/10.1038/s41587-019-0192-1.
Chuntova P, Hou Y, Naka R, Yamamichi A, Chen T, Goretsky Y, et al. Novel EGFRvIII-CAR transgenic mice for rigorous preclinical studies in syngeneic mice. Neuro Oncol. 2021. https://doi.org/10.1093/neuonc/noab182.
Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.aaw2672.
Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7–H3 as a novel CAR-t therapeutic target for glioblastoma. Mol Ther Oncolytics. 2019;14:279–87. https://doi.org/10.1016/j.omto.2019.07.002.
Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. 2018;20(1):55–65. https://doi.org/10.1093/neuonc/nox116.
Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. https://doi.org/10.1038/s41467-019-11869-4.
Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3(10):541–51. https://doi.org/10.1158/1541-7786.Mcr-05-0056.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–37. https://doi.org/10.1038/mt.2012.210.
Lin Q, Ba T, Ho J, Chen D, Cheng Y, Wang L, et al. First-in-human trial of EphA2-redirected CAR T-cells in patients with recurrent glioblastoma: a preliminary report of three cases at the starting dose. Front Oncol. 2021;11: 694941. https://doi.org/10.3389/fonc.2021.694941.
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23(7):2130–41. https://doi.org/10.1016/j.celrep.2018.04.051.
Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20(13):3025–33. https://doi.org/10.1016/j.celrep.2017.09.002.
Woroniecka K, Fecci PE. T-cell exhaustion in glioblastoma. Oncotarget. 2018;9(82):35287–8. https://doi.org/10.18632/oncotarget.26228.
Tudor T, Binder ZA, O’Rourke DM. CAR T cells. Neurosurg Clin N Am. 2021;32(2):249–63. https://doi.org/10.1016/j.nec.2020.12.005.
Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE. 2011;6(1): e16195. https://doi.org/10.1371/journal.pone.0016195.
Agrawal NS, Miller R, Jr., Lal R, Mahanti H, Dixon-Mah YN, DeCandio ML, et al. Current studies of immunotherapy on glioblastoma. J Neurol Neurosurg. 2014;1(1)
Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22(8):1865–74. https://doi.org/10.1158/1078-0432.Ccr-15-1507.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. https://doi.org/10.1126/science.aaa6204.
Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216(1):15–24. https://doi.org/10.1002/path.2370.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. https://doi.org/10.1038/nrclinonc.2016.217.
Agarwal P, Pajor MJ, Anson DM, Guda MR, Labak CM, Tsung AJ, et al. Elucidating immunometabolic targets in glioblastoma. Am J Cancer Res. 2017;7(10):1990–5.
Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844–59. https://doi.org/10.1182/blood-2008-12-195941.
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–85. https://doi.org/10.1158/0008-5472.Can-08-2281.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9. https://doi.org/10.1038/ni.2035.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99. https://doi.org/10.1038/nri3862.
Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4(3):235–40. https://doi.org/10.1038/ni887.
Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics. 2017;9:59. https://doi.org/10.1186/s13148-017-0358-y.
Pandiyan K, You JS, Yang X, Dai C, Zhou XJ, Baylin SB, et al. Functional DNA demethylation is accompanied by chromatin accessibility. Nucleic Acids Res. 2013;41(7):3973–85. https://doi.org/10.1093/nar/gkt077.
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–9. https://doi.org/10.1126/science.aae0491.
Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat Immunol. 2017;18(5):573–82. https://doi.org/10.1038/ni.3706.
Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021. https://doi.org/10.1126/science.aba1786.
Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. 2022;40(1):53-69.e9. https://doi.org/10.1016/j.ccell.2021.12.005.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59. https://doi.org/10.1056/NEJMoa1709919.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54. https://doi.org/10.1056/NEJMoa1708566.
The Lancet O. CAR T-cell therapy for solid tumours. Lancet Oncol. 2021;22(7):893. https://doi.org/10.1016/s1470-2045(21)00353-3.
Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol. 2019;15(8):813–22. https://doi.org/10.1080/1744666x.2019.1629904.
Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63(1):60–72. https://doi.org/10.1007/s00018-005-5390-y.
Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15. https://doi.org/10.1038/s41408-020-0280-y.
Topp M, Van Meerten T, Houot R, Minnema MC, Milpied N, Lugtenburg PJ, et al. Earlier steroid use with axicabtagene ciloleucel (Axi-Cel) in patients with relapsed/refractory large B cell lymphoma. Blood. 2019;134(Supplement_1):243. https://doi.org/10.1182/blood-2019-126081.
Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med. 2018;66(2):50–2. https://doi.org/10.1016/j.retram.2018.03.003.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–19. https://doi.org/10.1158/2159-8290.Cd-17-0698.
Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958–71. https://doi.org/10.1158/2159-8290.Cd-17-1319.
Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11:1973. https://doi.org/10.3389/fimmu.2020.01973.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. https://doi.org/10.1182/blood-2014-05-552729.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5. https://doi.org/10.1126/science.aar6711.
Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev. 2019;290(1):60–84. https://doi.org/10.1111/imr.12773.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32(9):1970–83. https://doi.org/10.1038/s41375-018-0065-5.
Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long terminal repeat CRISPR-CAR-coupled “universal” T cells mediate potent anti-leukemic effects. Mol Ther. 2018;26(5):1215–27. https://doi.org/10.1016/j.ymthe.2018.02.025.
Sutherland AR, Owens MN, Geyer CR. Modular chimeric antigen receptor systems for universal CAR T cell retargeting. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21197222.
Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1):132. https://doi.org/10.1186/s13045-018-0677-2.
Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in universal CAR-T cell therapy. Front Immunol. 2021;12: 744823. https://doi.org/10.3389/fimmu.2021.744823.
Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75(18):3853–64. https://doi.org/10.1158/0008-5472.Can-14-3321.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–7. https://doi.org/10.1038/cr.2016.142.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–66. https://doi.org/10.1158/1078-0432.Ccr-16-1300.
Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014;23(R1):R40–6. https://doi.org/10.1093/hmg/ddu125.
Guo Y, Xu B, Wu Z, Bo J, Tong C, Chen D, et al. Mutant B2M-HLA-E and B2M-HLA-G fusion proteins protects universal chimeric antigen receptor-modified T cells from allogeneic NK cell-mediated lysis. Eur J Immunol. 2021;51(10):2513–21. https://doi.org/10.1002/eji.202049107.
Riteau B, Menier C, Khalil-Daher I, Martinozzi S, Pla M, Dausset J, et al. HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol. 2001;13(2):193–201. https://doi.org/10.1093/intimm/13.2.193.
Rouas-Freiss N, Marchal RE, Kirszenbaum M, Dausset J, Carosella ED. The alpha1 domain of HLA-G1 and HLA-G2 inhibits cytotoxicity induced by natural killer cells: is HLA-G the public ligand for natural killer cell inhibitory receptors? Proc Natl Acad Sci USA. 1997;94(10):5249–54. https://doi.org/10.1073/pnas.94.10.5249.
Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122(8):1341–9. https://doi.org/10.1182/blood-2013-03-478255.
Tykodi SS, Fujii N, Vigneron N, Lu SM, Mito JK, Miranda MX, et al. C19orf48 encodes a minor histocompatibility antigen recognized by CD8+ cytotoxic T cells from renal cell carcinoma patients. Clin Cancer Res. 2008;14(16):5260–9. https://doi.org/10.1158/1078-0432.Ccr-08-0028.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8. https://doi.org/10.1038/nm.4441.
Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–26. https://doi.org/10.1158/2159-8290.Cd-18-0442.
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–52. https://doi.org/10.1172/jci83416.
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20(4):506–18. https://doi.org/10.1093/neuonc/nox182.
Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology. 2017;6(2): e1273302. https://doi.org/10.1080/2162402x.2016.1273302.
Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunol. 2021;10(4): e1274. https://doi.org/10.1002/cti2.1274.
Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 2019;40(2):142–58. https://doi.org/10.1016/j.it.2018.12.003.
Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning Innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683. https://doi.org/10.3389/fimmu.2019.02683.
Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2016;5(4): e1119354. https://doi.org/10.1080/2162402x.2015.1119354.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483. https://doi.org/10.1038/srep11483.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38(5):197–210. https://doi.org/10.1097/cji.0000000000000082.
Murakami T, Nakazawa T, Natsume A, Nishimura F, Nakamura M, Matsuda R, et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018;38(9):5049–56. https://doi.org/10.2187/anticanres.12824.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016. https://doi.org/10.1093/jnci/djv375.
Ramanathan A, Lorimer IAJ. Engineered cells as glioblastoma therapeutics. Cancer Gene Ther. 2021. https://doi.org/10.1038/s41417-021-00320-w.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53. https://doi.org/10.1038/s41587-020-0462-y.
Author information
Authors and Affiliations
Contributions
VDN, LG, and EF conceived the idea and contributed to editing of the manuscript. AAB, SB, and AT contributed to reviewing the manuscript and to editing the final draft.
Corresponding author
Ethics declarations
Funding
No external funding was used to assist in the preparation of this manuscript.
Conflicts of interest
Vincenzo Di Nunno, Enrico Franceschi, Alicia Tosoni, Lidia Gatto, Stefania Bartolini, and Alba Ariela Brandes declare they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Gatto, L., Di Nunno, V., Franceschi, E. et al. Pharmacotherapeutic Treatment of Glioblastoma: Where Are We to Date?. Drugs 82, 491–510 (2022). https://doi.org/10.1007/s40265-022-01702-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01702-6