
Abstract
Despite their transfusion-independence, non-transfusion-dependent thalassemia (NTDT) patients experience a variety of serious clinical complications that require prompt and comprehensive management. Transfusion therapy may still be an important part of management of this disease, in cases of acute stress, to support growth and development in childhood, or to prevent clinical morbidities stemming from ineffective erythropoiesis or hemolytic anemia. Although splenectomy is associated with improvements in hemoglobin levels, it leads to several short- and long-term adverse events, warranting caution in application of this intervention. Fetal hemoglobin induction therapy has been evaluated in non-randomized studies, with benefits extending beyond hematologic improvements to lowering morbidity risk. Effective and safe iron chelation therapy is now available for NTDT patients in whom iron overload develops, irrespective of transfusions, due to increased intestinal absorption, ultimately leading to clinically high iron burden levels and subsequent morbidity. Optimal management of NTDT patients requires a holistic approach targeting all hallmarks of the disease to ensure favorable patient outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Non-transfusion-dependent thalassemia (NTDT) encompasses three clinically distinct forms: β-thalassemia intermedia, hemoglobin E/β-thalassemia, and α-thalassemia intermedia (hemoglobin H disease). |
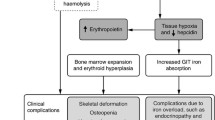
Ineffective erythropoiesis alongside chronic hemolytic anemia are the hallmarks of disease process in NTDT; which in turn lead to increased intestinal iron absorption (iron overload) and a hypercoagulable state. |
NTDT is associated with a multitude of serious clinical complications involving almost every organ system. |
Splenectomy should only be performed in indicated situations as it is associated with a variety of adverse events; while beneficial roles of tailored transfusion therapy, iron chelation therapy, and fetal hemoglobin induction justify use of these modalities to manage patients with NTDT. |
1 Introduction
The thalassemias are a group of inherited disorders of hemoglobin synthesis characterized by various degrees of defective production of the α- or β-globin chains of adult hemoglobin A, leading to the α- and β-thalassemias, respectively. When thalassemias mutations are inherited in homozygous or compound heterozygous forms, they lead to several phenotypes, although the hallmark of disease remains ineffective erythropoiesis and a chronic hemolytic anemia of varying severity. The thalassemias can also be co-inherited with structural hemoglobin variants, like hemoglobin S, C, and E, further adding to the heterogeneity and phenotypic diversity of thalassemia syndromes [1, 2]. Transfusion-dependence in these congenital anemias has been classically utilized as a factor to distinguish the various thalassemia phenotypes and their severity. Non-transfusion-dependent thalassemias (NTDT) are those phenotypes that do not require lifelong regular transfusions for survival, although they may require occasional or even frequent transfusions in certain clinical settings and for defined periods of time [3]. Although the NTDT classification can include a variety of thalassemia phenotypes, three forms have been the focus of most recent research studies and management guidelines owing to high prevalence and similarities in clinical manifestations: β-thalassemia intermedia, hemoglobin E/β-thalassemia (mild and moderate forms), and α-thalassemia intermedia (hemoglobin H disease) [3]. This review focuses on practical management considerations of these three NTDT forms, which are further described in Figs. 1 and 2 [3].

Genetic profiles most commonly associated with non-transfusion-dependent thalassemia. HPFH hereditary persistence of fetal hemoglobin

Main clinical differences between non-transfusion-dependent thalassemia (NTDT) and transfusion-dependent thalassemias (TDT)
2 Global Epidemiology of Non-Transfusion-Dependent Thalassemias (NTDT)
The thalassemia syndromes are the most common monogenetic disease worldwide, with around 68,000 children born with various phenotypes each year [4–6]. It is estimated that 80–90 million people are carriers of a β-thalassemia mutation across the world (1.5 %). Approximately half of these carriers originate from South East Asia [7]. Around 23,000 children are born with transfusion-dependent β-thalassemia each year, while a smaller number, distributed around parts of the Eastern Mediterranean and Africa [1], have β-thalassemia intermedia (NTDT) [1, 4, 6]. A high prevalence of hemoglobin E mutations is observed across India, Bangladesh, Thailand, Laos, and Cambodia (carrier frequencies up to 80 %); as well as through regions of China, Malaysia, Indonesia, and Sri Lanka [4, 8–10]. More than 19,000 children are born each year with the hemoglobin E/β-thalassemia phenotype (50 % fitting into the definition of NTDT) [4, 6]. In fact, it is the most common form of β-thalassemia identified in many newborn screening programs in the USA [11, 12]. It is estimated that 1,000,000 people currently live with hemoglobin E/β-thalassemia [13]. α-Thalassemia remains the most common among all inherited disorders of hemoglobin, with around 5 % of the world’s population being carriers, and 1,000,000 people affected with an α-thalassemia syndrome [14, 15]. It is most commonly observed in areas from sub-Saharan Africa through the Mediterranean region and Middle East, to the Indian sub-continent and East and Southeast Asia [1]. An increasing incidence of the disease is also being noted in North Europe and North America, owing to the high influx of migrants into large cities [16, 17]. Around 10,000 births are recorded each year for the NTDT form of α-thalassemia, α-thalassemia intermedia, or hemoglobin H disease [4, 6]. These numbers might even be underestimated since there are limited genetic epidemiology studies of thalassemia syndromes in parts of the world where the disease is known to be most prevalent.
3 Practical Disease Management
The following sections review management considerations in patients with NTDT that primarily target the pathophysiologic manifestations of the disease. Management of specific complications (endocrine and bone disease, fertility and pregnancy, hepatic pathology, thrombosis, pulmonary hypertension, extramedullary hematopoietic pseudotumors, and acute hemolytic crisis) is not covered and has been reviewed elsewhere [18, 19].
3.1 Targeting the Hematologic Abnormalities
In NTDT, erythropoiesis is ineffective because of an imbalance in the α- and β-globin chains production. Unstable globin chain tetramers ultimately precipitate and undergo oxidation into methemoglobin and hemichromes, with separation of heme from globin. The free iron released eventually catalyzes the formation of reactive oxygen species, which lead to oxidation of membrane proteins, structural membrane defects, and exposure of red-cell senescence antigens like phosphatidylserine, causing premature cell death within the bone marrow (termed ineffective erythropoiesis) or within the peripheral circulation (termed hemolysis) [3, 20]. While hemolysis is primarily associated with a high incidence of gall bladder stones [21], both ineffective erythropoiesis and hemolysis result in a state of chronic anemia and hypoxia, leading to erythroid marrow expansion and subsequent hepatosplenomegaly, extramedullary hematopoietic pseudotumors, as well as bone deformities and osteoporosis [3]. More importantly, ineffective erythropoiesis and hemolysis are also associated with a hypercoagulable state attributed to thrombogenicity of hemolyzed red blood cells; which ultimately leads to a high incidence of thromboembolic and cerebrovascular events as well as pulmonary hypertension, especially in patients with other contributing factors (e.g. high activated platelet counts) [22–24]. Ineffective erythropoiesis and chronic hypoxia also lead to iron overload due to increased intestinal absorption, with subsequent hepatic, endocrine, and vascular morbidities [25]; as will be discussed in a later section. This entails that without appropriate treatment, patients with NTDT will continue to experience various morbidities related to their underlying condition [26–28].
3.1.1 Transfusion Therapy
Transfusion therapy supplies patients with normal red blood cells, balancing the hemolytic anemia, and thus suppressing ineffective erythropoiesis [29–31]. Thus, although NTDT patients do not require lifelong regular transfusion therapy for survival, there is still merit in considering blood transfusions for the prevention or management of complications resulting from ineffective erythropoiesis or hemolytic anemia.
NTDT patients should not immediately be placed on regular transfusion programs for the mere fact of having anemia, unless the hemoglobin level is persistently and severely low (<5 g/dl). For example, should a patient show low hemoglobin levels during the course of an infection, pregnancy, or surgery, transfusions should only be given during the period of acute stress and do not necessarily need to become regular and lifelong. In young patients, observing the child for signs of poor growth and skeletal deformities can indicate the need for transfusions during this period of development [32, 33]. It should be noted that some children with hemoglobin E/β-thalassemia have a remarkable ability for adaptation to low hemoglobin levels [34, 35]. Children with non-deletional hemoglobin H disease are more likely to show signs of poor growth necessitating transfusions than children with the deletional form [36]. Observational studies have also provided evidence towards the role of blood transfusions in several clinical morbidities frequently encountered in adult NTDT patients, like leg ulcers, thrombotic events, pulmonary hypertension, silent brain infarcts, and extramedullary hematopoietic pseudotumors [24, 37, 38]. In the absence of randomized trials, there are no specific guidelines on the amount and duration of transfusions needed for the effective prevention or management of such morbidities and, thus, therapy should be tailored based on individual patient status and need and should be withdrawn when the desired outcomes are achieved. It should also be noted that the risk of alloimmunization is highest in minimally transfused and newly transfused patients, in splenectomized patients, and during pregnancy [39]. Such risk can be reduced by comprehensive genotype and antibody screening, and fully phenotyped matched blood should be given. Figure 3 highlights the practical use of transfusion therapy in the management course of patients with NTDT.

Practical guide to management of non-transfusion-dependent thalassemia using transfusion therapy, splenectomy, and hydroxyurea
Patients having regular transfusions for extended periods of time should be managed as per the guidelines for transfusion-dependent β-thalassemia major.
3.1.2 Splenectomy
Splenectomy has been a common practice in NTDT patients to increase the total hemoglobin level by 1–2 g/dl and avoid blood transfusions [3]. However, recent and cumulative evidence confirms that splenectomy is associated with a variety of adverse outcomes in patients with NTDT. As will be noted later, abnormalities of platelets and hemolyzed red blood cells are believed to be the key factors causing a hypercoagulable state in patients with NTDT [22]. These abnormalities become more prominent following splenectomy and increase the risk of vascular events [40, 41]. Splenectomized NTDT patients have a fivefold increased risk of venous thromboembolism, fourfold increased risk of pulmonary hypertension and leg ulcers, as well as a higher risk of silent cerebral infarction than nonsplenectomized patients [24, 37]. The median time to thrombosis following splenectomy is around 8 years [42]. A higher rate of iron overload-related morbidity has also been noted in splenectomized compared with nonsplenectomized NTDT patients [37], which may be attributed to lower ability to scavenge toxic iron species like non-transferrin-bound iron (NTBI) [43, 44]. Splenectomy also places NTDT patients of all ages at risk of infection, with associated morbidity and mortality [45]. Thus, a conservative approach to the use of splenectomy in NTDT patients is usually recommended [45], and the procedure should be reserved for specific cases. Figure 3 highlights the practical use of splenectomy in the management course of patients with NTDT.
When performed, laparoscopic splenectomy is preferred to the open technique, since the procedure is associated with lower intra-operative blood loss, postoperative morbidity and mortality, a shorter length of hospital stay, and a more favorable body image and cosmesis [46]. Partial splenectomy has been used to preserve some immune function while reducing the degree of hypersplenism, although the long-term success of this approach is still under evaluation. The likelihood of splenic re-growth and the volume of splenic tissue required to preserve immune function remain unknown [45]. The gall bladder should be inspected and removed during splenectomy if there is evidence of gallstones [45]. Post-splenectomy sepsis remains a risk in all patients, especially in children <2 years, between 1–4 years after surgery, and in patients with poor immune health [45]. Therefore, febrile splenectomized patients should undergo rapid evaluation and prompt treatment. Equally important is that splenectomized patients receive appropriate vaccinations and prophylactic antibiotics [45], as well as anticoagulation therapy, especially in those patients with high platelet counts and nucleated red blood cell levels [42].
3.1.3 Fetal Hemoglobin Induction
Increased production of γ-globin chains in patients with β-thalassemia can bind the excess α-chains (thus producing fetal hemoglobin), ameliorating the α/β-globin chain imbalance and ineffective erythropoiesis. This can occur developmentally through inheritance of certain mutations and polymorphisms [47] or through pharmacologic therapy as reviewed in this section. Irrespectively, there is now evidence that increased fetal hemoglobin levels are associated with lower morbidity rates in NTDT patients [48].
The first agent to be evaluated for its effect on inducing fetal hemoglobin in thalassemia patients was 5-azacytidine, a DNA methylation inhibitor that was associated with an unfavorable safety profile. More recently, another demethylating agent, decitabine, was evaluated in a pilot study in five patients with β-thalassemia intermedia using a subcutaneous 0.2 mg/kg dose two times per week for 12 weeks. The drug was able to increase total hemoglobin level by an average of 1 g/dl, with a manageable safety profile [49]. Favorable fetal and total hemoglobin responses to butyrate derivatives have also been observed in a few studies including small samples of NTDT patients, although effects were less notable on the long term [50]. The use of erythropoietic-stimulating agents has also been evaluated in small studies of NTDT patients, both in monotherapy [51] and in combination with other fetal hemoglobin stimulants like hydroxyurea or butyrate derivatives. In such combinations, an additive effect on total hemoglobin level increase was noted with high doses of the erythropoietic-stimulating agent [52, 53].
Hydroxyurea (also known as hydroxycarbamide) is the fetal hemoglobin-inducing agent that has been most studied in NTDT patients. Hydroxyurea is a cytotoxic, anti-metabolic, and anti-neoplastic agent with apparent potent fetal hemoglobin inducer properties, as noted in studies in patients with sickle cell disease [54]. In addition to its known effects in stimulating γ-globin production during stress erythropoiesis, hydroxyurea may have a more general role in augmenting globin synthesis, including β-globin in some NTDT patients who maintain the capacity to express normal β-globin chains [55]. There is also evidence that hydroxyurea diminishes the hypercoagulability of red cells in NTDT patients who have undergone splenectomy [56].
Despite the availability of a good amount of evidence on hydroxyurea use in patients with NTDT, randomized clinical trials have not been conducted to date. At doses of 10–20 mg/kg/day, mean increases in total hemoglobin level of 0.5 and 2.5 g/dl (average of around 1.5 g/dl) have been reported [50], with the portion of patients having increases of >1.0 g/dl ranging between 40 and 70 % [50]. The effects may be dependent on age, co-inheritance of certain mutations and polymorphisms (especially homozygosity for the XmnI polymorphism −158 C→T Gγ), and baseline hematologic values. According to a recent review, they are most commonly noted in the first 3–6 months, with further improvements noted up to 12 months of therapy, and sustained responses observed over long-term follow-up in several studies [50]. However, some studies noted a decline in hematological response on long-term therapy (beyond 12 months), and it has been theorized that long-term treatment with hydroxyurea may result in an impairment in the ability of certain hematopoietic stem cells to give rise effectively to erythroid lineage cells [50]. Regardless, increases in the range of 1–2 g/dl in total hemoglobin attributed to hydroxyurea are of clinical significance and may shift patients to a lower disease severity grouping [57], especially as they were usually associated with better exercise tolerance, appetite, and sense of well-being [50]. The benefit of hydroxyurea extends beyond correcting anemia. A beneficial role in patients with pulmonary hypertension was suggested, especially upon combination with the antioxidant l-carnitine [58–60]. Hydroxyurea therapy was also associated with improvements in leg ulcers [61] and extramedullary hematopoietic pseudotumors [38] in smaller studies. In a cross-sectional study of 584 β-thalassemia intermedia patients, hydroxyurea therapy was associated with reduced adjusted odds of extramedullary hematopoietic pseudotumors, pulmonary hypertension, leg ulcers, hypothyroidism, and osteoporosis [37].
According to a recent review, hydroxyurea therapy was generally well tolerated in most published studies with NTDT patients [50]. The rate of myelotoxicity ranged between 0 and 30 % and was usually dose-dependent and reversible. No renal or hepatic side effects were reported with hydroxyurea therapy, while data on dermatologic, neurologic, or endocrine adverse effects are heterogeneous and, in many instances, conflicting [50]. Figure 3 highlights the practical use of hydroxyurea in the management course of patients with NTDT.
3.1.4 On the Horizon
Pre-clinical studies have suggested that two agents, sotatercept (ACE-011) and ACE-536, that block the activity of certain transforming growth factor (TGF)-β family cytokines involved in late stages of erythropoiesis have the ability of increasing hemoglobin production in thalassemia [62]. Studies in thalassemic mice have also shown that a short treatment with a JAK2 inhibitor (TG101209) can ameliorate ineffective erythropoiesis and decrease spleen size [63, 64]. Interference with (reversal of) developmental fetal hemoglobin silencing corrected the anemia in animal studies [65], while gene therapy has shown similar benefits in human case reports [66]. Data from clinical trials exploring those novel therapies are awaited. Bone marrow transplantation is the only definitive curative therapy for the thalassemias. Data specific to patients with NTDT are lacking and called for. Until then, the decision between transplantation and medical therapy should be highly individualized and patient-specific, while also dependent on donor availability.
3.2 Targeting Iron Overload
Ineffective erythropoiesis in NTDT leads to inappropriately low hepcidin levels, increased intestinal iron absorption, and increased release of recycled iron from the reticuloendothelial system [67]. This in turn leads to depletion of macrophage iron, relatively lower levels of serum ferritin, increased liver iron concentration (LIC), and release into the circulation of free iron species that can cause target-organ damage [44, 68].
Without treatment, iron overload in NTDT patients continues to accumulate, and a considerable proportion of NTDT patients eventually reach LIC thresholds of clinical significance [3], with iron-related morbidity appearing beyond 10 years of age (or later at 15 years in deletional hemoglobin H disease) [26, 69, 70]. Although cardiac siderosis does not seem to be a concern in NTDT patients [71–74], several vital organs may be affected, with the iron overload state leading to significant morbidity and mortality [75]. An association between iron overload and hepatic fibrosis in non-chelated patients with NTDT has been observed [76], and the occurrence of hepatocellular carcinoma in hepatitis-negative patients is increasingly being reported [77]. In a recent study of 168 non-chelated patients with NTDT, higher LIC values were associated with a significantly increased risk of developing thrombosis, pulmonary hypertension, hypothyroidism, hypogonadism, and osteoporosis [78]. Liver iron concentration levels ≥5 mg Fe/g dry weight (dw) were associated with a considerable morbidity risk increase [79]. A more recent longitudinal follow-up over a 10-year period confirmed these findings, and a serum ferritin level of ≥800 ng/ml was the threshold after which all patients became at risk of developing morbidity [69]. Renal tubular dysfunction as evident from proteinuria has also been recently reported in iron-overloaded NTDT patients [80]. Iron overload has also been associated with a high prevalence of silent brain infarction, large cerebral vessel disease, and decreased neuronal function in NTDT patients [24, 81, 82]. Prompt diagnosis and management in NTDT is essential to prevent the occurrence of serious, sometimes irreversible, clinical morbidities.
Assessment of LIC remains the gold standard for quantification of total body iron [83]. Owing to limitations with performing a liver biopsy (invasiveness, sampling error, variability), and the limited availability of methods such as superconducting quantum interference device (SQUID) and magnetic iron detector (MID), magnetic resonance imaging (MRI) has become the preferred method to assess LIC in NTDT patients [25]. MRI using either R2 or T2* techniques are reliable and internationally reproducible, and have been validated against liver biopsy [84]. The upper limit to reliably estimate LIC by MRI is 30–40 mg Fe/g dw, depending on the scanner specifications [84]. In resource-poor countries, serum ferritin measurement may be the only method available for the assessment of iron overload. There is positive correlation between serum ferritin level and LIC in NTDT patients [70, 85–87], and the technique is still a reliable approach to follow serial changes in iron overload. However, one caution when using single/spot serum ferritin levels for decision making is that the ratio of serum ferritin to LIC is lower relative to patients with transfusion-dependent β-thalassemia [72, 86–88]. Thus, serum ferritin levels used to guide iron chelation decisions in transfusion-dependent β-thalassemia (e.g. 1,000 and 2,500 ng/ml) do not exactly reflect the same LIC levels or morbidity risk in NTDT, and NTDT-specific thresholds should always be used.
3.2.1 Iron Chelation Therapy
Considering phlebotomy is not an option in NTDT patients who are already complicated with anemia, iron chelation therapy remains the cornerstone of therapy for iron overload. Current recommendations are to initiate iron chelation therapy in NTDT patients who are 10 years or older (15 years in hemoglobin H disease) and have an LIC of ≥5 mg Fe/g dw, considering these are the thresholds when iron-related morbidity manifests, while therapy should be interrupted and patients observed when they reach a target LIC of ≤3 mg Fe/g dw to avoid over chelation [19, 68, 89, 90]. The 5 and 3 mg Fe/g dw values for starting and stopping iron chelation therapy were successfully used in the context of clinical trials [85]. However, LIC assessment is not always feasible and alternate serum ferritin level thresholds have been suggested [19, 68, 89, 90], partly since they have good predictive power for LIC (≤300 ng/ml for LIC of ≤3 mg Fe/g dw and ≥800 ng/ml for LIC of ≥5 mg Fe/g dw) [91, 92] and also since the two thresholds are associated with 0 and 100 % risk of morbidity in NTDT, respectively [69]. These findings encourage the use of serum ferritin to guide iron chelation therapy when LIC measurement is unavailable or when the need for careful resource allocation arises. However, it should be noted that close to 50 % of patients with a serum ferritin level <800 ng/ml may still have an LIC ≥5 mg Fe/g dw [91, 92]; highlighting the need for LIC assessment or other confirmatory means in this subgroup of patients before the decision not to initiate iron chelation therapy.
Subcutaneous deferoxamine therapy was the first iron chelator to be studied in NTDT patients since the 1980s in two small case series totaling 14 patients [93, 94]. Deferoxamine showed urinary iron excretion in the majority of patients (β-thalassemia intermedia). Aside from a lack of solid evidence from large studies on the benefit of deferoxamine in NTDT patients, the main limitation is its cumbersome subcutaneous administration, which might cause compliance challenges for NTDT patients in whom iron overload is diagnosed at a later stage (early adolescence) than transfusion-dependent patients. Data from recent studies of deferasirox in patients with NTDT [95, 96] noted that recruited patients have not been successfully treated previously with deferoxamine due to poor compliance (sporadic use). Therefore, the use of oral iron chelators for iron overload management in NTDT remains a more practical option.
Similar to experiences with deferoxamine, that with the oral chelator deferiprone is limited to small studies and case series in NTDT patients. Reductions in serum ferritin level in transfusion-independent hemoglobin E/β-thalassemia patients were noted in a study of 39 patients [97, 98]. However, only nine patients were shown to have reductions in LIC by direct liver biopsy [98]. Published data in β-thalassemia intermedia are restricted to a total of four patients [99, 100]. There has also been only one study of iron chelation using deferiprone in patients with hemoglobin H disease (n = 17) [101]. Although a reduction of serum ferritin was observed from 6 months of therapy, interpretation of changes in liver iron are difficult considering the nature of the assessment technique and the control group.
More recently, data from the largest and first randomized clinical trial of iron chelation therapy in 166 patients with NTDT became available (THALASSA) [85]. The once-daily oral iron chelator deferasirox resulted in a significant reduction of LIC compared with placebo following 12 months of therapy in patients ≥10 years of age with LIC ≥5 mg Fe/g dw. LIC decreased by a mean of 2.33 ± 0.70 and 4.18 ± 0.69 mg Fe/g dw in patients receiving starting doses of 5 mg/kg/day (p = 0.001) and 10 mg/kg/day (p < 0.001), respectively. Doses were doubled after 6 months for patients with LIC >7 mg Fe/g dw and <15 % reduction from baseline; and were interrupted when LIC was <3 mg Fe/g dw. The frequency of adverse events in patients receiving deferasirox was similar to that with placebo. The most common drug-related adverse events were nausea (6.6 %), rash (4.8 %), and diarrhea (3.6 %) [85]. Sub-analyses from the study showed that reduction in LIC with deferasirox 5 and 10 mg/kg/day starting dose groups is consistent irrespective of baseline LIC/serum ferritin, age, gender, race, splenectomy status, and underlying NTDT form. The analyses also showed that greater reductions in LIC were achieved in patients dose-escalated at 6 months from deferasirox 10 mg/kg/day starting dose to 20 mg/kg/day [102]. An extension of the core study showed that deferasirox progressively decreases iron overload over 2 years in NTDT patients, with a safety profile consistent with that in the core study [103]. The deferasirox safety profile remains consistent as patients approach the chelation interruption target of LIC 3 mg Fe/g dw [104]. On 23 January 2013, deferasirox received US Food and Drug Administration (FDA) approval as first-line therapy for the management of iron overload in NTDT patients 10 years and older, making it the first drug specifically approved for use in NTDT patients. Deferasirox had also received European Medicines Agency approval on 16 November 2012 for the treatment of chronic iron overload requiring chelation therapy when deferoxamine therapy is contraindicated or inadequate in patients aged 10 years and older with NTDT syndromes. However, it should be noted that deferasirox remains the only iron chelator ‘specifically’ approved for NTDT patients. The deferoxamine label in most European countries allows for its use in NTDT patients, although this was not based on any specific clinical studies from NTDT patient populations [19]. Figure 4 presents a practical guideline for iron overload assessment and chelation therapy in NTDT patients.

Practical guide to iron overload assessment and management in non-transfusion-dependent thalassemia. d day; HbH hemoglobin H, LIC liver iron concentration, MRI magnetic resonance imaging, NTDT non-transfusion-dependent thalassemia, Qx mo every x month, Qx yr every x year, SF serum ferritin
A beneficial effect of iron chelation therapy in reducing clinical morbidity risk in patients with NTDT is also suggested by observational studies, and further long-term studies in this direction are awaited [37, 76].
3.2.2 On the Horizon
Studies in NTDT mouse models suggested a role for hepcidin and transferrin therapy to decrease iron overload [105, 106]. Reducing transmembrane protease serine-6 has also been shown to ameliorate iron overload in mice [107]. Human studies evaluating the role of such modalities, alone or in combination with iron chelators, are of high merit, especially with the noted hematologic improvements associated with such therapies.
4 Conclusion
The complexity of pathophysiologic mechanisms and multiplicity of risk factors for morbidity in NTDT patients entail a holistic approach to management where several interventions may be necessary to prevent or manage clinical complications and thus improve outcomes and quality of life in this patient population.
References
Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4th ed. Oxford: Blackwell Science; 2001.
Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of hemoglobin : genetics, pathophysiology, and clinical management. Cambridge medicine. 2nd ed. New York: Cambridge University Press; 2009.
Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833–44.
Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–6.
Christianson A, Howson CP, Modell B. March of dimes global report on birth defects. New York: March of Dimes Brith Defects Foundation; 2006.
Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–7.
Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of beta-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010;3(1):103–17.
Weatherall DJ. Keynote address: The challenge of thalassemia for the developing countries. Ann N Y Acad Sci. 2005;1054:11–7.
Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704–12.
Olivieri NF, Pakbaz Z, Vichinsky E. HbE/beta-thalassemia: basis of marked clinical diversity. Hematol Oncol Clin North Am. 2010;24(6):1055–70.
Lorey F, Cunningham G, Vichinsky EP, Lubin BH, Witkowska HE, Matsunaga A, et al. Universal newborn screening for Hb H disease in California. Genet Test. 2001;5(2):93–100.
Lorey F. Asian immigration and public health in California: thalassemia in newborns in California. J Pediatr Hematol Oncol. 2000;22(6):564–6.
Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134:522–31.
Cohen AR, Galanello R, Pennell DJ, Cunningham MJ, Vichinsky E. Thalassemia. Hematology Am Soc Hematol Educ Program;2004. 14–34.
Vichinsky E. Complexity of alpha thalassemia: growing health problem with new approaches to screening, diagnosis, and therapy. Ann N Y Acad Sci. 2010;1202:180–7.
Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13.
Michlitsch J, Azimi M, Hoppe C, Walters MC, Lubin B, Lorey F, et al. Newborn screening for hemoglobinopathies in California. Pediatr Blood Cancer. 2009;52(4):486–90.
Taher AT, Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev. 2012;26(Suppl 1):S24–7.
Taher A, Vichinsky E, Musallam K, Cappellini MD. Viprakasit V. In: Weatherall D, editor. Guidelines for the management of non transfusion dependent thalassaemia (NTDT). Cyprus: Nicosia; 2013.
Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S12–5.
Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37(1):12–20.
Cappellini MD, Musallam KM, Poggiali E, Taher AT. Hypercoagulability in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S20–3.
Cappellini MD, Poggiali E, Taher AT, Musallam KM. Hypercoagulability in beta-thalassemia: a status quo. Expert Rev Hematol. 2012;5(5):505–11 quiz 12.
Musallam KM, Taher AT, Karimi M, Rachmilewitz EA. Cerebral infarction in beta-thalassemia intermedia: breaking the silence. Thromb Res. 2012;130(5):695–702.
Musallam KM, Cappellini MD, Wood JC, Taher AT. Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev. 2012;26(Suppl 1):S16–9.
Taher AT, Musallam KM, El-Beshlawy A, Karimi M, Daar S, Belhoul K, et al. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br J Haematol. 2010;150(4):486–9.
Musallam KM, Cappellini MD, Daar S, Karimi M, El-Beshlawy A, Taher AT. Serum ferritin levels and morbidity in β-thalassemia intermedia: a 10-year cohort study [abstract]. Blood. 2012;120(21):1021.
Musallam KM, Taher AT, Duca L, Cesaretti C, Halawi R, Cappellini MD. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with beta thalassemia intermedia. Blood Cells Mol Dis. 2011;47(4):232–4.
Cazzola M, Finch CA. Evaluation of erythroid marrow function in anemic patients. Haematologica. 1987;72(3):195–200.
Cazzola M, Pootrakul P, Huebers HA, Eng M, Eschbach J, Finch CA. Erythroid marrow function in anemic patients. Blood. 1987;69(1):296–301.
Cazzola M, De Stefano P, Ponchio L, Locatelli F, Beguin Y, Dessi C, et al. Relationship between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol. 1995;89(3):473–8.
Taher AT, Musallam KM, Cappellini MD, Weatherall DJ. Optimal management of beta thalassaemia intermedia. Br J Haematol. 2011;152(5):512–23.
Olivieri NF, Muraca GM, O’Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in haemoglobin E beta-thalassaemia. Br J Haematol. 2008;141(3):388–97.
O’Donnell A, Premawardhena A, Arambepola M, Allen SJ, Peto TE, Fisher CA, et al. Age-related changes in adaptation to severe anemia in childhood in developing countries. Proc Natl Acad Sci USA. 2007;104(22):9440–4.
Allen A, Fisher C, Premawardhena A, Peto T, Allen S, Arambepola M, et al. Adaptation to anemia in hemoglobin E-ss thalassemia. Blood. 2010;116(24):5368–70.
Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2012;26(Suppl 1):S31–4.
Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–92.
Haidar R, Mhaidli H, Taher AT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J. 2010;19(6):871–8.
Chou ST, Liem RI, Thompson AA. Challenges of alloimmunization in patients with haemoglobinopathies. Br J Haematol. 2012;159(4):394–404.
Musallam KM, Taher AT. Thrombosis in thalassemia: why are we so concerned? Hemoglobin. 2011;35(5–6):503–10.
Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111(2):467–73.
Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8(10):2152–8.
Tavazzi D, Duca L, Graziadei G, Comino A, Fiorelli G, Cappellini MD. Membrane-bound iron contributes to oxidative damage of beta-thalassaemia intermedia erythrocytes. Br J Haematol. 2001;112(1):48–50.
Taher A, Musallam KM, El Rassi F, Duca L, Inati A, Koussa S, et al. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146(5):569–72.
Saad GS, Musallam KM, Taher AT. The surgeon and the patient with beta-thalassaemia intermedia. Br J Surg. 2011;98(6):751–60.
Musallam KM, Khalife M, Sfeir PM, Faraj W, Safadi B, Abi Saad GS, et al. Postoperative outcomes after laparoscopic splenectomy compared with open splenectomy. Ann Surg. 2013;257(6):1116–23.
Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643.
Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood. 2012;119(2):364–7.
Olivieri NF, Saunthararajah Y, Thayalasuthan V, Kwiatkowski J, Ware RE, Kuypers FA, et al. A pilot study of subcutaneous decitabine in beta-thalassemia intermedia. Blood. 2011;118(10):2708–11.
Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood. 2013;121(12):2199–212 quiz 372.
Singer ST, Vichinsky EP, Sweeters N, Rachmilewitz E. Darbepoetin alfa for the treatment of anaemia in alpha- or beta- thalassaemia intermedia syndromes. Br J Haematol. 2011;154(2):281–4.
Loukopoulos D, Voskaridou E, Stamoulakatou A, Papassotiriou Y, Kalotychou V, Loutradi A, et al. Hydroxyurea therapy in thalassemia. Ann N Y Acad Sci. 1998;850:120–8.
Singer ST, Kuypers FA, Olivieri NF, Weatherall DJ, Mignacca R, Coates TD, et al. Fetal haemoglobin augmentation in E/beta(0) thalassaemia: clinical and haematological outcome. Br J Haematol. 2005;131(3):378–88.
Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74(2):652–6.
Zeng YT, Huang SZ, Ren ZR, Lu ZH, Zeng FY, Schechter AN, et al. Hydroxyurea therapy in beta-thalassaemia intermedia: improvement in haematological parameters due to enhanced beta-globin synthesis. Br J Haematol. 1995;90(3):557–63.
Singer ST, Vichinsky EP, Larkin S, Olivieri N, Sweeters N, Kuypers FA. Hydroxycarbamide-induced changes in E/beta thalassemia red blood cells. Am J Hematol. 2008;83(11):842–5.
Sripichai O, Makarasara W, Munkongdee T, Kumkhaek C, Nuchprayoon I, Chuansumrit A, et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83(6):482–4.
Amoozgar H, Farhani N, Khodadadi N, Karimi M, Cheriki S. Comparative study of pulmonary circulation and myocardial function in patients with beta-thalassemia intermedia with and without hydroxyurea, a case-control study. Eur J Haematol. 2011;87(1):61–7.
Karimi M, Borzouee M, Mehrabani A, Cohan N. Echocardiographic finding in beta-thalassemia intermedia and major: absence of pulmonary hypertension following hydroxyurea treatment in beta-thalassemia intermedia. Eur J Haematol. 2009;82(3):213–8.
Karimi M, Mohammadi F, Behmanesh F, Samani SM, Borzouee M, Amoozgar H, et al. Effect of combination therapy of hydroxyurea with l-carnitine and magnesium chloride on hematologic parameters and cardiac function of patients with beta-thalassemia intermedia. Eur J Haematol. 2010;84(1):52–8.
Gamberini MR, Fortini M, De Sanctis V. Healing of leg ulcers with hydroxyurea in thalassaemia intermedia patients with associated endocrine complications. Pediatr Endocrinol Rev. 2004;2(Suppl 2):319–22.
Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408–14.
Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–85.
Melchiori L, Gardenghi S, Guy EG, Rachmilewitz E, Giardina PJ, Grady RW, et al. Use of JAK2 inhibitors to limit ineffective erythropoiesis and iron absorption in mice affected by β-thalassemia and other disorders of red cell production [abstract]. Blood. 2009;114(22):2020.
Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058):993–6.
Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–22.
Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118(16):4321–30.
Musallam KM, Cappellini MD, Taher AT. Iron overload in beta-thalassemia intermedia: an emerging concern. Curr Opin Hematol. 2013;20(3):187–92.
Musallam KM, Cappellini MD, Daar S, Kairmi M, El-Beshlawy A, Graziadei G et al. Serum ferritin level and morbidity risk in transfusion-independent patients with beta-thalassemia intermedia: the ORIENT study. Haematologica. Epub 2014 Jul 4.
Lal A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364(8):710–8.
Origa R, Barella S, Argiolas GM, Bina P, Agus A, Galanello R. No evidence of cardiac iron in 20 never- or minimally-transfused patients with thalassemia intermedia. Haematologica. 2008;93(7):1095–6.
Taher AT, Musallam KM, Wood JC, Cappellini MD. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am J Hematol. 2010;85(4):288–90.
Roghi A, Cappellini MD, Wood JC, Musallam KM, Patrizia P, Fasulo MR, et al. Absence of cardiac siderosis despite hepatic iron overload in Italian patients with thalassemia intermedia: an MRI T2* study. Ann Hematol. 2010;89(6):585–9.
Mavrogeni S, Gotsis E, Ladis V, Berdousis E, Verganelakis D, Toulas P, et al. Magnetic resonance evaluation of liver and myocardial iron deposition in thalassemia intermedia and b-thalassemia major. Int J Cardiovasc Imaging. 2008;24(8):849–54.
Matta BN, Musallam KM, Maakaron JE, Koussa S, Taher AT. A killer revealed: 10-year experience with beta-thalassemia intermedia. Hematology. 2014;19(4):196–8.
Musallam KM, Motta I, Salvatori M, Fraquelli M, Marcon A, Taher AT, et al. Longitudinal changes in serum ferritin levels correlate with measures of hepatic stiffness in transfusion-independent patients with beta-thalassemia intermedia. Blood Cells Mol Dis. 2012;49(3–4):136–9.
Maakaron JE, Cappellini MD, Graziadei G, Ayache JB, Taher AT. Hepatocellular carcinoma in hepatitis-negative patients with thalassemia intermedia: a closer look at the role of siderosis. Ann Hepatol. 2013;12(1):142–6.
Musallam KM, Cappellini MD, Wood JC, Motta I, Graziadei G, Tamim H, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica. 2011;96(11):1605–12.
Musallam KM, Cappellini MD, Taher AT. Evaluation of the 5 mg/g liver iron concentration threshold and its association with morbidity in patients with beta-thalassemia intermedia. Blood Cells Mol Dis. 2013;51(1):35-38.
Ziyadeh FN, Musallam KM, Mallat NS, Mallat S, Jaber F, Mohamed AA, et al. Glomerular Hyperfiltration and Proteinuria in Transfusion-Independent Patients with beta-Thalassemia Intermedia. Nephron Clin Pract. 2012;121(3–4):c136–43.
Musallam KM, Beydoun A, Hourani R, Nasreddine W, Raad R, Koussa S, et al. Brain magnetic resonance angiography in splenectomized adults with beta-thalassemia intermedia. Eur J Haematol. 2011;87(6):539–46.
Musallam KM, Nasreddine W, Beydoun A, Hourani R, Hankir A, Koussa S, et al. Brain positron emission tomography in splenectomized adults with beta-thalassemia intermedia: uncovering yet another covert abnormality. Ann Hematol. 2012;91(2):235–41.
Angelucci E, Brittenham GM, McLaren CE, Ripalti M, Baronciani D, Giardini C, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000;343(5):327–31.
Wood JC. Impact of iron assessment by MRI. Hematology Am Soc Hematol Educ Program. 2011;2011:443–50.
Taher AT, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120(5):970–7.
Pakbaz Z, Fischer R, Fung E, Nielsen P, Harmatz P, Vichinsky E. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49(3):329–32.
Taher A, El Rassi F, Isma’eel H, Koussa S, Inati A, Cappellini MD. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93(10):1584–6.
Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–8.
Taher AT, Musallam KM, Viprakasit V, Porter JB, Cappellini MD. Iron chelation therapy for non-transfusion-dependent thalassemia (NTDT): a status quo. Blood Cells Mol Dis. 2014;52(2–3):88–90.
Taher AT, Viprakasit V, Musallam KM, Cappellini MD. Treating iron overload in patients with non-transfusion-dependent thalassemia. Am J Hematol. 2013;88(5):409–15.
Taher A, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Estimation of liver iron concentration by serum ferritin measurement in non-transfusion-dependent thalassemia patients: analysis from the 1-year THALASSA study [abstract]. Haematologica. 2012;96(S1):0927.
Taher A, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Serum ferritin for predicting clinically relevant LIC thresholds to guide management of patients with nontransfusion-dependent thalassemia treated with deferasirox: THALASSA study extension analysis [abstract]. Haematologica. 2013;98(S1):486.
Cossu P, Toccafondi C, Vardeu F, Sanna G, Frau F, Lobrano R, et al. Iron overload and desferrioxamine chelation therapy in beta-thalassemia intermedia. Eur J Pediatr. 1981;137(3):267–71.
Pippard MJ, Weatherall DJ. Iron balance and the management of iron overload in beta-thalassemia intermedia. Birth Defects Orig Artic Ser. 1988;23(5B):29–33.
Ladis V, Berdousi H, Gotsis E, Kattamis A. Deferasirox administration for the treatment of non-transfusional iron overload in patients with thalassaemia intermedia. Br J Haematol. 2010;151(5):504–8.
Voskaridou E, Plata E, Douskou M, Papadakis M, Delaki EE, Christoulas D, et al. Treatment with deferasirox (Exjade) effectively decreases iron burden in patients with thalassaemia intermedia: results of a pilot study. Br J Haematol. 2010;148(2):332–4.
Akrawinthawong K, Chaowalit N, Chatuparisuth T, Siritanaratkul N. Effectiveness of deferiprone in transfusion-independent beta-thalassemia/HbE patients. Hematology. 2011;16(2):113–22.
Pootrakul P, Sirankapracha P, Sankote J, Kachintorn U, Maungsub W, Sriphen K, et al. Clinical trial of deferiprone iron chelation therapy in beta-thalassaemia/haemoglobin E patients in Thailand. Br J Haematol. 2003;122(2):305–10.
Rombos Y, Tzanetea R, Konstantopoulos K, Simitzis S, Zervas C, Kyriaki P, et al. Chelation therapy in patients with thalassemia using the orally active iron chelator deferiprone (L1). Haematologica. 2000;85(2):115–7.
Olivieri NF, Koren G, Matsui D, Liu PP, Blendis L, Cameron R, et al. Reduction of tissue iron stores and normalization of serum ferritin during treatment with the oral iron chelator L1 in thalassemia intermedia. Blood. 1992;79(10):2741–8.
Chan JC, Chim CS, Ooi CG, Cheung B, Liang R, Chan TK, et al. Use of the oral chelator deferiprone in the treatment of iron overload in patients with Hb H disease. Br J Haematol. 2006;133(2):198–205.
Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox demonstrates a dose-dependent reduction in liver iron concentration and consistent efficacy across subgroups of non-transfusion-dependent thalassemia patients. Am J Hematol. 2013;88(6):503–6.
Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–93.
Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Approaching low liver iron burden in chelated patients with non-transfusion-dependent thalassemia: the safety profile of deferasirox. Eur J Haematol. 2014;92(6):521–6.
Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120(12):4466–77.
Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177–82.
Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123(4):1531–41.
Acknowledgments
The authors thank Kaivan Khavandi MD of EVIDA MEDICAL DMCC, Dubai, UAE, for medical editorial assistance. Financial support for medical editorial assistance was provided by the Hemoglobinopathy Research Initiative, Reading, UK, who received an unrestricted grant from Novartis Pharma AG, Basel, Switzerland. The grant provider did not have any role in the design or writing of, or the decision to submit, this manuscript for publication.
Conflicts of interest
AT Taher reports receiving research funding and honoraria from Novartis. MD Cappellini reports receiving honoraria from Novartis, Genzyme, and Celgene.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Taher, A.T., Cappellini, M.D. Management of Non-Transfusion-Dependent Thalassemia: A Practical Guide. Drugs 74, 1719–1729 (2014). https://doi.org/10.1007/s40265-014-0299-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0299-0