
Abstract
Beta thalassemia major (TM) is the most frequent form of transfusion-dependent inherited anemia in India. The thalassemia syndromes exhibit enormous variability in their genetic basis and phenotypic expression. The authors recommend that the diagnosis of TM or non-transfusion-dependent thalassemia (NTDT) should not be based on a one-time assessment. Many patients have a chronic anemia that is not severe enough to justify regular transfusions, but the clinical course can evolve with age. Continued observation may reveal that some patients who are considered NTDT will benefit from transfusions later in life. Clinical decision making can be influenced by the perceived difficulty in access to a safe blood supply and the cost of therapy. Here, authors present selected case scenarios that address common issues in the management of TM or NTDT. The recommendations are based on published evidence where available or on the authors’ shared experience. Among the topics under discussion are deciding when to start regular transfusions, the role of hydroxyurea in TM, the procedure for blood administration, the use of deferasirox for chelation and monitoring of side effects, the role of splenectomy, and the prospects for gene therapy. In order to achieve an optimal outcome with blood transfusions and chelation therapy over the lifetime, it is essential to adhere to the current guidelines for the management of thalassemia.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Query 1. An 18-mo-old child with anemia and hepatosplenomegaly has never been transfused. Hemoglobin is varying between 7 to 8 g/dl and HbF is 95% on high performance liquid chromatography (HPLC). Both parents have beta thalassemia trait. Should this patient be treated as non-transfusion-dependent thalassemia (NTDT) or as thalassemia major? Should one try hydroxyurea (HU) in such a case?
Comment: Let’s start with general recommendations for the use of chronic transfusions in thalassemia that are presented in Table 1 [1,2,3]. The twin goals of transfusion therapy are to relieve symptomatic anemia and control ineffective erythropoiesis. These two aspects are linked, such that lower hemoglobin levels would usually have increased erythropoietic drive. The threshold to begin transfusions when hemoglobin drops below 7 g/dl is appropriate for most patients, but symptoms can appear at higher hemoglobin level as well. When hemoglobin is >7 g/dl, the guidelines in Table 1 can be useful to determine the need for transfusions on an individual basis. If a decision is made to continue observation, it should be kept in mind that the hemoglobin level is not stable at the age of 18 mo because fetal hemoglobin synthesis can decline further. The use of hydroxyurea should be deferred until 3 y of age to determine if chronic transfusions are needed based on the criteria in the table. Children meeting these criteria would benefit from an earlier initiation of transfusions to prevent growth failure and complications of ineffective erythropoiesis. Delaying HU allows appreciation of the phenotype which is helpful in deciding whether to initiate regular transfusions. The age of 3 y is a generally accepted threshold to say if a patient has thalassemia intermedia who was placed on regular transfusions due to symptoms or has thalassemia major [4, 5]. These terms have been discarded in favour of the current nomenclature of transfusion-dependent thalassemia (TDT) and NTDT, but they remain relevant in assessing endogenous hemoglobin production.
Query 2. Often patients treated as NTDT have some degree of hemolytic facies, borderline growth failure, and occasional drop in hemoglobin to below 6–7 g/dl. When should the diagnosis of a patient being treated as NTDT be changed to thalassemia major and treated with regular transfusions instead?
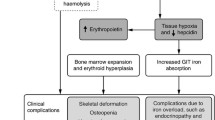
Comment: Hemoglobin levels in thalassemia exist along a continuum so that a simple classification of patients into transfusion dependent (TD) and NTDT is not feasible [3, 6]. The criteria for transfusions stated in Table 1 are aimed at reducing subjectivity in decision-making, but there will always be a group of patients like this who do not fall into one or the other group. An occasional drop in hemoglobin below 7 g/dl can be managed with intermittent transfusions and mild skeletal changes and growth deficits may be acceptable if the other criteria such as progressive splenomegaly and quality of life are not a concern [7]. In borderline cases the improvement in symptoms can be assessed with a limited 6-mo period of transfusions. Splenic enlargement is frequent and difficult problem in NTDT. Progressive splenomegaly indicates that the thalassemia is severe. These patients are likely to develop hypersplenism which will further worsen anemia. In the past, splenectomy was routinely performed in such cases, but the long-term complications are quite significant. Patients who undergo splenectomy and then stop regular transfusions are at high risk for severe complications as adults, including pulmonary hypertension, extra-medullary masses, osteoporosis, and cerebrovascular disease. For this reason, rapid enlargement of spleen (Table 1) is considered an indication to begin chronic transfusions [8,9,10]. In all cases, it should be emphasized to the family that the decision for starting or avoiding transfusions is based upon the current assessment but can change in the future. One particularly common issue is the decrease in tolerance to anemia in adults that makes it necessary to start transfusions much later in life.
Query 3. How do I decide when to increase transfusions from a single to 2 units of packed red cells in a patient with thalassemia major? At what age does this typically occur? How should one balance the number of units and the frequency of transfusions?
Comment: The aim is to sustain pre-transfusion hemoglobin in the desired range of 9.5–10.5 g/dl. To calculate volume of transfusion, consider that (i) on an average, children need 4-5 ml/kg packed red blood cells (PRBC) per week, (ii) one unit of PRBC is 325 ml, (iii) the transfusion interval should be 3–4 wk, and (iv) the maximum volume transfused at one visit should not exceed 20 ml/kg [1, 2]. The final consideration is whether the blood bank can provide half-units, which is possible in large infusion centres that can share compatible units between patients. Given these parameters, a single unit of blood would be sufficient for children up to a weight of 20 kg or 6 y if they are on a 3-wk schedule, and for younger children on a 4-wk schedule. Beyond this weight (or age), a second unit will be needed when pre-transfusion hemoglobin levels are consistently below target. As an example, a child whose weight is 25 kg can receive 1 ½ units every 4 wk, but when the weight approaches 30 kg, 2 full units can be transfused. The frequency of transfusions can be adjusted between 3 to 4 wk to keep the hemoglobin level at target for children with intermediate weight. The ability to transfuse partial units requires good communication between the blood bank and infusion centre to avoid the need for discarding blood. In medium to large infusion centres, patients who can share a unit of blood can be paired together for same day transfusion.
Query 4. Is hydroxyurea useful in patients with beta-thalassemia major?
Comment: When patients are receiving adequate blood to meet hemoglobin targets (9.5 to 10.5 g/dl before transfusion), there is no benefit of adding hydroxyurea. It is unclear, however, if hydroxyurea may have a role where access to blood is limited and hemoglobin frequently drops below 9 g/dl in TM [11]. Could there be a role for hydroxyurea in these cases to boost hemoglobin, suppress bone marrow expansion or slow the enlargement of the spleen? These questions have not been addressed in a clinical trial, so it is not possible to endorse the use of hydroxyurea in thalassemia major [12]. Hydroxyurea should also not be used to delay transfusion therapy if the criteria for transfusion-dependence are met, such as hemoglobin level consistently below 7 g/dl.
Query 5. Deferasirox is currently the most popular iron chelating drug administered to patients with thalassemia major. Is it necessary to administer it empty stomach? Is absorption poorer with meals?
Comment: The absorption of deferasirox is increased by dietary fat and studies have shown greater bioavailability when deferasirox is given with food. There was initially a concern that peak plasma levels of deferasirox are more variable when given with meals, but this was not associated with any increase in toxicity [13]. Conversely, deferasirox is better tolerated by patients when taken with meals, which can improve adherence with chelation. For these reasons, it is recommended to take deferasirox with food.
Query 6. In an individual patient, can one consider exceeding the recommended maximum dose of 40 mg/kg/d of deferasirox for iron chelation? Could this be considered in a patient who is severely iron overloaded and deferoxamine is not an option due to cost?
Comment: The dispersible tablet (DT) of deferasirox has a dose range of 20–40 mg/kg/d. Another formulation of deferasirox is the film coated tablet (FCT) which has improved bioavailability and therefore the corresponding dose range is 14–28 mg/kg/d. Most patients will achieve negative iron balance with a deferasirox DT dose of 30 mg/kg/d, such that exceeding the maximum recommended dose of 40 mg/kg/d should never be necessary [14]. In case of lack of efficacy, consider if adherence with treatment is being compromised or if the patient is receiving very high transfusion volume (>300 ml/kg/y) [15]. There are certain scenarios where iron overload needs to be lowered rapidly with combined chelation therapy. These patients benefit from combination of deferasirox with deferoxamine [16, 17], or deferasirox with deferiprone [18] to increase chelation intensity.
Query 7. Is there an evidence to suggest that administering deferasirox in two divided doses is more efficacious than a single dose per day?
Comment: Use of twice daily instead of once daily dosing leads to lower peak and higher trough plasma concentrations of deferasirox. Some studies suggest that patients who are refractory to once daily deferasirox can respond to taking the same total amount as two divided doses [19]. This approach has also been tried in patients who have creatinine elevation with a single daily dose. However, most patients are not expected to benefit from a trial of twice-daily dosing. The routine use of twice-daily dosing is also not recommended because of adherence with prescribed therapy is lower when a drug is given in multiple daily doses.
Query 8. How frequently should liver enzymes be monitored in a patient receiving deferasirox? What is the protocol for stopping and restarting the drug if the enzymes become elevated?
Comment: Interpretation of elevated liver enzyme concentrations in thalassemia is complex due to multiple potential etiologies. Besides deferasirox, high liver iron concentration, chronic viral hepatitis, and any intercurrent illness can induce transaminitis. A general guideline for monitoring of potential toxicity from deferasirox is provided in Table 2 [2, 20,21,22].
Query 9. A patient with NTDT begins to show signs of growth failure near adolescence with hemoglobin hovering around 7 to 8 g/dl. I wish to start him on regular transfusions but fear the development of alloimmunization at this relatively older age. What should I do?
Comment: Development of antibodies to donor red blood cells is a serious complication of transfusion therapy in thalassemia [23, 24]. Extended phenotypic matching of red blood cell units would markedly reduce the risk of alloimmunization. However, due to the cost involved in finding phenotypically matched donors, the current recommendation is that patients who are not alloimmunized receive blood matched only to Rh and Kell antigens. This is based on the observation that most alloantibodies are directed against Rh (C,c,E,e) and Kell antigens. The prevalence of alloimmunization in thalassemia in India has been reported to be 3–10% [25, 26], which is lower than reported in the United States (20%). This variation may reflect the difference in ethnicity between patients and donors which increases the potential for mismatch of red cell antigens. In India, population mobility in the metropolitan centres may increase the risk of alloimmunization in the future if red cell phenotypes are distinct in the different geographical regions of the country. Among patient variables, initiating transfusions at an older age or during pregnancy increases the risk of developing alloimmunization. In such cases, at a minimum, the blood should be matched to Rh/Kell, but preferably include Duffy, Kidd and S antigens as well.
Query 10. Should an allogeneic stem cell transplant ever be considered in a patient being treated as NTDT?
Comment: Inherent in the decision to not transfuse a patient with thalassemia is the assessment that he or she will have an acceptable quality of life without the development of chronic complications that can affect longevity [7]. Stem cell transplant cannot be recommended in this setting. However, if the decision to not transfuse is based on the lack of availability of blood or the development of red blood cell alloimmunization, then the patient does not truly fall in the NTDT group and transplant can be considered as an option.
Query 11. What is the duration over which a unit of blood should be transfused in everyday practice in thalassemia day care centre?
Comment: The maximum rate of transfusion is 5 ml/kg per hour. Transfusion is started at a very low rate for the first 15 min to confirm compatibility, then either increased to the maximum rate or at the patient’s preferred rate. From start to finish, a typical transfusion of 16 ml/kg should take less than 4 h if only one unit is transfused or 5–6 h if two units are given. Blood bank policy requires that a unit of blood be transfused in under 4 h from the time it is issued.
Query 12. A 13-y-old patient with thalassemia major requires 2 units at each visit. Should the 2 units be transfused consecutively on a single day or over 2 d?
Comment: Both units should be transfused on a single day if the volume of transfusion does not exceed 20 ml/kg. The entire transfusion can be completed in less than 6 h once the units have arrived at the infusion centre.
Query 13. At what temperature should blood be transfused? Should one wait for it to reach room temperature?
Comment: The transfusion should be initiated at the temperature at which blood was issued by the blood bank, usually 1-6 °C. Bringing blood to room temperature is neither necessary nor desirable as it extends the time since its release from the blood bank and increases the risk of growth of any bacterial contaminants. In cold weather, the infusion centre can be kept warm or the patient can be offered extra blankets if needed to increase their comfort.
Query 14. An 8-y-old patient with thalassemia major has increased transfusion requirement for 6 mo from one unit every 21–25 d to lately every 10–14 d. Pre-transfusion hemoglobin is typically 6–8 g/dl. Liver is 5 cm and spleen 8 cm below the costal margin. Father owns a small ‘daily-needs’ shop in a village. Will you recommend splenectomy?
Comment: Splenomegaly in thalassemia major is a consequence of inadequate transfusion therapy. Patients who maintain hemoglobin >9.5 g/dl prior to transfusion do not develop splenomegaly. In this child, the transfusion of a single unit of blood every 3–4 wk did not meet the recommended targets due to growth and weight gain. The recommended approach is to intensify transfusions for 6–9 mo to maintain hemoglobin in the 10–11 g/dl range, which would reduce the hepato-splenomegaly and after which the lower target of 9.5 to 10.5 g/dl can be resumed. This often requires transfusion of 20 ml/kg at each transfusion visit, every 2 wk initially and then every 3 wk. Since spleen size contributes to the intravascular volume, the transfusion requirement decreases as splenomegaly is reversed. If such an approach is impractical due to the cost of care, then splenectomy would aid in reducing the transfusion needs. However, prevention of splenic enlargement with adequate transfusions is by far the better approach. The risks associated with splenectomy in thalassemia major are sepsis, thrombosis, pulmonary hypertension, and an increase in an extra-hepatic iron deposition.
Query 15. I deal with patients with beta thalassemia as well as E beta thalassemia. What are the common differences in treatment in the two conditions?
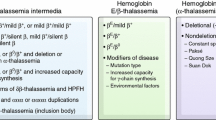
Comment: E beta thalassemia is generally less severe than beta thalassemia, although both conditions can vary between mild anemia to transfusion-dependence [10, 27]. HbE is a functional hemoglobin with better oxygen carrying characteristics than HbF. Even when combined with a beta 0 mutation, the variable synthesis of HbE and HbF affects the patient’s phenotype [28]. It is less common for patients with HbE to become transfusion-dependent during infancy. Additionally, a hemoglobin of 6–7 g/dl can be well tolerated in some patients with E beta thalassemia. Thus, the decision to begin regular transfusions cannot be based on hemoglobin alone. The symptoms that would prompt transfusions are provided in Table 1. Hydroxyurea seems to be less effective in E beta than in beta thalassemia. The co-inheritance of alpha thalassemia (1 or 2 gene deletion) significantly ameliorates anemia in E beta thalassemia. The spectrum of complications is similar between the two conditions. The major issue is to recognize that indications for starting transfusions and targets for pre-transfusion hemoglobin are somewhat different, so that standard guidelines for beta thalassemia major should not be uniformly adopted for E beta thalassemia. One could aim for a lower pre-transfusion hemoglobin in E beta thalassemia based on adequate growth and lack of symptoms of anemia. The customization of treatment plan is important in this group of patients.
Query 16. Do you think gene therapy would be accessible to most patients with beta-thalassemia major in the low and middle-income countries?
Comment: One must begin by emphasizing that prevention of thalassemia remains the central pillar of any attempt to reduce the number of individuals affected by thalassemia major [29]. If new births with thalassemia are decreased, more resources will be available to care for the current patients. These patients will benefit from curative therapy, whether in the form of bone marrow transplant or gene therapy. The expense of the current version of gene therapy will be prohibitive until cost reductions can be found in the manufacturing process [30]. It is also essential that the current efforts to develop gene therapy directed at the common thalassemia mutations in India is expanded as this can provide a lower cost alternative. Alternative approaches such as performing the procedure in utero or at a young age, or the development of in vivo gene therapy may eventually prove to be more accessible [31, 32]. Research into improving outcomes for alternative donor bone marrow transplants should also be intensified [33, 34]. In the wider picture, gene therapy is only one of several approaches that deserve attention to reduce the burden of thalassemia major in India and other resource-constrained countries.
Query 17. Should patients with thalassemia major and NTDT be supplemented daily with calcium and vitamin D3? If yes, how much? What if the vitamin D3 levels are normal?
Comment: Osteopenia in thalassemia major or NTDT occurs from multiple causes, such as anemia, bone marrow expansion, poor nutrition, low calcium intake, vitamin D deficiency, zinc deficiency, poor physical activity, low body weight, delayed puberty, and hypogonadism. Suboptimal peak bone mass is a serious issue in adults with thalassemia as it predisposes to fracture risk and chronic pain later in life. Ensuring adequate calcium and vitamin D intake is an important modifiable risk factor. It is recommended to maintain plasma 25-OH vitamin D3 level in the 30–40 ng/ml range. This test should be done once or twice annually to determine vitamin D status. If the level is low, then vitamin D3 should be supplemented at a dose of 2000 IU daily [35]. Patients with 25-OH D3 level < 15 ng/ml should be given 60,000 units orally once a month (this dose can be administered during the transfusion visits for convenience) for 4–8 doses. Patients with normal vitamin D status should take vitamin D3 1000 IU daily and encouraged to spend more time in outside play or other activities to increase sun exposure. The goal of calcium intake is to meet the requirement from diet and use supplements if needed to provide up to 50% of daily need. The recommended dietary allowance for calcium is as follows: 1–3 y: 700 mg, 4–8 y: 1000 mg, 9–18 y: 1300 mg, and adults: 1000 mg [36]. Patients are directed to increase dietary sources of calcium, particularly dairy products, vegetables such as mustard and cabbage, and beans. Calcium supplements should not be used as the main source to meet daily requirement due to the risk of nephrolithiasis.
Query 18. Is the measurement of HbA1c reliable in patients with thalassemia?
Comment: HbA1c or glycated hemoglobin is formed in a time-dependent manner as the red cells are exposed to glucose in the plasma over their life span of 120 d. Therefore, the use of HbA1c is unreliable in thalassemia or any other hemolytic anemia that shortens red cell survival. In thalassemia major, the presence of transfused red cells is another variable affecting the interpretation of A1c. Fructosamine can be used instead of HbA1c to assess glycemic control in thalassemia major.
Query 19. A patient with thalassemia major has had a splenectomy. What are the recommendations for booster vaccines in the follow-up?
Comment: Assuming that all vaccines were up to date at the time of splenectomy (specifically including meningococcal ACWY and B, Haemophilus influenzae type b, and the pneumococcal polysaccharide and conjugate vaccines), the following are recommended [37]: Patients should receive the influenza vaccine every year prior to onset of flu season, one booster dose of pneumococcal polysaccharide vaccine 5 y after the first dose, and a booster dose of meningococcal ACWY every 5 y. Patients over the age of 65 y should receive another booster dose of pneumococcal polysaccharide vaccine.
References
Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT) [Internet], 3rd ed. Nicosia (CY): Thalassaemia International Federation; 2014. Available at: http://www.ncbi.nlm.nih.gov/books/NBK269382/. Accessed 30 Oct 2017.
Lal A, Sheth S, Gilbert S, Kwiatkowski JL. Thalassemia management checklists: quick reference guides to reduce disparities in the care of patients with transfusion-dependent thalassemia. Blood. 2018;132:2233.
Olivieri NF, Brittenham GM. Management of the thalassemias. Cold Spring Harb Perspect Med. 2013;3:a011767.
Badens C, Joly P, Agouti I, et al. Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011;96:1712–4.
Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12:61–76.
Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2:245–55.
Taher AT, Musallam KM, Cappellini MD, Weatherall DJ. Optimal management of β thalassaemia intermedia. Br J Haematol [Internet]. 2011. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21250971. Accessed 3 Feb 2011.
Piga A, Serra M, Longo F, et al. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am J Hematol. 2011;86:808–10.
Bansal D. Splenectomy for β-thalassemia major in resource challenged settings: often a hobson’s choice! Indian J Pediatr. 2015;82:1082–3.
Olivieri NF, Muraca GM, O’Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in haemoglobin E beta-thalassaemia. Br J Haematol. 2008;141:388–97.
Bradai M, Abad MT, Pissard S, Lamraoui F, Skopinski L, de Montalembert M. Hydroxyurea can eliminate transfusion requirements in children with severe {beta}-thalassemia. Blood. 2003;102:1529–30.
Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood. 2013;121:2199–212.
Goldberg SL, Giardina PJ, Chirnomas D, Esposito J, Paley C, Vichinsky E. The palatability and tolerability of deferasirox taken with different beverages or foods. Pediatr Blood Cancer. 2013;60:1507–12.
Taher A, Cappellini MD, Vichinsky E, et al. Efficacy and safety of deferasirox doses of >30 mg/kg per d in patients with transfusion-dependent anaemia and iron overload. Br J Haematol. 2009;147:752–9.
Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in -thalassemia major. Blood. 2008;111:583–7.
Aydinok Y, Kattamis A, Cappellini MD, et al; HYPERION Investigators. Effects of deferasirox-deferoxamine on myocardial and liver iron in patients with severe transfusional iron overload. Blood. 2015;125:3868–77.
Lal A, Porter J, Sweeters N, et al. Combined chelation therapy with deferasirox and deferoxamine in thalassemia. Blood Cells Mol Dis. 2013;50:99–104.
Totadri S, Bansal D, Bhatia P, Attri SV, Trehan A, Marwaha RK. The deferiprone and deferasirox combination is efficacious in iron overloaded patients with β-thalassemia major: a prospective, single center, open-label study. Pediatr Blood Cancer. 2015;62:1592–6.
Chang H-H, Lu M-Y, Liao Y-M, et al. Improved efficacy and tolerability of oral deferasirox by twice-daily dosing for patients with transfusion-dependent β-thalassemia. Pediatr Blood Cancer. 2011;56:420–4.
Cappellini MD. Long-term efficacy and safety of deferasirox. Blood Rev. 2008;22:S35–41.
Díaz-García JD, Gallegos-Villalobos A, Gonzalez-Espinoza L, Sanchez-Niño MD, Villarrubia J, Ortiz A. Deferasirox nephrotoxicity—the knowns and unknowns. Nat Rev Nephrol. 2014;10:574–86.
Food and Drug Adminstration. Exjade: full prescribing information [Internet]. 2019. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021882s031lbl.pdf. Accessed 28 Jul 2019.
Lal A, Wong TE, Andrews J, et al. Transfusion practices and complications in thalassemia. Transfusion. 2018;58:2826–35.
Vichinsky E, Neumayr L, Trimble S, et al. Transfusion complications in thalassemia patients: a report from the Centers for Disease Control and Prevention (CME). Transfusion. 2014;54:972–81.
Dhawan HK, Kumawat V, Marwaha N, et al. Alloimmunization and autoimmunization in transfusion dependent thalassemia major patients: study on 319 patients. Asian J Transfus Sci. 2014;8:84–8.
Pahuja S, Pujani M, Gupta SK, Chandra J, Jain M. Alloimmunization and red cell autoimmunization in multitransfused thalassemics of Indian origin. Hematology. 2010;15:174–7.
Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2:a011734.
Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134:522–31.
Colah RB, Gorakshakar A. Control of thalassemia in India. Thalassemia Reports [Internet] 2014;4. Available at: http://www.pagepressjournals.org/index.php/thal/article/view/thal.2014.1955. Accessed 30 Sept 2014.
Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. New Engl J Med. 2018;378:1479–93.
McClain LE, Flake AW. In utero stem cell transplantation and gene therapy: recent progress and the potential for clinical application. Best Pract Res Clin Obstet Gynaecol. 2016;31:88–98.
Wang H, Georgakopoulou A, Psatha N, et al. In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia. J Clin Invest. 2019;129:598–615.
Anurathapan U, Hongeng S, Pakakasama S, et al. Hematopoietic stem cell transplantation for homozygous β-thalassemia and β-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant. 2016;51:813–8.
Sun Q, Wu B, Lan H, et al. Haploidentical haematopoietic stem cell transplantation for thalassaemia major based on an FBCA conditioning regimen. Br J Haematol. 2018;182:554–8.
Fung EB, Aguilar C, Micaily I, Haines D, Lal A. Treatment of vitamin D deficiency in transfusion-dependent thalassemia. Am J Hematol. 2011;86:871–3.
Office of Dietary Supplements. Calcium: fact sheet for health professionals [Internet]. Available at: https://ods.od.nih.gov/factsheets/Calcium-HealthProfessional/. Accessed 28 Jul 2019.
Centers for Disease Control. Vaccination of Adults with Asplenia [Internet]. 2019. Available at: https://www.cdc.gov/vaccines/adults/rec-vac/health-conditions/asplenia.html. Accessed 28 Jul 2019.
Author information
Authors and Affiliations
Contributions
The authors contributed to formulation of questions and discussion of recommended management. DB is the guarantor of this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lal, A., Bansal, D. Thalassemia: Common Clinical Queries in Management. Indian J Pediatr 87, 75–81 (2020). https://doi.org/10.1007/s12098-019-03065-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-019-03065-5