
Abstract
Epilepsy is one of the most prevalent neurological diseases and although numerous novel anticonvulsants have been approved, the proportion of patients who are refractory to medical treatment of seizures and have progressive co-morbidities such as cognitive impairment and depression remains at about 20–30%. In the last decade, extensive research has identified a therapeutic capacity of the components of the brain renin–angiotensin system (RAS) in seizure- and epilepsy-related phenomena. Alleviating the activity of RAS in the central nervous system is considered to be a potential adjuvant strategy for the treatment of numerous detrimental consequences of epileptogenesis. One of the main advantages of RAS is associated with its modulatory influence on different neurotransmitter systems, thereby exerting a fine-tuning control mechanism for brain excitability. The most recent scientific findings regarding the involvement of the components of brain RAS show that angiotensin II (Ang II), angiotensin-converting enzyme (ACE), Ang II type 1 (AT1) and type 2 (AT2) receptors are involved in the control of epilepsy and its accompanying complications, and therefore they are currently of therapeutic interest in the treatment of this disease. However, data on the role of different components of brain RAS on co-morbid conditions in epilepsy, including hypertension, are insufficient. Experimental and clinical findings related to the involvement of Ang II, ACE, AT1, and AT2 receptors in the control of epilepsy and accompanying complications may point to new therapeutic opportunities and adjuvants for the treatment of common co-morbid conditions of epilepsy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
There is a link between the brain renin–angiotensin system (RAS) and seizure activity, behavioral changes and neuropathology, hypertension and epilepsy, and possible treatment of co-morbid conditions in epilepsy. |
Overexpression of RAS components in the central nervous system is considered a risk factor for seizure and epilepsy development and the accompanying complications. |
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 (AT1) receptor antagonists have anticonvulsant, anti-inflammatory, antioxidant, behavioral, and neuroprotective properties in the epileptic state. |
1 Introduction
As a multifactorial neurological disorder, epilepsy is characterized by spontaneous epileptiform activity and accompanying complications such as depression, cognitive impairment, autism, and attention deficit and hyperactivity disorder [1,2,3,4]. In patients with temporal lobe epilepsy (TLE), hippocampal formation is the most affected brain structure with detected histopathological changes in the hilus of the dentate gyrus (DG) and CA1 and CA3 subfields, as well as altered expression and function of different neuropeptide receptors [5, 6]. Antiepileptic drugs (AEDs) mainly suppress the symptoms of epilepsy but are unable to prevent epileptogenesis or the development of a chronic epileptic state, characterized by detrimental neurochemical, morphological and behavioral consequences, comprising oxidative stress, disturbed equilibrium between excitatory and inhibitory neurotransmitter systems, inflammation and neuronal loss in the limbic system [2, 7,8,9,10,11].
In addition to the renin–angiotensin system (RAS), the components of which are found in most organs and tissues and are crucial for cardiovascular control, the discovery of an independent RAS in the central nervous system (CNS) expanded the functions of this system beyond its classical physiologies, including seizure susceptibility, learning and memory, stress, and depression [12, 13]. The role of brain RAS in the regulation of neuronal plasticity and its beneficial impact in neurological disorders such as epilepsy, Alzheimer’s disease, Parkinson’s disease, and Huntington’s chorea has been the subject of research and interpretation during the last decade [12,13,14,15]. The octapeptide angiotensin II (Ang II), generated by angiotensin-converting enzyme (ACE) or tonin, is the main biologically active peptide, acting as a neuromodulator in the CNS with dual effects on neuronal excitability: excitatory, mediated by Ang II type 1 (AT1) receptors, and inhibitory, mediated by Ang II type 2 (AT2) receptors [16,17,18].
Over the last few years, accumulated scientific evidence has provided strong support for the involvement of brain RAS components in seizure- and epilepsy-related phenomena. Recent studies from several laboratories, including ours, have reported altered levels of angiotensin peptides and receptor expression in limbic brain regions characterized by low seizure threshold, both in experimental models and in patients with TLE [19,20,21,22]. Most of the findings demonstrated that inhibition of RAS components, in particular Ang II and AT1 receptors, attenuated seizure susceptibility [20, 23,24,25,26]. Currently, data on the role of different components of brain RAS on co-morbid conditions in epilepsy, including hypertension, are insufficient. This review summarizes experimental and clinical findings related to the involvement of Ang II, ACE, AT1, and AT2 receptors in the control of epilepsy and accompanying complications, which might point to new therapeutic opportunities and adjuvants for the treatment of common co-morbid conditions of epilepsy.
2 Experimental Findings: The Brain Renin–Angiotensin System (RAS) and Epilepsy
The experimental findings regarding the effects of RAS components in epilepsy are summarized in Table 1.
2.1 Seizure Activity
Initial studies, launched by our team, demonstrated that the biologically active angiotensin peptides Ang II, Ang III, and Ang IV exhibited anticonvulsant activity, both in acute seizure tests in naïve mice and in pentylenetetrazol-kindled mice [27,28,29,30,31]. However, long-term intracerebroventricular (ICV) infusion of Ang II, starting after kainate-induced status epilepticus (SE) in rats, exacerbated epileptogenesis, shortened the latent period and increased the number of spontaneous motor seizures (SMS) [32]. These findings suggest that, depending on whether the brain is in a physiological or pathophysiological state, Ang II can exert dual effects on seizure susceptibility: anticonvulsant in acute seizure tests in naïve animals or proconvulsant in epileptogenesis. The latter is associated with complex plastic phenomena and network reorganizations leading to a decrease in the seizure threshold and impaired balance between the classical excitatory and inhibitory neurotransmitter system. The increased seizure susceptibility is closely linked with neuronal loss in limbic structures and γ-aminobutyric acid (GABA)-ergic interneurons in the hilus of the DG, recruitment of new synapses, and axonal sprouting in glutamatergic neurons [33,34,35,36]. Epileptogenesis is also associated with aberrant neurogenesis, severe inflammation (activated microglia and destroyed astrocyte function), and altered expression and function of important receptors closely related to seizure susceptibility [37]. Experimental results in models of epilepsy revealed that the levels of RAS components were increased in the brain. Thus, upregulation of Ang II levels, ACE, and AT1 receptors was reported in rat hippocampus both in a genetic-based model [20] and in an acquired model of TLE induced by pilocarpine [21, 24]. In accordance with these findings, we recently reported that upregulation of the AT1 receptors is a common pathway associated with epileptogenesis in normotensive Wistar and spontaneously hypertensive rats (SHRs) [22]. The proconvulsant effect of Ang II in epileptogenesis might involve excitatory influences via AT1 receptors related to facilitated release of glutamate and activation of excitatory N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in brain structures such as the hippocampus [38, 39], the paraventricular nucleus of the hypothalamus [40], as well as the basolateral complex and the central and cortical amygdala [41].
2.2 Anticonvulsant Activity
During the last decade, data were accumulated in support of the hypothesis that inhibition of RAS exerts anticonvulsant effects in both naïve animals and models of epilepsy, which confirm the proconvulsant action of Ang II in the brain. In the maximal electroshock (MES) test in mice, enalapril and losartan potentiated the anticonvulsant activity of AEDs without affecting their plasma and brain concentrations, thereby exhibiting pharmacodynamic effects [42,43,44]. Similarly, two ACE inhibitors, fosinopril and zofenopril, not only potentiated the effect of AEDs, but exerted direct effects on seizures in the DBA/2 (Dilute Brown Non-Agouti) mice model of generalized tonic–clonic seizures [45]. Further, the new-generation antihypertensive drug aliskiren, as well as the AT1 receptor antagonists telmisartan and olmesartan, had an anticonvulsant effect against pentylenetetrazol-induced clonic seizures and in the MES test in mice [46, 47]. Another study showed that long-term treatment with either enalapril or losartan significantly reduced seizure intensity, the effect being accompanied by a blood pressure decrease in a genetic model of epilepsy Wistar audiogenic rats [20]. Russo et al. [48] showed that long-term enalapril pretreatment in SHRs inhibited the occurrence of small-vessel disease and concurrently stabilized the rapid kindling process. Our results are in agreement with the reports revealing that long-term losartan administration exerted an anticonvulsant effect in the acute pentylenetetrazol seizure test in naïve rats [49]. In support of our finding, the AT1 receptor antagonists telmisartan and olmesartan in a dose-dependent manner decreased the seizure duration in the MES test and increased the seizure latency in the pentylenetetrazol test in mice [50]. Bar-Klein et al. [23] reported that treatment with losartan reduced the frequency and intensity of seizures, accompanied by transforming growth factor (TGF)-β in a model of albumin-induced vascular injury in Wistar rats, resulting in vessel damage and subsequent epileptiform activity. Long-term treatment with losartan alleviated the frequency of SMS in a kainate-induced post-SE model of TLE in Wistar rats [25]. In contrast, the AT1 receptor antagonist ZD7155 injected only once was unable to suppress SE both in SHRs and in Wistar-Kyoto (WKY) rats [51]. Thus, continuous treatment was superior over a single administration in the prevention of seizure-related consequences of SE. More recently, we found that losartan was able to blunt the SE-induced protein expression increase of AT1 receptors in the CNS [22]. These findings confirmed the putative role of the AT1 receptors and ACE inhibitors in seizure phenomena and epilepsy. It is well-known that the AT1 receptors are distributed mostly in brain regions that are responsible for the control of the cardiovascular functions, such as the hypothalamus, brain stem, and anterior pituitary [52]. However, although found in low levels, these receptors were co-expressed with GABAA receptor complexes in limbic brain structures vulnerable to epileptiform activity such as the hippocampus, the amygdala, and the piriform cortex [53]. The close link of RAS with the classical inhibitory neurotransmission was also supported by the fact that the GABAergic pathway had an inhibitory regulatory influence on specific neuronal networks related to important functions of Ang II in brain, such as thirst and blood pressure, as well as control of the hormonal release involved in these phenomena [54, 55].
2.3 Behavioral Changes
Ang II, the main effector peptide of RAS, has been reported to exert effects on motor activity, anxiety, depressive-like behavior, and memory consolidation in experimental rats [13, 53].
Several laboratories demonstrated that rats with spontaneous seizures are characterized by lack of anxiety and fear along with hyperlocomotion, which suggests increased emotionality and impulsivity [3, 56,57,58]. This inadequate behavior correlated with impaired links between crucial limbic regions such as the amygdala, ventral hippocampus, and entorhinal cortex [57]. Recently, we reported that continuous exposure to Ang II impaired some of the concomitant behavioral changes in epileptic rats [32]. In contrast, AT1 receptor blockade attenuated epilepsy-enhanced locomotion and affected the emotional state of anxiety in a phase-dependent manner, suggesting that activation of the AT1/AT2 receptors underlies the diurnal behavioral variations in kainate-treated normotensive Wistar rats [25]. Similarly, Villapol et al. [59] found that candesartan improved motor and spatial memory functions in a mouse model of traumatic brain injury (TBI). While AT1 receptor antagonists and ACE inhibitors exhibit anxiolytic effects [50, 60,61,62,63], the anxiogenic effect of long-term infusion of Ang II was suggested to be under the control of the AT1 receptor [32, 64].
Long-term Ang II administration evoked depression-like behavior in naïve rats [32]. In epileptic rats, Ang II exacerbated the co-morbid depression [32], while losartan treatment alleviated this emotional disturbance [25]. These findings are in line with numerous experimental and clinical data, suggesting that suppression of Ang II AT1 receptor signaling pathways leads to antidepressant-like activity [65,66,67,68,69].
In general, many reports have demonstrated that Ang II is able to alter spatial memory in naïve animals. Although endogenous Ang II was stated as being a non-participant in spatial memory formation [70], short- or long-term administration of the octapeptide impaired the hippocampus-dependent spatial memory in intact mice and rodents tested in the Morris water maze test [71]. Moreover, most of the reports indicated that Ang II worsened the cognitive processes via AT1 receptor stimulation [71, 72]. In accordance with the literature data, we showed that long-term ICV infusion of Ang II delayed the learning capacity of naïve rats in the radial arm maze test [32]. It could be speculated that damage to some specific limbic circuits, with a key role in memory impairment, could be affected by Ang II. However, this octapeptide had no impact on the epilepsy-induced deficit in spatial memory, which could be due to the exacerbating effect of Ang II on impulsive-like behavior with increased locomotion.
In the kainate model of TLE, losartan treatment had beneficial effects on memory in both SHRs and Wistar rats [73]. Likewise, spatial learning and memory were improved by losartan in the Morris water maze following pilocarpine-induced SE in rats [24]. Interestingly, naïve and epileptic SHRs showed better performance in the radial maze test than normotensive rats [73, 74], which might be explained by increased motor activity and impulsive-like behavior, typical for this strain. However, clinical data revealed a close relationship between hypertension and memory deficit in middle-aged patients [75]. Thus, experimental data from our laboratory and those of others [73, 76] suggest that SHRs are not an appropriate strain to study deficits in cognition associated with co-morbid hypertension in epilepsy.
2.4 Neuropathology Changes
Although Ang II exacerbated epileptogenesis and behavioral co-morbidities, this octapeptide exerted a strong neuroprotection mostly in the CA1 field of the dorsal hippocampus in epileptic rats [32]. The beneficial effect of Ang II on SE-induced neuronal damage might be mediated by the AT2 receptors. In support of this assumption are results from other investigators who used different paradigms and also reported the neuroprotective effect of Ang II via stimulation of the AT2 receptors [77, 78]. Alternatively, as a modulator of classical neurotransmitters, Ang II might act indirectly via stimulation of classical inhibitory GABAergic neurotransmission [38, 39, 79].
In addition to the anticonvulsant activity and favorable behavioral outcomes, several studies disclosed anti-inflammatory and neuroprotective effects of RAS inhibition in chronic epileptic conditions. The AT1 receptor antagonists were reported to exert strong anti-inflammatory and neuroprotective effects in a TBI model in mice. Thus, while candesartan was able to activate peroxisome proliferator-activated receptor (PPAR)-γ and reduced the TGF-β1 cytokine expression in cortical and hippocampal astrocytes, candesartan mitigated TBI provoked neuronal loss and activated microglia in mice [59]. In the pilocarpine model of TLE, Sun et al. [24] showed that losartan attenuated inflammatory responses through inhibition of tumor necrosis factor-α in activated glial cells. Furthermore, neuroprotection was demonstrated in the CA1 and CA3 areas of the hippocampus and in the hilus of the DG after long-term treatment with losartan. Recently, we demonstrated that long-term treatment with losartan during epileptogenesis exerted strong neuroprotection in the CA1 area of the hippocampus in rats [25]. Interestingly, long-term ICV infusion of Ang II after SE also reduced the kainate-induced damage in the CA1 area of the hippocampus in rats [32]. We can speculate that this specific neuroprotection mainly in the CA1 area of the hippocampus exerted by either losartan or Ang II treatments during epileptogenesis could be attributed to the response of the endogenous Ang II as a result of AT2 receptor subtype activation. While the latter is known to be negligible in physiological conditions, it was upregulated both in a rat model of epilepsy [20] and in patients with epilepsy [19].
3 Clinical Findings of Brain RAS and Epilepsy
Clinical findings regarding the activity of RAS components in human epilepsy are summarized in Table 2. Argañaraz et al. [19] originally reported that the expression and synthesis of AT1 receptors are increased in the cortex and hippocampus of patients with TLE. These authors also observed elevated levels of AT2 receptors in the hippocampus of epileptic patients, but without an increase in the messenger RNA (mRNA), proposing involvement of both receptor subtypes in the pathophysiology of TLE. The anticonvulsant drug carbamazepine was found to inhibit ACE in TLE patients, confirming the relationship between epilepsy and RAS [80].
A recent study revealed that losartan failed to suppress human epileptiform activity in brain slices from patients with pharmacoresistant TLE [81]. A possible reason for this lack of efficacy was attributed to the mechanisms involved in seizure generation used in the in vitro experimental approach, which could not reflect the real situation under in vivo conditions. This issue warrants the need for further detailed clinical investigations, which could allow us to better understand the link between epilepsy and RAS.
4 Co-Morbid Hypertension and Epilepsy
Few studies consider the bidirectional relationship between hypertension and epilepsy. Hypertension predisposes to lower seizure threshold and is thereby is a high-risk factor that can trigger epileptogenesis and development of epilepsy [82, 83]. In support of this presumption are our results showing that the kainate-induced seizure activity and neurotoxicity are more severe in hypertensive conditions [84]. A similar electroencephalogram (EEG) profile was observed between rats with congenital hypertension and kainate-treated normotensive rats [85].
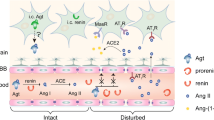
The revised investigations identified a common profile of some abnormalities related to epilepsy and hypertension, as shown in Fig. 1.

Abnormalities related to epilepsy and hypertension. RAS renin–angiotensin system
4.1 Overexpression
Like in normotensive rats, losartan alleviated seizure frequency both during the treatment period and after its discontinuation in SHRs [84]. In general, the AT1 and AT2 receptors were missing or expressed in small amounts in limbic structures in mammals [51], but under pathological conditions overexpression of components of RAS was observed in experimental animals and in patients with either epilepsy or hypertension [19, 86]. In agreement with these authors we reported that compared with normotensive Wistar rats, the AT1 receptors in the hippocampus of SHRs were overexpressed not only under epileptic conditions but also in naïve rats [22] (Table 1). Losartan exerted stronger and more structure-dependent suppression of the upregulated AT1 receptors in epileptic SHRs than in normotensive rats.
4.2 Blood–Brain Barrier Dysfunction and Vascular Damage
An extreme increase of the arterial blood pressure resulted in blood–brain barrier (BBB) dysfunction [87,88,89]. On the other hand, seizures predisposed to BBB damage, which caused extravasation of plasma constituents and vasogenic brain edema [88, 90]. Thus, impairment of the BBB might initiate epileptogenesis through direct neuronal depolarization or excessive leakage [90]. Previous reports demonstrated that blood pressure suppression [90, 91] during convulsions enabled the BBB disruption to be avoided. Alongside this, repeated losartan treatment was effective in the prevention of BBB permeability in hypertensive rats [92, 93], suggesting that this drug might be protective against barrier leakage in epileptic conditions, as well as in epilepsy accompanied with hypertension. Still, losartan was shown to decrease central sympathetic nerve activity in hypertensive rats [94].
Other investigations postulated that disregarded hypertension, a cerebrovascular risk factor, could indirectly elicit and accelerate the development of epilepsy. Russo et al. [48] found that in the amygdala kindling model of TLE, SHRs with hypertension-related cerebral small vessel disease attained Racine’s stage 5 quicker than controls. Another study reported that vascular damage predisposed to epilepsy and mainly affected the frontal and central lobes in stroke patients, while leukoaraiosis was mostly distributed among patients with TLE [83, 95, 96]. However, at present, the risk factors for leukoaraiosis remain unclear.
4.3 Neuroinflammation and Glial Sensitization
Both epilepsy and hypertension promoted neuroinflammation, including astrocytes and microglial activation leading to the release of inflammatory cytokines and chemokines [23, 24, 97, 98]. The elevated Ang II levels, observed in epileptic and hypertensive conditions, altered migration, differentiation, and proliferation of neuronal and glial cells and caused dysregulation of the neuronal–microglial signaling and the production of proinflammatory factors [24, 97]. Positive astrocytes were detected in two hypertensive strains: SHRs and deoxycorticosterone acetate-salt-treated rats [98]. Like epileptic rats, naïve SHRs were characterized by increased levels of inflammatory interleukins and decreased levels of anti-inflammatory cytokines [97]. While chronic inflammation promoted the development of epilepsy and essential hypertension, RAS inhibitors could diminish these conditions, illustrating an anti-inflammatory activity [23, 24, 97].
4.4 Oxidative Stress
Hyperactive RAS and AT1 receptor-mediated Ang II responses, in particular, included increased release of inflammatory and oxidant substances such as leukotrienes, free radicals, C-reactive protein and prostaglandins, and thus initiated oxidative stress with a subsequent disturbance in the antioxidant defense system [99,100,101]. Hypertension associated with excessive release of free radicals could be a risk factor for the development of epileptogenic foci. Like hypertension, seizure and epilepsy also lead to excitotoxicity and generation of reactive oxygen/nitrogen species and oxidative stress [84, 102, 103]. In 2014, we tested subchronic losartan infusion before SE in the kainate model of TLE, which prevented oxidative stress and neurotoxicity in normotensive Wistar rats and SHRs [84]. Losartan attenuated the kainate-induced increase in lipid peroxidation both in SHRs and Wistar rats and produced higher expression of heat shock protein 72 in the hippocampus. In agreement with our previous data and those of other researchers, SHRs were characterized by a disturbed oxidative defense system compared with normotensive rats in physiological conditions [11, 101]. Previously, Haugen et al. [104] reported that hyperactivity of RAS triggered oxidative stress in SHRs. In this regard, several studies supported the suggestion that Ang II-dependent generation of superoxide and oxygen species presents a key mechanism of cardiac and renal impairment, secondary to diverse pathologies [105,106,107]. Yet, some of the beneficial effects associated with RAS inhibition can be linked to the prevention of oxidant-mediated damage [108]. Experimental studies evidenced that antagonists of AT1 receptors function as free radical-scavenging antioxidants in SHRs. Thus, losartan treatment, at the same dose as in our studies (10 mg/kg/day in drinking water for a period of 14 days), increased superoxide dismutase (SOD) activity and glutathione peroxidase to protect brain, kidney, and liver in SHRs, but did not change these parameters in normotensive WKY rats [101]. We found that losartan pretreatment reduced the kainate-induced adaptive increase in the cytosolic SOD activity in the frontal cortex of both normotensive Wistar rats and SHRs [84]. In the latter, the antioxidant effect was also detected in the hippocampus, which could result from the differences in the antioxidant buffering capacities of both strains in the above-mentioned brain regions. Interestingly, we did not detect any changes in the mitochondrial SOD activity in kainate-induced limbic seizures, which suggests that the cytosolic defense system is more sensitive to the acute kainate seizure model than the mitochondrial antioxidant enzyme system in the two strains. Our results support the presumption of an indirect effect of the AT1 receptor antagonist on the antioxidant system, since data on a direct interaction between RAS inhibition and ROS are still lacking.
4.5 Circadian Rhythm
Findings in animal models and patients with a family history of hypertension revealed a positive correlation between Ang II production in the CNS and the circadian rhythm of arterial pressure [109, 110]. Disturbed diurnal rhythms of the cerebral and systemic circulation were observed in epileptic patients [111].
4.6 Hippocampal Neuropathology and Neuronal Loss
In addition to the neuropathological changes described in Sect. 2.4, further literature findings suggested that the close relationship between hypertension and epilepsy involved hippocampal neuropathology and neuronal loss [25, 26, 32, 83, 98, 112,113,114]. Patients with hypertensive encephalopathy were characterized by hippocampal sclerosis, which led to the development of TLE [83]. A reduced number of neurons was observed in the hilus of the DG of SHRs and in hypertensive rats treated with deoxycorticosterone [98]. Neuronal damage is a known feature in experimental [35, 114] and human epilepsy [5, 6]. Our results showed that continuous AT1 receptor blocking was effective against neuronal loss not only in normotensive rats with epilepsy, but also exerted neuroprotection mostly in the CA3 area of the hippocampus and the septo-temporal hilus of the DG in epileptic SHRs [25, 26, 32].
4.7 Neurochemical Changes
Neurochemical changes in hypertension and epilepsy diseases have been established [3, 112, 115]. Administration of Ang II could influence the levels of central neurotransmitters in rats [116]. Like in epileptic Wistar rats, concentrations of the monoamines serotonin, tryptophan, and dopamine were decreased in the hippocampus of naïve and epileptic SHRs [3]. While epilepsy is associated with noradrenergic, serotonergic, and GABAergic deficits [117], elevated levels of extracellular neurotransmitters in the hippocampus were identified during seizure activity in humans and in animal seizure models, as an adaptive response to reducing seizures [115]. Recently, we reported that the kainate-evoked changes of hippocampal monoamine release distinguished between SHRs and normotensive rats [51]. The kainate-induced increase of the hippocampal monoamine levels in SHRs and WKY rats was abolished by the AT1 receptor antagonist ZD7155, reflecting the association of the monoamines and RAS in the mechanisms of epilepsy and hypertension.
4.8 Behavioral Changes
Data from the literature showed that both hypertension and epilepsy have various behavioral disturbances (as discussed early, including in Sect. 2.3), and most of them were positively affected by RAS inhibition.
All these data imply that the burden of co-morbidity with epilepsy is high. Inhibitors of AT1 receptors were proven to be effective not only in the treatment of cardiovascular and metabolic disorders plus hypertension and diabetes mellitus, but also in neurodegenerative disorders such as epilepsy [10, 20, 110]. Moreover, the combination of antihypertensives and AEDs was reported to be a relatively safe strategy for treatment of epilepsy associated with hypertension [43,44,45,46,47]. We also showed that blocking of the AT1 receptors was more pronounced in a model of epilepsy with co-morbid hypertension [22, 84].
5 Conclusion and Future Perspectives
Generally, experimental and clinical data confirmed the involvement of the brain RAS in the pathophysiology of epilepsy and its accompanying complications. The alteration of the function and expression of RAS components in many brain structures involved in seizure susceptibility have been crucial for the development of epileptogenesis and its consequences both in animal models of epilepsy and in patients with mesial TLE [19,20,21,22, 32], including epilepsy with co-morbid hypertension [26, 84]. The RAS inhibitors have been shown to possess anticonvulsant, anti-inflammatory, antioxidant, behavioral, and neuroprotective properties in an epileptic state [20, 23,24,25,26, 59, 84]. The use of AEDs has been shown only to suppress the symptoms but not to influence epilepsy and its concomitant deleterious changes, which warrants the need for developing new therapeutic approaches [118, 119]. Besides, most patients using AEDs are at risk of severe and long-term adverse effects and drug interactions, and develop pharmacoresistance at a later stage, reducing their quality of life [120, 121]. The excessive release of Ang II under pathological conditions exerted proconvulsant properties and aggravated the development of a chronic epileptic phase and its concomitant behavioral changes, including hyperlocomotion and depression, but also elicited neuroprotection [32], which suggests a fine-tuning modulatory mechanism through activation of the AT1 or AT2 receptors that might be considered potential drug targets. Thus, RAS inhibitors, commonly used as a strategy for prevention of high blood pressure, alone or in combination with AEDs, have been shown to be effective in the treatment of other neurological disorders, as well as epilepsy and its concomitant alterations, either by direct a decrease in blood pressure or indirect reduction of free radicals and/or proinflammatory cytokines and modulation of the neuronal and vascular circuit [20, 23,24,25,26, 43, 44, 46, 47, 59, 84]. Moreover, RAS inhibition, in particular as a result of blockade of the AT1 receptors, was more pronounced in a model of epilepsy with co-morbid hypertension [22, 26, 84].
These studies suggest that the ACE inhibitors and blockade of the AT1 receptors may be useful to reduce the detrimental consequences resulting from epilepsy, as an adjunctive treatment in epilepsy with co-morbid hypertension. Yet, these findings highlight the importance of experimental and clinical studies focused on the inhibition of central RAS components to better understand the pathological mechanism of epilepsy under hypertensive conditions.
References
Quigg M, Straume M, Smith T, Menaker M, Bertram EH. Seizures induce phase shifts of rat circadian rhythms. Brain Res. 2001;913:165–9. https://doi.org/10.1016/S0006-8993(01)02780-9.
Shin HW, Davis R. Review of levetiracetam as a first line treatment in status epilepticus in the adult patients—what do we know so far? Front Neurol. 2013;5(4):111. https://doi.org/10.3389/fneur.2013.00111.
Tchekalarova J, Pechlivanova D, Atanasova T, Markova P, Lozanov V, Stoynev A. Diurnal variations in depression-like behavior of Wistar and spontaneously hypertensive rats in the kainate model of temporal lobe epilepsy. Epilepsy Behav. 2011;20:277–85. https://doi.org/10.1016/j.yebeh.2010.12.021.
Watanabe M, Forsgren L, Tomson T, Mathern GW, Glynn M, Engel J, et al. ILAE official report: a practical clinical definition of epilepsy. Brain Res. 2014;55:475–82. https://doi.org/10.1111/epi.12550.
Sloviter RS. Apoptosis: a guide for the perplexed. Trends Pharmacol Sci. 2002;23:19–24. https://doi.org/10.1016/S0165-6147(00)01867-8.
Dobolyi A, Kékesi KA, Juhász G, Székely AD, Lovas G, Kovács Z. Receptors of peptides as therapeutic targets in epilepsy research. Curr Med Chem. 2014;21:764–87.
Kanner AM, Balabanov A. Valproate: a practical review of its uses in neurological and psychiatric disorders. Expert Rev Neurother. 2002;2:151–65. https://doi.org/10.1586/14737175.2.2.151.
Gaitatzis A, Carroll K, Majeed A, Sander JW. The epidemiology of the comorbidity of epilepsy in the general population. Brain Res. 2004;45:1613–22. https://doi.org/10.1111/j.0013-9580.2004.17504.x.
Lacey CJ, Salzberg MR, Roberts H, Trauer T, D’Souza WJ. Psychiatric comorbidity and impact on health service utilization in a community sample of patients with epilepsy. Brain Res. 2009;50:1991–4. https://doi.org/10.1111/j.1528-1167.2009.02165.x.
Kovac S, Walker MC. Neuropeptides in epilepsy. Neuropeptides. 2013;47:467–75. https://doi.org/10.1016/j.npep.2013.10.015.
Atanasova M, Petkova Z, Pechlivanova D, Dragomirova P, Blazhev A, Tchekalarova J. Strain-dependent effects of long-term treatment with melatonin on kainic acid-induced status epilepticus, oxidative stress and the expression of heat shock proteins. Pharmacol Biochem Behav. 2013;111:44–50. https://doi.org/10.1016/j.pbb.2013.08.006.
Wright JW, Harding JW. Brain renin-angiotensin—a new look at an old system. Prog Neurobiol. 2011;95:49–67. https://doi.org/10.1016/j.pneurobio.2011.07.001.
Wright JW, Harding JW. The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch. 2013;465:133–51. https://doi.org/10.1007/s00424-012-1102-2.
De Bundel D, Smolders I, Vanderheyden P, Michotte Y. Ang II and Ang IV: unraveling the mechanism of action on synaptic plasticity, memory, and epilepsy. CNS Neurosci Ther. 2008;14:315–39. https://doi.org/10.1111/j.1755-5949.2008.00057.x.
Stragier B, De Bundel D, Sarre S, Smolders I, Vauquelin G, Dupont A, et al. Involvement of insulin-regulated aminopeptidase in the effects of the renin–angiotensin fragment angiotensin IV: a review. Heart Fail Rev. 2008;13:321–37. https://doi.org/10.1007/s10741-007-9062-x.
Dzau VJ, Bernstein K, Celermajer D, Cohen J, Dahlöf B, Deanfield J, et al. The relevance of tissue angiotensin-converting enzyme: manifestations in mechanistic and endpoint data. Am J Cardiol. 2001;88:1–20. https://doi.org/10.1016/S0002-9149(01)01878-1.
Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88.
Carey RM. Update on the role of the AT2 receptor. Curr Opin Nephrol Hypertens. 2005;14:67–71. https://doi.org/10.1097/00041552-200501000-00011.
Argañaraz GA, Konno AC, Perosa SR, Santiago JFC, Boim MA, Vidotti DB, et al. The renin-angiotensin system is upregulated in the cortex and hippocampus of patients with temporal lobe epilepsy related to mesial temporal sclerosis. Brain Res. 2008;49:1348–57. https://doi.org/10.1111/j.1528-1167.2008.01581.x.
Pereira MGAG, Becari C, Oliveira JAC, Salgado MCO, Garcia-Cairasco N, Costa-Neto CM. Inhibition of the renin–angiotensin system prevents seizures in a rat model of epilepsy. Clin Sci. 2010;119:477–82. https://doi.org/10.1042/CS20100053.
Gouveia TLF, Frangiotti MIB, De Brito JMV, De Castro Neto EF, Sakata MM, Febba AC, et al. The levels of renin-angiotensin related components are modified in the hippocampus of rats submitted to pilocarpine model of epilepsy. Neurochem Int. 2012;61:54–62. https://doi.org/10.1016/j.neuint.2012.04.012.
Atanasova D, Tchekalarova J, Ivanova N, Nenchovska Z, Pavlova E, Atanassova N, et al. Losartan suppresses the kainate-induced changes of angiotensin AT1receptor expression in a model of comorbid hypertension and epilepsy. Life Sci. 2018;193:40–6. https://doi.org/10.1016/j.lfs.2017.12.006.
Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014;75:864–75. https://doi.org/10.1002/ana.24147.
Sun H, Wu HQ, Yu X, Zhang GL, Zhang R, Zhan SQ, et al. Angiotensin II and its receptor in activated microglia enhanced neuronal loss and cognitive impairment following pilocarpine-induced status epilepticus. Mol Cell Neurosci. 2015;65:58–67. https://doi.org/10.1016/j.mcn.2015.02.014.
Tchekalarova JD, Ivanova NM, Pechlivanova DM, Atanasova D, Lazarov N, Kortenska L, et al. Antiepileptogenic and neuroprotective effects of losartan in kainate model of temporal lobe epilepsy. Pharmacol Biochem Behav. 2014;127:27–36. https://doi.org/10.1016/j.pbb.2014.10.005.
Tchekalarova JD, Ivanova N, Atanasova D, Pechlivanova DM, Lazarov N, Kortenska L, et al. Long-term treatment with losartan attenuates seizure activity and neuronal damage without affecting behavioral changes in a model of co-morbid hypertension and epilepsy. Cell Mol Neurobiol. 2016;36:927–41. https://doi.org/10.1007/s10571-015-0278-3.
Tchekalarova J, Kambourova T, Georgiev V. Long-term theophylline treatment changes the effects of angiotensin II and adenosinergic agents on the seizure threshold. Brain Res Bull. 2000;52:13–6. https://doi.org/10.1016/S0361-9230(99)00254-3.
Tchekalarova J, Georgiev V. Ang II and Ang III modulate PTZ seizure threshold in non-stressed and stressed mice: possible involvement of noradrenergic mechanism. Neuropeptides. 2006;40:339–48. https://doi.org/10.1016/j.npep.2006.07.005.
Tchekalarova J, Georgiev V. Adenosine-angiotensin II interactions in pentylenetetrazol seizure threshold in mice. J Physiol Paris. 1999;93:191–7. https://doi.org/10.1016/S0928-4257(99)80151-X.
Tchekalarova J, Georgiev V. Angiotensin peptides modulatory system: how is it implicated in the control of seizure susceptibility? Life Sci. 2005;76:955–70. https://doi.org/10.1016/j.lfs.2004.10.012.
Tchekalarova J, Kambourova T, Georgiev V. Effects of angiotensin III and angiotensin IV on pentylenetetrazol seizure susceptibility (threshold and kindling): interaction with adenosine A1 receptors. Brain Res Bull. 2001;56:87–91. https://doi.org/10.1016/S0361-9230(01)00568-8.
Ivanova NM, Atanasova D, Pechlivanova DM, Mitreva R, Lazarov N, Stoynev AG, et al. Long-term intracerebroventricular infusion of angiotensin II after kainate-induced status epilepticus: effects on epileptogenesis, brain damage, and diurnal behavioral changes. Epilepsy Behav. 2015;51:1–12. https://doi.org/10.1016/j.yebeh.2015.06.036.
Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol. 1999;408:449–60. https://doi.org/10.1002/(SICI)1096-9861(19990614)408:4%3c449:AID-CNE1%3e3.0.CO;2-R.
Tsunashima K, Schwarzer C, Kirchmair E, Sieghart W, Sperk G. GABAA receptor subunits in the rat hippocampus III: altered messenger RNA expression in kainic acid-induced epilepsy. Neuroscience. 1997;80:1019–32. https://doi.org/10.1016/S0306-4522(97)00144-9.
Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–6. https://doi.org/10.1126/science.2879352.
Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–22. http://www.ncbi.nlm.nih.gov/pubmed/3981241.
Klein P, Dingledine R, Aronica E, Bernard C, Blümcke I, Boison D, et al. Commonalities in epileptogenic processes from different acute brain insults: do they translate? Brain Res. 2018;59:37–66. https://doi.org/10.1111/epi.13965.
Albrecht D, Broser M, Krüger H. Excitatory action of angiotensins II and IV on hippocampal neuronal activity in urethane anesthetized rats. Regul Pept. 1997;70:105–9. https://doi.org/10.1016/S0167-0115(97)00015-3.
Haas HL, Felix D, Celio MR, Inagami T. Angiotensin II in the hippocampus. A histochemical and electrophysiological study. Experientia. 1980;36:1394–5. https://doi.org/10.1007/BF01960117.
Latchford KJ, Ferguson AV. ANG II-induced excitation of paraventricular nucleus magnocellular neurons: a role for glutamate interneurons. Am J Physiol Regul Integr Comp Physiol. 2004;286:R894–902. https://doi.org/10.1152/ajpregu.00603.2003.
Albrecht D, Nitschke T, Von Bohlen und Halbach O. Various effects of angiotensin II on amygdaloid neuronal activity in normotensive control and hypertensive transgenic [TGR(mREN-2)27] rats. FASEB J. 2000;14:925–31. https://doi.org/10.1096/fasebj.14.7.925.
Łukawski K, Janowska A, Jakubus T, Tochman-Gawda A, Czuczwar SJ. Angiotensin AT1 receptor antagonists enhance the anticonvulsant action of valproate in the mouse model of maximal electroshock. Eur J Pharmacol. 2010;640:172–7. https://doi.org/10.1016/j.ejphar.2010.04.053.
Łukawski K, Janowska A, Jakubus T, Raszewski G, Czuczwar SJ. Combined treatment with gabapentin and drugs affecting the renin-angiotensin system against electroconvulsions in mice. Eur J Pharmacol. 2013. https://doi.org/10.1016/j.ejphar.2013.02.054.
Łukawski K, Janowska A, Jakubus T, Czuczwar SJ. Interactions between angiotensin AT1 receptor antagonists and second-generation antiepileptic drugs in the test of maximal electroshock. Fundam Clin Pharmacol. 2014;28:277–83. https://doi.org/10.1111/fcp.12023.
De Sarro G, Di Paola ED, Gratteri S, Gareri P, Rispoli V, Siniscalchi A, et al. Fosinopril and zofenopril, two angiotensin-converting enzyme (ACE) inhibitors, potentiate the anticonvulsant activity of antiepileptic drugs against audiogenic seizures in DBA/2 mice. Pharmacol Res. 2012;65:285–96. https://doi.org/10.1016/j.phrs.2011.11.005.
Łukawski K, Raszewski G, Czuczwar SJ. Effect of aliskiren, a direct renin inhibitor, on the protective action of antiepileptic drugs against pentylenetetrazole-induced clonic seizures in mice. Fundam Clin Pharmacol. 2019;33:191–8. https://doi.org/10.1111/fcp.12421.
Łukawski K, Raszewski G, Czuczwar SJ. Interactions of aliskiren, a direct renin inhibitor, with antiepileptic drugs in the test of maximal electroshock in mice. Eur J Pharmacol. 2018;819:108–13. https://doi.org/10.1016/j.ejphar.2017.11.037.
Russo E, Leo A, Scicchitano F, Donato A, Ferlazzo E, Gasparini S, et al. Cerebral small vessel disease predisposes to temporal lobe epilepsy in spontaneously hypertensive rats. Brain Res Bull. 2017;130:245–50. https://doi.org/10.1016/j.brainresbull.2017.02.003.
Pechlivanova DM, Stoynev AG, Tchekalarova JD. The effects of chronic losartan pretreatment on restraint stress-induced changes in motor activity, nociception and pentylenetetrazol generalized seizures in rats. Folia Med. 2011;53:69–73. https://doi.org/10.2478/v10153-010-0040-z.
Pushpa VH. Evaluation and comparison of anticonvulsant activity of t elmisartan and olmesartan in experimentally induced animal models of epilepsy. J Clin Diagn Res. 2014;8:8–12. https://doi.org/10.7860/jcdr/2014/9455.5061.
Tchekalarova J, Loyens E, Smolders I. Effects of AT1 receptor antagonism on kainate-induced seizures and concomitant changes in hippocampal extracellular noradrenaline, serotonin, and dopamine levels in Wistar-Kyoto and spontaneously hypertensive rats. Epilepsy Behav. 2015;46:66–71. https://doi.org/10.1016/j.yebeh.2015.03.021.
Wright JW, Yamamoto BJ, Harding JW. Angiotensin receptor subtype mediated physiologies and behaviors: new discoveries and clinical targets. Prog Neurobiol. 2008;84:157–81. https://doi.org/10.1016/j.pneurobio.2007.10.009.
Jöhren J, Saavedra O. Expression messenger of AT 1 ~ and ATlB angiotensin RNA in forebrain of 2-week-old 11 receptor rats. Am J Physiol. 1996;271:E104–12. https://doi.org/10.1152/ajpendo.1996.271.1.E104.
Chen QH, Toney GM. Responses to GABA-A receptor blockade in the hypothalamic PVN are attenuated by local AT 1 receptor antagonism. Am J Physiol Regul Integr Comp Physiol. 2003;85:R1231–9. https://doi.org/10.1152/ajpregu.00028.2003.
Unger T, Bles F, Ganten D, Lang RE, Rettig R, Schwab NA. Gabaergic stimulation inhibits central actions of angiotensin II: pressor responses, drinking and release of vasopressin. Eur J Pharmacol. 1983;90:1–9. https://doi.org/10.1016/0014-2999(83)90207-8.
Brandt C, Gastens AM, Sun M, Hausknecht M, Löscher W. Treatment with valproate after status epilepticus: effect on neuronal damage, epileptogenesis, and behavioral alterations in rats. Neuropharmacology. 2006;51:789–804. https://doi.org/10.1016/j.neuropharm.2006.05.021.
Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium–pilocarpine model in adult rats. Brain Res. 2005;46:499–508. https://doi.org/10.1111/j.0013-9580.2005.38704.x.
Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Brain Res. 1993;34:420–32. https://doi.org/10.1111/j.1528-1157.1993.tb02582.x.
Villapol S, Yaszemski AK, Logan TT, Sánchez-Lemus E, Saavedra JM, Symes AJ. Candesartan, an angiotensin II at 1-receptor blocker and PPAR-γ agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacology. 2012;37:2817–29. https://doi.org/10.1038/npp.2012.152.
Nayak V, Patil PA. Antidepressant activity of fosinopril, ramipril and losartan, but not of lisinopril in depressive paradigms of albino rats and mice. Ind J Exp Biol. 2008;46:180–4.
Saavedra JM, Ando H, Armando I, Baiardi G, Bregonzio C, Juorio A, et al. Anti-stress and anti-anxiety effects of centrally acting angiotensin II AT1 receptor antagonists. Regul Pept. 2005;128:227–38. https://doi.org/10.1016/j.regpep.2004.12.015.
Saavedra JM, Benicky J, Zhou J. Angiotensin II: multitasking in the brain. J Hypertens. 2006;24:131–7. https://doi.org/10.1097/01.hjh.0000220418.09021.ee.
Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36:1–18. https://doi.org/10.1016/j.psyneuen.2010.10.001.
de los Marinzalda MA, Bregonzio C, Baiardi G, Casarsa BS, Gargiulo PA, Pérez PA. Fear-potentiated behaviour is modulated by central amygdala angiotensin II AT1 receptors stimulation. Biomed Res Int. 2014;2014:183248. https://doi.org/10.1155/2014/183248.
Giardina WJ, Ebert DM. Positive effects of captopril in the behavioral despair swim test. Biol Psychiatry. 1989;25:697–702. https://doi.org/10.1016/0006-3223(89)90240-0.
Ayyub M, Najmi AK, Akhtar M. Protective effect of irbesartan an angiotensin (AT 1) receptor antagonist in unpredictable chronic mild stress induced depression in mice. Drug Res (Stuttg). 2017;67:59–64. https://doi.org/10.1055/s-0042-118172.
Ping G, Qian W, Song G, Zhaochun S. Valsartan reverses depressive/anxiety-like behavior and induces hippocampal neurogenesis and expression of BDNF protein in unpredictable chronic mild stress mice. Pharmacol Biochem Behav. 2014;124:5–12. https://doi.org/10.1016/j.pbb.2014.05.006.
Martin P, Massol J, Puech AJ. Captopril as an antidepressant? Effects on the learned helplessness paradigm in rats. Biol Psychiatry. 1990;27:968–74. https://doi.org/10.1016/0006-3223(90)90034-Y.
Aswar U, Chepurwar S, Shintre S, Aswar M. Telmisartan attenuates diabetes induced depression in rats. Pharmacol Rep. 2017;69:358–64. https://doi.org/10.1016/j.pharep.2016.12.004.
Chalas A, Conway EL. No evidence for involvement of angiotensin II in spatial learning in water maze in rats. Behav Brain Res. 1996;81:199–205. https://doi.org/10.1016/S0166-4328(96)00062-9.
Tota S, Goel R, Pachauri SD, Najmi AK, Hanif K, Nath C. Effect of angiotensin II on spatial memory, cerebral blood flow, cholinergic neurotransmission, and brain derived neurotrophic factor in rats. Psychopharmacology. 2013;226:357–69. https://doi.org/10.1007/s00213-012-2913-8.
Mogi M, Iwanami J, Horiuchi M. Roles of brain angiotensin II in cognitive function and dementia. Int J Hypertens. 2012;2012:169649. https://doi.org/10.1155/2012/169649.
Ivanova N, Tchekalarova J, Atanasova D, Pechlivanova D, Lazarov N. Strain-dependent effects of AT1 receptor antagonist losartan on spatial memory performance of wistar and spontaneously hypertensive rats in kainate model of temporal epilepsy. Epilepsy Behav. 2018;71:839–46. https://doi.org/10.7546/CRABS.2018.06.15.
Herrero AI, Sandi C, Venero C. Individual differences in anxiety trait are related to spatial learning abilities and hippocampal expression of mineralocorticoid receptors. Neurobiol Learn Mem. 2006;86:150–9. https://doi.org/10.1016/j.nlm.2006.02.001.
Schmidt R, Payer F, Niederkorn K, Offenbacher H, Horner S, Blematl B, et al. Magnetic resonance imaging white matter lesions and cognitive impairment in hypertensive individuals. Arch Neurol. 1991;48:417–20. https://doi.org/10.1001/archneur.1991.00530160087019.
Wyss JM, Kadish I, Van Groen T. Age-related decline in spatial learning and memory: attenuation by captopril. Clin Exp Hypertens. 2003;7:455–74. https://doi.org/10.1081/CEH-120024988.
Grammatopoulos TN, Ahmadi F, Jones SM, Fariss MW, Weyhenmeyer JA, Zawada WM. Angiotensin II protects cultured midbrain dopaminergic neurons against rotenone-induced cell death. Brain Res. 2005;1045:64–71. https://doi.org/10.1016/j.brainres.2005.03.038.
Wilms H, Rosenstiel P, Unger T, Deuschl G, Lucius R. Neuroprotection with angiotensin receptor antagonists: a review of the evidence and potential mechanisms. Am J Cardiovasc Drugs. 2005;5:245–53. https://doi.org/10.2165/00129784-200505040-00004.
Armstrong DL, Garcia EA, Ma T, Quinones B, Wayner MJ. Angiotensin II blockade of long-term potentiation at the perforant path-granule cell synapse in vitro. Peptides. 1996;17:689–93. https://doi.org/10.1016/0196-9781(96)00030-7.
Almeida SS, Naffah-Mazzacoratti MG, Guimarães PB, Wasinski F, Pereira FE, Canzian M, et al. Carbamazepine inhibits angiotensin I-converting enzyme, linking it to the pathogenesis of temporal lobe epilepsy. Transl Psychiatry. 2012;2:93. https://doi.org/10.1038/tp.2012.21.
Reyes-Garcia SZ, Scorza CA, Ortiz-Villatoro NN, Cavalheiro EA. Losartan fails to suppress epileptiform activity in brain slices from resected tissues of patients with drug resistant epilepsy. J Neurol Sci. 2019;397:169–71. https://doi.org/10.1016/j.jns.2019.01.008.
Delanty NVC. Seizures, hypertension, and posterior leukoencephalopathy. New York: Springer Science; 2002. p. 251–60.
Gasparini S, Ferlazzo E, Sueri C, Cianci V, Ascoli M, Cavalli SM, Epilepsy Study Group of the Italian Neurological Society. Hypertension, seizures, and epilepsy: a review on pathophysiology and management. Neurol Sci. 2019;40(9):1775–83. https://doi.org/10.1007/s10072-019-03913-4.
Tchekalarova J, Ivanova N, Pechlivanova D, Ilieva K, Atanasova M. Strain-dependent effects of sub-chronically infused losartan against kainic acid-induced seizures, oxidative stress, and heat shock protein 72 expression. Cell Mol Neurobiol. 2014;34:133–42. https://doi.org/10.1007/s10571-013-9994-8.
Vorobyov V, Schibaev N, Kaptsov V, Kovalev G, Sengpiel F. Cortical and hippocampal EEG effects of neurotransmitter agonists in spontaneously hypertensive vs. kainate-treated rats. Brain Res. 2011;1383:154–68. https://doi.org/10.1016/j.brainres.2011.01.107.
Romero-Nava R, Rodriguez JE, Reséndiz-Albor AA, Sánchez-Munõz F, Ruiz-Hernandéz A, Huang F, et al. Changes in protein and gene expression of angiotensin II receptors (AT1 and AT2) in aorta of diabetic and hypertensive rats. Clin Exp Hypertens. 2016;38:56–62. https://doi.org/10.3109/10641963.2015.1060984.
Johansson B. Indomethacin and cerebrovascular permeability to albumin in acute hypertension and cerebral embolism in the rat. Exp Brain Res. 1980;42:331–6.
Ndode-Ekane XE, Hayward N, Gröhn O, Pitkänen A. Vascular changes in epilepsy: functional consequences and association with network plasticity in pilocarpine-induced experimental epilepsy. Neuroscience. 2010;166:312–32. https://doi.org/10.1016/j.neuroscience.2009.12.002.
Cornford EM, Oldendorf W. Epilepsy and the blood–brain barrier. Adv Neurol. 1986;44:787–812.
Gorter JA, Van Vliet EA, Aronica E. Status epilepticus, blood–brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav. 2015;49:13–6. https://doi.org/10.1016/j.yebeh.2015.04.047.
Oztaş B, Kaya M. The effect of acute hypertension on blood–brain barrier permeability to albumin during experimentally induced epileptic seizures. Pharmacol Res. 1991;23(1):41–6. https://doi.org/10.1016/s1043-6618(05)80104-5.
Kucuk M, Kaya M, Kalayci R, Cimen V, Kudat H, Arican N, et al. Effects of losartan on the blood–brain barrier permeability in long-term nitric oxide blockade-induced hypertensive rats. Life Sci. 2002;71:937–46. https://doi.org/10.1016/S0024-3205(02)01772-1.
Kaya M, Kalayci R, Küçük M, Arican N, Elmas I, Kudat H, et al. Effect of losartan on the blood–brain barrier permeability in diabetic hypertensive rats. Life Sci. 2003;73:3235–44. https://doi.org/10.1016/j.lfs.2003.06.014.
Ye S, Zhong H, Duong VN, Campese VM. Losartan reduces central and peripheral sympathetic nerve activity in a rat model of neurogenic hypertension. Hypertension. 2002;39:1101–6. https://doi.org/10.1161/01.HYP.0000018590.26853.C7.
Gasparini S, Ferlazzo E, Beghi E, Sofia V, Mumoli L, Labate A. Epilepsy associated with leukoaraiosis mainly affects temporal lobe: a casual or causal relationship? Epilepsy Res. 2015;109:1–8. https://doi.org/10.1016/j.eplepsyres.2014.10.012.
Ferlazzo E, Gasparini S, Beghi E, Sueri C, Russo E, Leo A. Epilepsy in cerebrovascular diseases: review of experimental and clinical data with meta-analysis of risk factors. Epilepsia. 2016;57(8):1205–14. https://doi.org/10.1111/epi.13448.
Haspula D, Clark MA. Neuroinflammation and sympathetic overactivity: mechanisms and implications in hypertension. Auton Neurosci. 2018;210:10–7. https://doi.org/10.1016/j.autneu.2018.01.002.
Pietranera L, Saravia F, Gonzalez Deniselle MC, Roig P, Lima A, De Nicola AF. Abnormalities of the hippocampus are similar in deoxycorticosterone acetate-salt hypertensive rats and spontaneously hypertensive rats. J Neuroendocrinol. 2006;18:466–74. https://doi.org/10.1111/j.1365-2826.2006.01436.x.
Van Den Buuse M. Circadian rhythms of blood pressure, heart rate, and locomotor activity in spontaneously hypertensive rats as measured with radio-telemetry. Physiol Behav. 1994;55:783–7. https://doi.org/10.1016/0031-9384(94)90060-4.
Das UN. Angiotensin-II behaves as an endogenous pro-inflammatory molecule. J Assoc Physicians India. 2005;53:472–6.
Polizio AH, Peña C. Effects of angiotensin II type 1 receptor blockade on the oxidative stress in spontaneously hypertensive rat tissues. Regul Pept. 2005;128:1–5. https://doi.org/10.1016/j.regpep.2004.12.004.
Ambrosio G, Tritto I, Chiariello M. The role of oxygen free radicals in preconditioning. J Mol Cell Cardiol. 1995;27:1035–9. https://doi.org/10.1016/0022-2828(95)90072-1.
Gupta YK, Briyal S. Protective effect of vineatrol against kainic acid induced seizures, oxidative stress and on the expression of heat shock proteins in rats. Eur Neuropsychopharmacol. 2006;16:85–91. https://doi.org/10.1016/j.euroneuro.2005.07.004.
Haugen EN, Croatt AJ, Nath KA. Angiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int. 2000;58:144–52. https://doi.org/10.1046/j.1523-1755.2000.00150.x.
Zhang H, Schmeißer A, Garlichs CD, Plötze K, Damme U, Mügge A, et al. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells. Role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res. 1999;44:215–22. https://doi.org/10.1016/S0008-6363(99)00183-2.
Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase–derived radicals. Circ Res. 2004;95:1019–26. https://doi.org/10.1161/01.RES.0000148637.85595.c5.
Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–6. https://doi.org/10.1161/01.RES.0000135483.12297.e4.
De Cavanagh EMV, Piotrkowski B, Fraga CG. Concerted action of the renin-angiotensin system, mitochondria, and antioxidant defenses in aging. Mol Asp Med. 2004;25:27–36. https://doi.org/10.1016/j.mam.2004.02.006.
Stoynev AG, Ikonomov OC, Minkova NK, Zacharieva SZ, Stoyanovsky VG. Circadian rhythms of arterial pressure: basic regulatory mechanisms and clinical value. Acta Physiol Pharmacol Bulg. 1999;24:43–51.
Campos LA, Bader M, Baltatu OC. Brain renin-angiotensin system in hypertension, cardiac hypertrophy, and heart failure. Front Physiol. 2012;2:115. https://doi.org/10.3389/fphys.2011.00115.
Beĭn G, Shakhotin VN, Muromtseva VM. Diurnal rhythm of the cerebral and systemic circulation in epileptic patients [in Russian]. Zh Nevropatol Psikhiatr Im S S Korsakova. 1978;78:561.
Greenwood RS, Meeker R, Sullivan H, Hayward JN. Kindling in spontaneous hypertensive rats. Brain Res. 1989;495:58–65. https://doi.org/10.1016/0006-8993(89)91217-1.
Hernandez CM, Høifødt H, Terry AV. Spontaneously hypertensive rats: further evaluation of age-related memory performance and cholinergic marker expression. J Psychiatry Neurosci. 2003;28:197–209.
Scorza FA, Arida RM, Cysneiros RM, Scorza CA, de Albuquerque M, Cavalheiro EA. Qualitative study of hippocampal formation in hypertensive rats with epilepsy [in Portuguese]. Arq Neuropsiquiatr. 2005;63:283–8.
Meurs A, Clinckersb R, Ebinger G, Michotte Y, Smolders I. Seizure activity and changes in hippocampal extracellular glutamate, GABA, dopamine and serotonin. Epilepsy Res. 2008;78(1):50–9. https://doi.org/10.1016/j.eplepsyres.2007.10.007.
Mendelsohn FA, Jenkins TA, Berkovic SF. Effects of angiotensin II on dopamine and serotonin turnover in the striatum of conscious rats. Brain Res. 1993;613(2):221–9. https://doi.org/10.1016/0006-8993(93)90902-y.
Jobe PC. Common pathogenic mechanisms between depression and epilepsy: an experimental perspective. Epilepsy Behav. 2003;4(Suppl 3):S14–24.
Litt B, Esteller R, Echauz J, D’Alessandro M, Shor R, Henry T, et al. Epileptic seizures may begin hours in advance of clinical onset: a report of five patients. Neuron. 2001;30:51–64. https://doi.org/10.1016/S0896-6273(01)00262-8.
McKeown MJ, McNamara JO. When do epileptic seizures really begin? Neuron. 2001;30:1–3. https://doi.org/10.1016/S0896-6273(01)00253-7.
Arroyo S, de la Morena A. Life-threatening adverse events of antiepileptic drugs. Epilepsy Res. 2001;47:155–74. https://doi.org/10.1016/S0920-1211(01)00306-0.
Berg AT, Langfitt J, Shinnar S, Vickrey BG, Sperling MR, Walczak T, et al. How long does it take for partial epilepsy to become intractable? Neurology. 2003;60:186–90. https://doi.org/10.1212/01.WNL.0000031792.89992.EC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No financial support was received for the publication of this review.
Conflict of interest
Natasha Ivanova and Jana Tchekalarova declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Ivanova, N., Tchekalarova, J. The Potential Therapeutic Capacity of Inhibiting the Brain Renin–Angiotensin System in the Treatment of Co-Morbid Conditions in Epilepsy. CNS Drugs 33, 1101–1112 (2019). https://doi.org/10.1007/s40263-019-00678-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00678-4