
Abstract
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease, which affects millions of people worldwide. The disease is characterized by chronically elevated blood glucose concentrations (hyperglycaemia), which result in comorbidities and multi-organ dysfunction. This is due to a gradual loss of glycaemic control as a result of increasing insulin resistance, as well as decreasing β-cell function. The objective of T2DM drug interventions is, therefore, to reduce fasting and postprandial blood glucose concentrations to normal, healthy levels without hypoglycaemia. Several classes of novel antihyperglycaemic drugs with various mechanisms of action have been developed over the past decades or are currently under clinical development. The development of these drugs is routinely supported by the application of pharmacokinetic/pharmacodynamic modelling and simulation approaches. They integrate information on the drug’s pharmacokinetics, clinically relevant biomarker information and disease progression into a single, unifying approach, which can be used to inform clinical study design, dose selection and drug labelling. The objective of this review is to provide a comprehensive overview of the quantitative approaches that have been reported since the 2008 review by Landersdorfer and Jusko in an increasing order of complexity, starting with glucose homeostasis models. Each of the presented approaches is discussed with respect to its strengths and limitations, and respective knowledge gaps are highlighted as potential opportunities for future drug–disease model development in the area of T2DM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The dynamic interplay between biological system, disease and drug effects, both beneficial and harmful, is usually complex, and its assessment requires the concerted use of experimental and model-based analysis techniques. These techniques can be used to evaluate the cause–effect relationship at different levels of organizational complexity and range from observational and descriptive (pharmacometric) to completely mechanistic (systems pharmacology) approaches via the use of biomarkers. |
Several quantitative modelling approaches have been developed since the 2008 review in Clinical Pharmacokinetics to assess the efficacy and safety profiles of novel antihyperglycaemic drugs. These models vary in their level of complexity, depending on the stage of drug development. Prior scientific knowledge, pathophysiological information and disease-specific predictive biomarkers are used to develop and qualify these models. |
There is no one-size-fits-all model. All drug–disease models should be fit for purpose, i.e. developed and qualified to provide answers to the question(s) of interest. |
Tailoring drug–disease models to long-term treatment outcomes and special populations, and accounting for genetic and non-genetic covariates, can further enhance their predictive performance for quantitative decision making and personalized medicine applications in therapy for type 2 diabetes mellitus. |
1 Introduction
The objective of pharmacokinetic/pharmacodynamic modelling and simulation approaches is to characterize and predict the effects of drugs in living organisms under physiological and pathophysiological conditions [1–3]. Pharmacokinetic/pharmacodynamic approaches have evolved in recent years from a descriptive, empirical discipline to a mechanistic science, which has increasingly been employed in all phases of drug development as the theoretical basis for (1) the selection of drug candidates; (2) lead optimization; (3) the optimization of early proof-of-concept clinical trials; (4) dose optimization; and (5) optimization of clinical trial design [1, 4–6]. Respective models can be established at different levels of spatial and temporal complexity, ranging from observational and descriptive (pharmacometric; drug-centric models) to completely mechanistic approaches (systems pharmacology; network-centric models) but should always be fit for purpose, i.e. provide answers to the question(s) of interest. They should also be developed with a well-characterized knowledge of basic physiology and basic biochemistry in order to sufficiently depict reality. As a consequence, mechanism-based or physiology-directed (enhanced pharmacokinetic/pharmacodynamic; pathway-centric) models have been gaining popularity in recent years. They differ substantially from descriptive models, as they contain specific expressions that characterize—in a strictly qualitative and quantitative fashion—processes along the causal pathway between drug administration and effect [2]. However, further advancement of the discipline is necessary to better inform drug development and regulatory decision making. The predictive performance of pharmacokinetic/pharmacodynamic models is reliant upon biomarker response analysis. A biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [7]. As a consequence, they can be used to study the dynamics of a disease system, to compare the effect of new and existing treatments, to develop new drug candidates and to inform regulatory decision making.
Drug–disease models developed for diabetes mellitus use a plethora of quantitatively predictive and clinically relevant biomarkers. The respective biomarker panel is not limited to the conventional biomarkers fasting glucose (FG), fasting serum insulin (FSI) and glycosylated haemoglobin (HbA1c) as the approvable endpoint; rather, it includes a whole variety of pharmacodynamic measurements, which are used to assess target engagement, glycaemic control, β-cell function and insulin sensitivity, as well as the impact of disease progression and different therapeutic interventions. The choice of biomarker depends on the specific research question, the drug’s mechanism of action and the duration of the trial, as well as the stage of the drug development program [8].
Landersdorfer and Jusko [9] summarized frequently employed type 2 diabetes mellitus (T2DM) modelling and simulation approaches in their review, published in 2008, which encompassed models for diagnostic purposes, glucose homeostasis models, ancillary biomarker models and drug–disease models. The objective of our review is to provide a comprehensive update on key drug–disease models developed for T2DM, with a particular focus on models published since 2008 (Table 1). To that end, we first discuss models that characterize glucose homeostasis at the glucose–insulin level before step-wise expanding the model scope using the underlying (patho)physiology. A respective literature search was performed in PubMed and primarily considered articles in the English language, published after 2008. A list of the drug classes with their proposed mechanisms of action and relevant biomarkers is shown in Table 2.
2 Glucose Homeostasis Models
T2DM is characterized by chronically elevated glucose concentrations (hyperglycaemia), which can result in the dysfunction of multiple organs over time [10]. In healthy subjects, the body maintains tight blood glucose control via the complementary effects of two hormones, insulin and glucagon, which interact via negative feedback loops [11, 12]. While an increase in insulin concentrations results in a decrease in glucose concentrations, an increase in glucagon concentrations results in an increase in glucose concentrations. Both hormones are produced in the pancreas: glucagon in α cells and insulin in β cells [13]. As a consequence, the functional activity of the pancreas plays a critical role in glycaemic control.
de Winter et al. [14] published a semi-mechanistic model to describe longitudinal changes in fasting plasma glucose (FPG), FSI and HbA1c in T2DM patients, using three turnover models: one each for FPG, FSI and HbA1c. These biomarkers are not independent of each other but are connected via feedback and feed-forward loops, i.e. (1) a rise in glucose concentrations gives rise to insulin secretion; (2) a rise in insulin concentrations results in a decrease in glucose concentrations; and (3) a sustained increase in glucose concentrations results in an increase in HbA1c concentrations. The model also accounted for disease progression through the use of two empirical monotonic functions, each characterizing the decrease in β-cell function and insulin sensitivity. In addition, the treatment effect was incorporated as step functions (EFB = treatment effect on β-cell function, EFS = treatment effect on insulin sensitivity). While the model by de Winter et al. [14] provides a physiology-directed framework for characterizing changes in biomarkers from baseline as a function of disease progression and three drug treatments (pioglitazone, metformin and gliclazide), it faces several limitations. The main limitations are that (1) drug input was estimated as a step function of time rather than via a dose–concentration–response model, as different dose levels of these drugs were used in clinical trials; (2) the monotonic disease progression functions that were employed are strictly empirical and may not accurately reflect the dynamics of the underlying disease processes; (3) the first-order elimination rate constant (k out) for FSI was set at 1, which is unlikely to accurately reflect the biomarker’s half-life and thus the system’s dynamics; and (4) the direct linear relationship between fast biomarker FPG and slow biomarker HbA1c is unlikely, since this relationship is based on the glycosylation of haemoglobin to HbA1c which is non-linear in nature. The model by de Winter et al. [14] was consequently expanded by several investigators to address these limitations by accounting for processes that drive the dynamics of the biomarkers.
For example, Choy et al. [15] expanded the de Winter model [14] by characterizing changes in HbA1c over time through the use of three transit compartments, which account for the delay in changes in HbA1c concentrations from baseline relative to those in FPG concentrations. Body weight is recognized to have an inverse relationship with insulin sensitivity and thereby affects disease progression, which was accounted for by a separate turnover model for body weight. The impact of diet and exercise was characterized as an inhibitory effect on the generation of body weight, and respective changes in body weight were used as a predictor of EFS. Other investigators took the glycosylation pattern of red blood cells (RBCs), i.e. the mechanistic basis for HbA1c formation, into stronger consideration. An earlier mechanistic model for the production of HbA1c proportional to plasma glucose concentrations was developed by Benincosa and Jusko [9, 16]. Later publications added more detail, such as differences in RBC aging. For example, Lledo-Garcia et al. [17] published an integrated glucose–RBC–HbA1c (IGRH) model, which quantitatively describes RBC aging. In addition, they included reduction in RBC lifespan with increasing glucose concentrations as an empirical power function in the model. Information on glycosylation rate constants and the mean RBC lifespan, as well as the mean lifespan of RBC precursor cells, was obtained from the literature and used as priors to compute respective average glucose (AG) concentrations. The authors assumed for their analysis that glycosylation is ongoing throughout the lifespan of RBCs at a rate that is proportional to AG and non-glycosylated haemoglobin concentrations. Their findings indicated that HbA1c is formed faster in diabetic individuals than in healthy subjects, which is due to higher AG concentrations and thus a higher glycosylation rate in diabetics (Fig. 1). The altered RBC dynamics between diabetic and healthy individuals are reflected by the fact that the corresponding mean age of RBCs was lower in patients (49 days) than in healthy subjects (59 days). It should also be noted that this inverse relationship between RBC lifespan and AG lacks a mechanistic basis and thus will face limitations with respect to their predictive performance beyond the studied AG range. The RBC lifespan model by Lledo-Garcia et al. [17] is conceptually similar to the model developed by Hamren et al. [18]. Both models differ in the number of transit compartments used (Lledo-Garcia et al.: 12; Hamren et al.: 4) but concur in that the relationship between glucose (FPG, AG) and HbA1c is nonlinear and that the fraction of glycosylated RBCs increases as they become older. It should be noted, though, that the number of transit compartments does not correspond to the number of cells at different stages of maturation. The transit compartments describe RBC aging and represent different age stages; each compartment will have a distribution of RBCs of different ages, which transition to the next one until the cell dies in the last compartment, giving the number of compartments the shape of the distribution of lifespans for an RBC cohort in an individual. At any given age stage, the cell can become glycosylated, resulting in a delay in cause and effect—in this case, between changes in FPG and changes in HbA1c.

Fractions of a red blood cell (RBC) cohort remaining in circulation at different times following their production and the glycosylated haemoglobin (HbA1c) formed, for a diabetic (diab.) patient (average glucose concentration 373 mg/dL) and a non-diabetic (Non-diab.) subject (average glucose concentration 85 mg/dL), based on simulation from the final model. HbA1c concentrations at steady state (HbA1css) are also illustrated. (Figure and legend modified from Lledo-Garcia et al. [17])
In addition to the RBC lifespan, these models can also be expanded on the basis of mechanisms involved in disease progression. While, for example, the de Winter model [14] assumed a monotonic decline in both β-cell function and insulin sensitivity, other investigators, such as Ribbing et al. [19], used more mechanistic approaches to characterize disease impact. In this example, Ribbing et al. incorporated information on FPG, FSI, insulin sensitivity and clinical measures of β-cell mass (BCM) into a unifying model to characterize changes in these biomarkers at various stages of disease. These interindividual differences in disease state contribute, at least in part, to the substantial interindividual variability in biomarker response to therapeutic intervention(s). These interindividual differences were handled in the Ribbing et al. model [19] by allowing for subpopulations of non-diabetic, insulin-resistant and long-term-treated T2DM patients. The impact of the disease state on β-cell adaptation, i.e. the ability of β cells to increase insulin output to maintain glycaemic control, was implemented as an offset in β-cell adaptation (OFFSET), which leads to a higher set point for FPG in T2DM patients, as shown in the following equation:
where d 0 is the glucose-independent rate of β-cell apoptosis; R 1 is the rate of change in β-cell replication as a function of glucose concentrations; FPG′ it is the offset-corrected FPG for the ith individual at time t; and R 2 is the glucose toxicity to β cells, i.e. the extent of β-cell dysfunction due to chronic exposure to high glucose concentrations.
The relationship between insulin clearance and insulin sensitivity (S) was incorporated according to:
where S it and k it are the insulin sensitivity and insulin elimination rate constants, respectively, in the ith individual at time t; S H and k H are the values for the typical healthy subject; and REL k–S is the nonlinear relation between S and k.
The developed model was then applied to data on tesaglitazar, a dual peroxisome proliferator–activated receptor (PPAR)-α/γ agonist, using clinical phase 2 and 3 data. Tesaglitazar increases insulin sensitivity in liver, fat and skeletal muscle cells, as well as peripheral glucose uptake, and decreases hepatic glucose output. The effect of tesaglitazar was incorporated into the model as an indirect stimulatory effect on insulin sensitivity and on β-cell adaptation. Given that only FPG and fasting insulin (FI) values were collected, it was assumed that (1) FPG and FI were at steady state at the subject’s first visit; and that (2) BCM and S it were in equilibrium. The authors also assumed that β-cell function per unit of BCM is the same in T2DM patients and healthy individuals in the fasted state. They also did not attempt to quantify disease progression, because of the short duration of the study (6 months). Another limitation of the model is the generalized assumption that the only difference between T2DM patients and normal subjects is an offset in β-cell adaptation, lower insulin sensitivity and lower insulin clearance. The employed hyperbolic function for BCM also indicates that healthy subjects, who have glucose concentrations below the physiological set point value, would experience a decline in BCM, which is of limited physiological meaningfulness. Nevertheless, the incorporation of β-cell dynamics is an important aspect of the model to enhance understanding of the interaction between the drug, disease and biological system.
In the models presented so far, β-cell function or BCM have been used as surrogates for disease progression. However, the pancreas itself is also a target for therapeutic interventions and/or drug effects. For example, G-protein-coupled receptor (GPR) agonists engage the upstream receptor present mainly on pancreatic β cells to increase insulin secretion, whereas glucokinase activators (GKAs) influence the glucokinase enzyme, which plays a role in regulating glucose concentrations [20].
2.1 G-Protein-Coupled Receptor Agonists
GPRs are highly expressed on pancreatic β cells and have been shown to play an important role in insulin secretion. There are a number of different GPRs, of which GPR40 and GPR119 have served as druggable targets.
GPR40 (free fatty acid receptor-1) downstream signalling by coupling to the Gaq/11 subunit of G-proteins results in increased intracellular calcium concentrations and phospholipase activation, which in turn results in increased insulin secretion [21, 22]. TAK-875, a GPR40 agonist drug candidate, was developed as an adjunct to diet and exercise to improve glycaemic control in T2DM patients. Naik et al. [23] published a pharmacokinetic/pharmacodynamic model for TAK-875 to guide dose selection for clinical trials. The pharmacokinetic model consisted of a two-compartment model with an oral absorption compartment. Indirect response (IDR) models were utilized for the turnover of glucose and HbA1c, wherein the rate of HbA1c production was directly linked to changes in glucose concentrations. Systemic drug concentrations were used to characterize pharmacodynamics, using a maximum effect (E max) model. The drug effect was modelled as a stimulatory effect on glucose utilization. Given that the drug primarily affects insulin secretion, an expansion of the model in order to account for insulin dynamics and food effects may have been worthwhile. The model may also face limitations with predicting long-term treatment effects, because it does not characterize the underlying disease status and progression in terms of β-cell deterioration or decreasing insulin sensitivity. Baseline FPG (BFPG) and aspartate aminotransferase (AST) were found to be influential covariates for the pharmacological response. E max was found to increase in an exponential manner with observed BFPG but was directly proportional to the AST concentrations. Subsequently, development of this drug was voluntarily discontinued by the sponsor because of hepatic safety concerns.
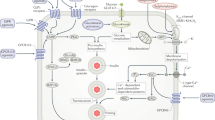
GPR119, a second G-protein-coupled β-cell receptor, mediates glucose-dependent insulin secretion via increasing intracellular cyclic adenosine monophosphate (cAMP) concentrations. It also results in increased secretion of glucagon-like peptide (GLP)-1 and gastric inhibitory peptide (GIP) in the gut [24]. Thus there is an increasing interest in developing therapeutic candidates that can take advantage of the insulin-secretory property of GPR119. The pharmacodynamic effect of a potential GPR119 agonist to induce insulin secretion can be evaluated by using IDR models for the various biomarkers involved in the process. In this regard, Tess et al. [25] presented a systems pharmacology model to describe the impact of modelling on GPR119 agonist drug development (Fig. 2). In the model, the effects of incretins, insulin, glucose and glucagon were linked via cascading turnover models with physiologically based feedback loops. GPR119 agonist activity was modelled in two parts: (1) stimulation of incretin, using a sigmoidal E max model; and (2) an agonistic effect, along with incretins, on the production of insulin. The increase in insulin secretion was subsequently linked to inhibition of gluconeogenesis and glucagon secretion. This is a representative example of how semi-mechanistic modelling approaches could be extended to include ancillary/interim biomarkers, as well as food effects, in the modelling strategy.

Pfizer–Rosa physiological model of diabetes. The model represents the physiology, pathophysiology and pharmacology relevant to G-protein-coupled receptor (GPR) 119 agonism. The model comprises multiple interconnected modules, which describe (1) glucose distribution, storage and utilization; (2) insulin secretion, distribution and elimination; (3) incretin secretion, distribution and elimination; (4) glucagon secretion, distribution and elimination; (5) nutrient absorption (e.g. meals) and glucose perturbations (e.g. oral glucose tolerance testing); and (6) pharmacokinetics and pharmacodynamics of relevant antidiabetic therapeutics (e.g. sitagliptin, exenatide, metformin and glyburide). (Figure and legend modified from Tess et al. [25])
2.2 Glucokinase Activators
Glucokinase, a glucose-phosphorylating enzyme, is another drug target of interest for developing antihyperglycaemic medicines, because of its pronounced impact on glucose homeostasis, which is the result of its glucose-sensing role on β cells. Signals obtained from this sensor mediate hepatic glucose clearance and glycogen synthesis. This control mechanism seems to be impaired in T2DM [26], which makes glucokinases an interesting drug target. Schneck et al. [27] and Zhang et al. [28] extended a previously published integrated glucose–insulin (IGI) model in order to characterize glucagon dynamics [29–31] and applied a mechanism-based integrated glucose–insulin–glucagon (GIG) model in order to prospectively select optimal oral doses and dosing regimens for a small-molecule GKA candidate (LY2599506) for phase 2 studies based on phase 1 data. The GIG model incorporated the feedback mechanisms between glucose, insulin and glucagon, which were based on drug pharmacokinetics, glucose, insulin, glucagon and meal data, obtained from multiple-ascending-dose studies in T2DM patients for up to 26 days. The drug effect was modelled as a stimulating effect on insulin secretion, as well as an inhibitory effect on hepatic glucose production (HGP). In addition, predicted glucose concentrations from the GIG model were used as a feed-forward input in a previously published RBC lifespan model [18] to predict HbA1c concentrations. The authors performed clinical trial simulations to test the optimal doses and dosing regimen to maximize the HbA1c-lowering effect with minimal hypoglycaemia risk. Subsequently, they confirmed the predictive performance of the model with actual clinical trial data, which demonstrates the predictive value of models that consider key hormones (insulin and glucagon) responsible for glucose homeostasis and hypoglycaemia.
2.3 Glucagon Receptor Antagonists
In addition to the processes described for β cells, pancreatic α cells play an important role in glucose homeostasis, as they secrete glucagon. Glucagon-mediated effects counterbalance insulin-mediated affects because they stimulate HGP via glycogenolysis. As a consequence, dysregulated glucagon secretion can lead to elevated HGP and thus contribute to hyperglycaemia in T2DM patients [32–35]. As a consequence, inhibition of glucagon signalling can reduce HGP and thereby help re-establish glycaemic control in diabetic patients [36]. Lau et al. [37] reported that a monoclonal antibody (mAb) that blocked the glucagon receptor had profound glucose-lowering effects in various animal models. The effect of glucose homeostasis mediated by the antibody was then studied in diabetic mice. A pair of semi-mechanistic turnover models was incorporated to describe the glucose–glucagon interrelationship. Glucagon stimulated the production of glucose via glycogenolysis, whereas a subsequent increase in glucose concentrations provided a negative feedback loop to inhibit glucagon secretion by the pancreas. The hypothesized inhibitory effect of the mAb on the glucagon receptor signalling pathway via competitive inhibition was included as inhibition of the glucagon stimulatory effect on HGP. This model was able to describe the stimulation of glucagon concentrations due to antagonism of the glucagon receptor by the mAb. Increase in glucagon concentrations with glucagon receptor antagonism (GRA), in spite of glucose-lowering effects, have also been observed in clinical trials, which is believed to be caused by blocking the negative feedback loop for glucagon secretion [36]. The dynamic and physiological interplay between insulin and glucagon is a vital component for maintaining glucose homeostasis. Even though the current model characterized the glucagon effect on glucose concentrations, it could be improved by also incorporating insulin effects on glucose production and utilization, as discussed for the GIG model in Sect. 3.2.
3 Models Characterizing Incretin Effects
In addition to the immediate effects of glucose, insulin and glucagon, maintenance of glycaemic control is heavily influenced by food intake, which triggers its own hormonal response in anticipation of rising glucose concentrations after food intake [38]. Incretins are gastrointestinal hormones that are particularly important for this regulatory process. They are released upon food intake and stimulate pancreatic β cells, resulting in the release of insulin in anticipation of rising glucose concentrations following meals. The dynamic interplay between glucose, insulin and incretins has been characterized by various investigators in order to appropriately capture the impact of food on glucose homeostasis. For example, Kjellsson et al. [39] combined the previously published IGI model for meal tolerance tests by Jauslin et al. [29] and the IGRH model (described in Sect. 2). The IGI model comprises system-, study- and drug-specific parameters, which provides the flexibility of using the IGI model for different studies and drugs. It characterized the dynamic interplay between glucose and insulin over 24 h following an oral glucose dose (input) considered equivalent to the carbohydrate content per meal. The incretin effect was modelled as a positive effect on insulin secretion. Circadian effects on variations in glucose production, insulin secretion, insulin sensitivity and insulin elimination were also taken into consideration during model development. Output from the IGI model (AG concentrations; C g,av) was used as input for the IGRH model. Parameters of the IGI model and drug effects were estimated using phase 1 data on a GKA. AG concentrations were simulated according to a phase 2 study design and used in the IGRH model for prediction of HbA1c concentrations. The performance of the approach was assessed by comparing the model‐based prediction with the actual outcome of the phase 2 study. The modelling approach was able to predict the longitudinal HbA1c response in a 12‐week study using only data from a 1‐week study where glucose and insulin concentrations were measured. However, Kjellsson et al. [39] demonstrated the predictive performance of a regulatively acceptable late-stage endpoint (HbA1c), using the short-term biomarkers glucose and insulin. Moreover, the dynamics of these three biomarkers have different timescales. Use of previously published models in combination with new data consequently represents a useful approach for establishing multiscale link models that integrate different aspects of glucose–insulin–HbA1c dynamics in order to evaluate long(er)-term drug effects on the basis of short-term biomarker data. However, it is important to note that models that use short-term (e.g. 12-week) data may face limitations with predicting long-term (e.g. 52-week) changes in HbA1c, as the impact of disease progression is not considered sufficiently. Insufficient information on and/or consideration of the T2DM trajectory may consequently result in an overly optimistic prediction of antiglycaemic effects. It should also be noted that there are different incretins, which serve both as feedback to the glucose–insulin system and as therapeutic targets at the same time. GLP-1 and GIP are two incretins that bind to specific GPRs, leading to an increase in intracellular cAMP concentrations and subsequent release of insulin from pancreatic β cells [38].
3.1 Incretin Mimetics or Glucagon-Like Peptide-1 Agonists
Incretin mimetics, also referred to as GLP-1 agonists, are a class of antihyperglycaemic drugs that mimic the effect of incretins on insulin secretion. Exenatide is an example of a subcutaneously administered GLP-1 agonist that is less susceptible to degradation by dipeptidyl peptidase (DPP)-4 enzymes than endogenous incretins. It was found to have a longer half-life, as well as enhanced potency, in comparison with endogenous GLP-1. It was reported that exenatide does not exhibit linear kinetics in preclinical species, seemingly because of secondary elimination pathways (e.g. target-mediated endocytosis) in addition to glomerular filtration, its main elimination route. The impact of this nonlinear clearance pathway on the elimination of exenatide was modelled by Chen et al. [40], using a target-mediated drug disposition (TMDD) model that was informed by preclinical data. Corresponding changes in glucose concentrations were described by a turnover model, and following drug–receptor complex formation, the drug effect was incorporated as stimulation of glucose elimination (Fig. 3a). Once developed and qualified, the preclinical model was used to predict human exenatide pharmacokinetics by using a body weight-based allometric scaling approach using three preclinical species (mouse, rat and monkey) [41]. It was found that the absorption kinetics following subcutaneous administration were not adequately described using a single first-order rate constant. Therefore, a Michaelis–Menten function using the maximum absorption rate (V max) and substrate amount of drug at the absorption site when the reaction rate was half of V max (K m) was used to characterize drug absorption. The scaled preclinical model was able to predict human exenatide pharmacokinetics following intravenous and subcutaneous administration reasonably well. It should be noted that while the majority of model parameters were similar and shared across species, the receptor density and respective binding affinities differed. It was observed that human pharmacokinetics following intravenous administration were best described by using all shared and monkey-specific parameter values for receptor density and binding affinity. However, human pharmacokinetic profiles following subcutaneous administration were best predicted by using all of the shared and rat-specific V max and K m parameter values. It would provide added insight if one were to use the predicted human exenatide pharmacokinetic information from the TMDD interspecies scaling approach as the pharmacokinetic input for a clinical pharmacodynamic model (e.g. Cirincione et al. [42]) to subsequently determine the clinical pharmacodynamic response. This would provide a seamless quantitative workflow from the preclinical setting to the clinical setting. Such a framework could also be employed for other drugs that exhibit target-mediated disposition.

a Pharmacokinetic–pharmacodynamic model used to characterize exenatide after 2-week treatment in streptozotocin and nicotinamide–induced diabetic rats. \( \mathop \smallint \nolimits_{0}^{t} {\text{RC}} \cdot {\text{d}}t \) represents the area under the drug–receptor complex concentration–time curve. f disease(k out,t) represents the change in the elimination rate constant (K out) over time as a function of the disease state. (Figure and legend modified from Chen et al. [40]). b Pharmacokinetic–pharmacodynamic model for sitagliptin. Dipeptidyl peptidase (DPP)-4 activity is inhibited by sitagliptin. Glucose from a meal is absorbed using transit compartments with a single transfer rate constant, K tr. The zero-order rate constant for the production of active glucagon-like peptide (GLP)-1 (K in) is increased by glucose in the gastrointestinal tract (glucose in the transit compartment), and the first-order rate constant for the loss of active GLP-1 (K out2) is decreased by DPP-4 inhibition. S(t) is the stimulation function, using the amount of glucose in the transit compartment, and l(t) is the inhibition function, using DPP-4 inhibition (Figure and legend modified from Kim et al. [43]). A T amount of free drug in peripheral compartment, C concentration, CL/F oral clearance, K a absorption rate constant, K deg degradation rate constant for free receptor, K el elimination rate constant, K int rate constant of internalization for drug-receptor complex, K m Michaelis–Menten constant, K off first-order dissociation rate constant, K on second-order association rate constant, K out1 first-order rate constant for the loss of plasma active GLP-1 by the non-DDP-4 pathway, K pt first-order distribution rate constant from plasma (central compartment) to peripheral compartment, K syn zero-order synthesis rate constant, K tp first-order distribution rate constant from peripheral compartment to plasma (central compartment), Q/F intercompartmental oral clearance, R free receptor, RC drug-receptor complex, S stimulation index of the drug-effect on glucose utilization, SC subcutaneous, V C volume of drug distribution in the central compartment, V max maximum absorption rate
3.2 Dipeptidyl Peptidase-4 Inhibitors
DPP-4 is an endogenous enzyme that inactivates GLP-1 and GIP, reducing their insulin-secretory effects. DPP-4 inhibitors, also called ‘gliptins’, were developed as a class of novel oral antihyperglycaemic drugs during the early 2000s. Sitagliptin is an example of a widely used oral DPP-4 inhibitor. Kim et al. [43] developed a pharmacokinetic/pharmacodynamic model for sitagliptin in healthy volunteers (Fig. 3b). The pharmacokinetics of sitagliptin were characterized by a two-compartment model with first-order oral drug input. Active GLP-1 concentrations were modelled as a turnover compartment with two first-order elimination rate constants, k out1 and k out2. The drug effect was incorporated into the model as an inhibitory function on k out2, using a sigmoidal E max equation, whereas the effect of food on GLP-1 was considered a stimulatory function on GLP-1 production. It was noted by the authors that active GLP-1 showed biphasic secretions, and because of lack of appropriate biomarkers for the delayed secretion of active GLP-1, the developed model for active GLP-1 could not capture the biphasic behaviour observed in the data. Since the objective of DPP-4 inhibitor therapy is indirect production of insulin, thereby lowering glucose concentrations, it would have been worthwhile to expand this model by linking it to the previously described glucose–insulin–HbA1c model. Such a link between GLP-1 and food effects on insulin was previously described by Landersdorfer et al. [44] in T2DM patients, wherein DPP-4 inhibition by oral vildagliptin was characterized by a TMDD model [45, 46], whereas the GLP-1 effect was modelled as stimulation of insulin-dependent utilization of plasma glucose.
4 Sodium/Glucose Cotransporter 2 Inhibitors
Given that glucose is an essential source of energy, its production, as well as its catalytic and unspecific clearance, is tightly controlled in the body. In healthy individuals, most of the glucose that is filtered via the kidneys is selectively reabsorbed via high-capacity sodium/glucose cotransporter (SGLT) 2, which is located in the proximal tubule. As a consequence, very little glucose is eliminated in the urine. However, if this transporter-mediated glucose reuptake is inhibited, most of the filtered glucose is eliminated via the urine. As such, SGLT2 inhibitors have gained popularity because of this potential benefit, not only for newly diagnosed T2DM patients but also for treating chronic diabetic patients who respond poorly to other antihyperglycaemic drugs that are dependent on the insulin-secretory capacity of β cells.
Canagliflozin was the first-in-class SGLT2 inhibitor to be approved by the US Food and Drug Administration (FDA) for the treatment of hyperglycaemia in patients with T2DM. de Winter et al. [47] presented a population pharmacokinetic/pharmacodynamic analysis of canagliflozin, which links the direct acute effects of the drug on renal glucose reabsorption to long-term glucose control (HbA1c), using 12- to 26-week, phase 2/3 clinical trial data. The steady-state change in HbA1c was modelled by using a relationship between systemic exposure to the drug and the observed reduction in HbA1c as a function of the primary effect of drug exposure on the renal threshold of glucose excretion (RTg):
where ∆HbA1c,ss is the steady-state change in HbA1c (%); ∆RTg,max is the maximal effect of canagliflozin on RTg; k is the scaling parameter; EC50 is the drug concentration producing a half-maximal reduction in RTg; and ∆PBO is the placebo-effect on HbA1c, including the effects of diet and exercise.
In addition, the authors characterized dynamic changes in HbA1c by using a turnover model:
where K in is the rate of haemoglobin glycation; K out is the rate of erythrocyte cell death; and Ef is the net combined treatment effect affecting mean plasma glucose exposure, and thereby haemoglobin glycation, expressed as:
where Efp is the effect of placebo, diet and exercise counselling; Efc is the primary effect of canagliflozin on RTg lowering; and Efg is the additional HbA1c-lowering effect in the gut for a high dose (300 mg).
It was determined that in addition to its main pharmacological action of SGLT2 inhibition, canagliflozin may also exert transient inhibition of intestinal SGLT1-mediated postprandial absorption of glucose. This effect was observed at canagliflozin doses of greater than 200 mg. Therefore, an additional HbA1c-lowering effect in the gut was included (Efg) for a 300 mg dose compared with a 100 mg dose of canagliflozin. In this study, the authors seem to have considered a baseline HbA1c value of 5 % as representative of a healthy normal population. Accordingly, Ef was proportional to the ratio of an individual patient baseline HbA1c value to the patient population baseline HbA1c value, both of which were considered as a change from a healthy HbA1c baseline value of 5 %. This reflected the impact of baseline hyperglycaemia on efficacy commonly observed with diabetes treatments. In addition, the estimated glomerular filtration rate was found to be a significant covariate on the maximal renal effect of canagliflozin, which indicates that the drug may have an altered effect in patients with renal impairment. It is important to note here that although Eq. 5 characterizes the change in the HbA1c value, the formation of HbA1c was not dependent on the concentrations of glucose and haemoglobin, and thus lacked a mechanistic basis. In addition to canagliflozin, several other SGLT2 inhibitors have been approved or are currently under development (Table 1). Published models for these drugs have evaluated other important aspects, such as the impact of hepatic and renal impairment on drug exposure [48]. However, there is a discrepancy between model-predicted and observed glucose reabsorption following administration of some SGLT2 inhibitors, which is possibly due to the presence of compensatory mechanisms of glucose reabsorption upon SGLT2 inhibition [49]. Thus it is important to integrate such compensatory feedback mechanisms in conjunction with other glucose reabsorption pathways in order to have a mechanistic yet physiologically relevant model.
5 Challenges and Future Directions
Many therapeutic interventions are inadvertently associated with adverse drug effects (ADEs). An increase in body weight is a typical example of an ADE in the T2DM arena. Gao and Jusko [50] developed a semi-mechanistic pharmacokinetic/pharmacodynamic model linking rosiglitazone systemic exposure to the homeostatic feedback between glucose and insulin. The drug effect was modelled as inhibition of glucose production, as well as enhancement of insulin sensitivity, thus indirectly stimulating the utilization of glucose. In addition, the model also included weight gain (which is commonly observed for rosiglitazone) as a separate turnover compartment, wherein the drug inhibits weight loss. Thus not only the desired therapeutic effect but also the undesired ADE, i.e. weight gain, were qualitatively and quantitatively characterized in this analysis. This study also outlines the possibility of establishing a drug–disease platform model, which, once established and qualified, can be used to characterize the pharmacological effects of single or combination therapy with drugs that have different mechanisms of action—such as DPP-4 inhibitors, incretin mimetics, GPR40, GPR119, GRAs, GKAs or SGLT2 inhibitors—by incorporating interim and ancillary biomarker data related to disease status and progression, as well as long-term treatment effects. However, given that several antihyperglycaemic drugs—such as DDP4 inhibitors, GLP-1 agonists and SGLT2 inhibitors—have only recently been approved, long-term and postmarketing safety data are frequently still lacking for these drugs. For example, recent reports suggest that the observed cardiovascular benefits of GLP-1 (an indirect result of DDP4 inhibition), based on immediate (short)-term data, may have been overly optimistic [51]. Scirica et al. [52] reported that even though DPP-4 inhibition by saxagliptin did not alter the rate of ischaemic events, the rate of hospitalization for heart failure was increased, according to the large, international SAVOR-TIMI 53 trial. More recently, a literature-based meta-analysis of randomized clinical trials of DDP4-inhibitors (the keywords for the literature search included ‘vildagliptin’, ‘sitagliptin’, ‘saxagliptin’, ‘alogliptin’, ‘linagliptin’ and ‘dutogliptin’) found that the overall risk of acute heart failure was higher in patients treated with DPP-4 inhibitors than in the placebo/active comparator arms [53]. Moreover, the current preferred method of empirical clinical research is hindered by the high development costs and lengthy timeline required to bring a new medicine to the market. Thus, there is a high unmet need to integrate long-term clinical outcomes data into short(er)-term drug–disease models in order to be able to predict desired and undesired long-term treatment outcomes as early as possible in the drug development process. There are already a number of promising approaches available—such as the Archimedes® model [54] (http://archimedesmodel.com/), the UKPDS model [55] (https://www.dtu.ox.ac.uk/outcomesmodel/) and ECHO-T2DM [56]—that attempt to meet this challenge by incorporating treatment-, trial-, health- and pharmacoeconomic-outcomes data into an overarching modeling and simulation framework.
Despite the progress made in the treatment of T2DM, large interindividual differences in responses to therapeutic interventions continue to pose a serious challenge for drug developers, regulatory decision makers and practitioners. While the sources of variability in the pharmacokinetics of anti-hyperglycaemic drugs are typically well characterized at the end of a drug development program, respective sources of variability impacting the pharmacodynamics of a drug, as well as the individual disease trajectory, are in many cases less well understood. However, sufficient understanding of these factors is critically important when attempting to optimally treat a patient suffering from T2DM, because of the chronic, progressive nature of the disease. In particular, a more detailed understanding of interindividual pharmacogenetic differences between patients and how they relate to differences in disease progression and treatment response is seen as an opportunity to optimize T2DM therapy [57]. It also highlights the fact that not all patients within a heterogeneous T2DM population are likely to respond equally well to a given therapeutic intervention (responders versus non-responders), which delineates the need for individualized single and combination therapy.
This challenge seems to be amplified in special patient populations, such as paediatric or geriatric populations, where, for example, enzyme or transporter ontogenies or comorbidities play a critical role. Regulatory agencies around the globe have responded to this challenge by updating their guidances. For example, the US FDA Safety and Innovation Act (FDASIA) 2012 requires sponsors to submit a paediatric drug development plan at the end of phase 2 studies [58], whereas a Paediatric Investigation Plan is required earlier in Europe at the end of phase 1 studies [59, 60]. The establishment of these paediatric development plans is challenging in many cases because of the lack of clinical data and the incomplete understanding of the drug’s pharmacokinetics/pharmacodynamics in children. Yet respective paediatric development programmes are likely to become more important in the T2DM arena because of the emergence of childhood diabetes since the 1990s and the limited treatment options [61]. Insulin and metformin are the only anti-hyperglycaemic medications approved by both the FDA and the European Medicines Agency (EMA) for use in children to date, since glimepiride failed non-inferiority testing when compared with metformin monotherapy [62]. This may be in part due to the fact that paediatric dosing regimens are frequently developed under the assumption that disease progression and response to therapeutic interventions are the same in adults and children. However, this may or may not be universally true for T2DM drugs, and it outlines the need for better understanding of the relevant pathophysiological processes in adults and children. One way of addressing this challenge is the use of physiologically based modelling and simulation platforms—such as SimCYP, GastroPlus™, PKSim® or SimBiology®—which integrate information on the underlying pathophysiology, as well as genetic information on drug-metabolizing enzymes, into a single, unifying approach. Once established and qualified, these physiologically based models can be used to evaluate clinically unstudied scenarios and thus to ‘de-risk’ a drug molecule prior to it reaching the bedside. In isolation, however, physiologically based pharmacokinetic models are of limited meaningfulness and need to be expanded to physiologically based pharmacokinetic/pharmacodynamic models when attempts are made to characterize and predict the impact of disease progression and therapeutic interventions over an extended period of time, particularly in special patient populations.
6 Conclusion
T2DM presents a huge socioeconomic burden and adversely affects patients’ quality and duration of life. Any improvement in alleviating this burden, and perhaps even preventing the onset or delaying the progression of the disease, would undoubtedly be highly beneficial for patients and society. Given the complexity of the underlying physiological processes, a plethora of factors arise that need to be considered when attempts are made to optimally treat a patient. Simultaneous assessment of all of these factors is difficult, if not impossible, to achieve in head-to-head clinical trials, because of the required sample size and cost. Mathematical and statistical modelling and simulation approaches have been gaining popularity in the last two decades for drug development and regulatory decision making, because they allow for the integration of in vitro, animal and clinical data into a single, unifying model, which can be used to characterize and predict the impact of disease progression and therapeutic interventions on clinically relevant biomarkers.
Respective models can be established at various levels of spatial and temporal complexity, ranging from observational and descriptive (pharmacometric; drug-centric) to completely mechanistic (systems pharmacology; network-centric) approaches. Both approaches have advantages and limitations. Pharmacometric models are routinely employed to characterize treatment effects (changes from placebo/baseline), support dose selection and inform clinical trial design. However, they face limitations with respect to their ability to characterize complex, multilevel (disease) processes and the impact of the patient’s disease status on the treatment response. Systems pharmacology models, on the other hand, are currently used to characterize the pertinent pathophysiology (comprising the key pathways or targets of interest), quantitatively integrate relevant biology across systems and explore the impact of (novel) therapeutic interventions on the system. They too face limitations—for example, with respect to their ability to identify and estimate respective model parameters from clinical data—because of the inherent complexity of the model. Quite frequently, systems pharmacology models are not set up to predict long-term clinical outcomes, which hinders their applications in late-stage drug development and regulatory decision making. In addition, their parameter values are frequently derived from the literature or from different experimental settings, which can result in high variability and thus increased uncertainty in systems pharmacology models. These advantages and limitations of pharmacometric and systems pharmacology models have given rise to mechanism-based (enhanced pharmacokinetic/pharmacodynamic; pathway-centric) approaches, which start out simple and become increasingly complex to sufficiently characterize changes in clinically relevant biomarkers as the result of disease progression and therapeutic interventions. Although these enhanced pharmacokinetic/pharmacodynamic models have improved properties with respect to their ability to characterize the dynamic interaction between the drug(s), the biological system and the disease at multiple biomarker levels, they too can face limitations with characterizing multiple pathways that contribute to a clinical condition, as well as capturing the heterogeneity of T2DM. As a consequence, there is no one-size-fits-all model. All drug–disease models should be fit for purpose and hence should be set up to provide answers to the questions of interest, which invariably are related to evaluating and establishing the balance between efficacy and safety. Figure 4 illustrates a paradigm wherein the uncertainty and variability associated with a trial—for example, uncertainty and variability in dose response, trial design and execution, and data analysis—are taken into consideration to set decision criteria and make informed decisions. Moreover, tailoring drug–disease models to long-term treatment outcomes and special populations, and accounting for genetic and non-genetic covariates, can further enhance their predictive performance for quantitative decision making and personalized medicine applications in T2DM therapy.

Schematic of application of modelling and simulation for optimum dose selection for an antihyperglycaemic agent. BMI body mass index, FBG fasting blood glucose, HbA 1c glycosylated haemoglobin. (Figure and legend modified from Garhyan et al. [8])
References
Schmidt S, Post TM, Boroujerdi M, Van Kesteren C, Ploeger B, Della Pasqua OE, et al. Disease progression analysis: towards mechanism-based models. In: Kimko HHC, Peck CC, editors. Clinical trial simulations—applications and trends. New York: Springer; 2010. p. 437–60.
Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G. Mechanism-based pharmacokinetic–pharmacodynamic modeling—a new classification of biomarkers. Pharm Res. 2005;22(9):1432–7. doi:10.1007/s11095-005-5882-3.
Breimer DD, Danhof M. Relevance of the application of pharmacokinetic–pharmacodynamic modelling concepts in drug development: the “wooden shoe” paradigm. Clin Pharmacokinet. 1997;32(4):259–67. doi:10.2165/00003088-199732040-00001.
Danhof M, de Jongh J, De Lange EC, Della Pasqua O, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic–pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol. 2007;47:357–400. doi:10.1146/annurev.pharmtox.47.120505.105154.
Miller R, Ewy W, Corrigan BW, Ouellet D, Hermann D, Kowalski KG, et al. How modeling and simulation have enhanced decision making in new drug development. J Pharmacokinet Pharmacodyn. 2005;32(2):185–97. doi:10.1007/s10928-005-0074-7.
Derendorf H, Lesko LJ, Chaikin P, Colburn WA, Lee P, Miller R, et al. Pharmacokinetic/pharmacodynamic modeling in drug research and development. J Clin Pharmacol. 2000;40(12 Pt 2):1399–418.
Caveney EJ, Cohen OJ. Diabetes and biomarkers. J Diabetes Sci Technol. 2011;5(1):192–7.
Garhyan P, Topp B, Chien JY, Sinha V, Danhof M, Schmidt S. Drug–disease model–based development of therapeutic agents for treatment of diabetes. In: Schmidt S, Derendorf H, editors. Applied pharmacometrics. New York: Springer; 2014. p. 139–59.
Landersdorfer CB, Jusko WJ. Pharmacokinetic/pharmacodynamic modelling in diabetes mellitus. Clin Pharmacokinet. 2008;47(7):417–48. doi:10.2165/00003088-200847070-00001.
Lin Y, Sun Z. Current views on type 2 diabetes. J Endocrinol. 2010;204(1):1–11. doi:10.1677/JOE-09-0260.
Farhy LS, McCall AL. Glucagon—the new ‘insulin’ in the pathophysiology of diabetes. Curr Opin Clin Nutr Metabol Care. 2015;18(4):407–14. doi:10.1097/MCO.0000000000000192.
Osundiji MA, Evans ML. Brain control of insulin and glucagon secretion. Endocrinol Metab Clin North Am. 2013;42(1):1–14. doi:10.1016/j.ecl.2012.11.006.
Quesada I, Tuduri E, Ripoll C, Nadal A. Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199(1):5–19. doi:10.1677/JOE-08-0290.
de Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, et al. A mechanism-based disease progression model for comparison of long-term effects of pioglitazone, metformin and gliclazide on disease processes underlying type 2 diabetes mellitus. J Pharmacokinet Pharmacodyn. 2006;33(3):313–43. doi:10.1007/s10928-006-9008-2.
Choy S, Kjellsson MC, Karlsson MO, de Winter W. Weight-HbA1c-insulin-glucose (WHIG) model for long term disease progression of type 2 diabetes; PAGANZ, Population Approach Group of Australia and New Zealand; 2013. http://www.paganz.org/abstracts/weight-hba1c-insulin-glucose-whig-model-for-long-term-disease-progression-of-type-2-diabetes/.
Benincosa L, Jusko WJ. Novel method of treatment. Geneva: World Intellectual Property Organization; 1999 (Publ. no. W0/2000/027341).
Lledo-Garcia R, Mazer NA, Karlsson MO. A semi-mechanistic model of the relationship between average glucose and HbA1c in healthy and diabetic subjects. J Pharmacokinet Pharmacodyn. 2013;40(2):129–42. doi:10.1007/s10928-012-9289-6.
Hamren B, Bjork E, Sunzel M, Karlsson M. Models for plasma glucose, HbA1c, and hemoglobin interrelationships in patients with type 2 diabetes following tesaglitazar treatment. Clin Pharmacol Ther. 2008;84(2):228–35. doi:10.1038/clpt.2008.2.
Ribbing J, Hamren B, Svensson MK, Karlsson MO. A model for glucose, insulin, and beta-cell dynamics in subjects with insulin resistance and patients with type 2 diabetes. J Clin Pharmacol. 2010;50(8):861–72. doi:10.1177/0091270009349711.
Liu S, Ammirati MJ, Song X, Knafels JD, Zhang J, Greasley SE, et al. Insights into mechanism of glucokinase activation: observation of multiple distinct protein conformations. J Biol Chem. 2012;287(17):13598–610. doi:10.1074/jbc.M111.274126.
Burant CF. Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes. Diabetes Care. 2013;36(Suppl 2):S175–9. doi:10.2337/dcS13-2037.
Ferdaoussi M, Bergeron V, Kebede M, Mancini A, Alquier T, Poitout V. Free fatty acid receptor 1: a new drug target for type 2 diabetes? Can J Diabetes. 2012;36(5):275–80. doi:10.1016/j.jcjd.2012.08.002.
Naik H, Lu J, Cao C, Pfister M, Vakilynejad M, Leifke E. Pharmacometric approaches to guide dose selection of the novel GPR40 agonist TAK-875 in subjects with type 2 diabetes mellitus. CPT Pharmacometr Syst Pharmacol. 2013;2:e22. doi:10.1038/psp.2012.23.
Zhu X, Huang W, Qi H. GPR119 agonists: a novel strategy for type 2 diabetes treatment. In: Oguntibeju OO, editor. Diabetes mellitus—insights and perspectives. Rijeka: InTech; 2013. doi:10.5772/48444.
Tess D, Ghosh A, Chen D, Cornelius P, Maurer T, Baillie R et al. Impact of modeling on GPR119 agonist development; ACoP, American Conference on Pharmacometrics Annual Meeting; 2011. http://www.rosaandco.com/posters/rosaACop2011PfizerPoster.pdf.
Matschinsky FM, Zelent B, Doliba N, Li C, Vanderkooi JM, Naji A, et al. Glucokinase activators for diabetes therapy: may 2010 status report. Diabetes Care. 2011;34(Suppl 2):S236–43. doi:10.2337/dc11-s236.
Schneck KB, Zhang X, Bauer R, Karlsson MO, Sinha VP. Assessment of glycemic response to an oral glucokinase activator in a proof of concept study: application of a semi-mechanistic, integrated glucose–insulin–glucagon model. J Pharmacokinet Pharmacodyn. 2013;40(1):67–80. doi:10.1007/s10928-012-9287-8.
Zhang X, Schneck K, Bue-Valleskey J, Yeo KP, Heathman M, Sinha V. Dose selection using a semi-mechanistic integrated glucose–insulin–glucagon model: designing phase 2 trials for a novel oral glucokinase activator. J Pharmacokinet Pharmacodyn. 2013;40(1):53–65. doi:10.1007/s10928-012-9286-9.
Jauslin PM, Frey N, Karlsson MO. Modeling of 24-hour glucose and insulin profiles of patients with type 2 diabetes. J Clin Pharmacol. 2011;51(2):153–64. doi:10.1177/0091270010362536.
Silber HE, Jauslin PM, Frey N, Gieschke R, Simonsson US, Karlsson MO. An integrated model for glucose and insulin regulation in healthy volunteers and type 2 diabetic patients following intravenous glucose provocations. J Clin Pharmacol. 2007;47(9):1159–71. doi:10.1177/0091270007304457.
Jauslin PM, Silber HE, Frey N, Gieschke R, Simonsson US, Jorga K, et al. An integrated glucose–insulin model to describe oral glucose tolerance test data in type 2 diabetics. J Clin Pharmacol. 2007;47(10):1244–55. doi:10.1177/0091270007302168.
Gastaldelli A, Miyazaki Y, Pettiti M, Buzzigoli E, Mahankali S, Ferrannini E, et al. Separate contribution of diabetes, total fat mass, and fat topography to glucose production, gluconeogenesis, and glycogenolysis. J Clin Endocrinol Metab. 2004;89(8):3914–21. doi:10.1210/jc.2003-031941.
Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, et al. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes. 2000;49(8):1367–73.
Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333(9):550–4. doi:10.1056/NEJM199508313330903.
Ward WK, Best JD, Halter JB, Porte DJ. Prolonged infusion of somatostatin with glucagon replacement increases plasma glucose and glucose turnover in man. J Clin Endocrinol Metab. 1984;58(3):449–53. doi:10.1210/jcem-58-3-449.
Kelly RP, Garhyan P, Raddad E, Fu H, Lim CN, Prince MJ, et al. Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes Metab. 2015;17(4):414–22. doi:10.1111/dom.12446.
Lau YY, Ma P, Gibiansky L, Komorowski R, Wang J, Wang G, et al. Pharmacokinetic and pharmacodynamic modeling of a monoclonal antibody antagonist of glucagon receptor in male ob/ob mice. AAPS J. 2009;11(4):700–9. doi:10.1208/s12248-009-9150-z.
Hayes MR, Mietlicki-Baase EG, Kanoski SE, De Jonghe BC. Incretins and amylin: neuroendocrine communication between the gut, pancreas, and brain in control of food intake and blood glucose. Annu Rev Nutr. 2014;34:237–60. doi:10.1146/annurev-nutr-071812-161201.
Kjellsson MC, Cosson VF, Mazer NA, Frey N, Karlsson MO. A model-based approach to predict longitudinal HbA1c, using early phase glucose data from type 2 diabetes mellitus patients after anti-diabetic treatment. J Clin Pharmacol. 2013;53(6):589–600. doi:10.1002/jcph.86.
Chen T, Kagan L, Mager DE. Population pharmacodynamic modeling of exenatide after 2-week treatment in STZ/NA diabetic rats. J Pharm Sci. 2013;102(10):3844–51. doi:10.1002/jps.23682.
Chen T, Mager DE, Kagan L. Interspecies modeling and prediction of human exenatide pharmacokinetics. Pharm Res. 2013;30(3):751–60. doi:10.1007/s11095-012-0917-z.
Cirincione B, Passareli J, Kothare P, Cummings M, Grasela T, Fineman M. Development of a steady-state exposure–response model for exenatide once weekly; ASCPT, American Society for Clinical Pharmacology and Therapeutics; 2009. http://www.cognigencorp.com/images/uploads/posters/poster95_ASCPT_exenatideQW_2009mar21.pdf.
Kim BH, Kim SE, Kang D, Lim KS, Kim JR, Jang IJ, et al. Pharmacokinetic–pharmacodynamic modelling of biomarker response to sitagliptin in healthy volunteers. Basic Clin Pharmacol Toxicol. 2013;113(2):113–25. doi:10.1111/bcpt.12068.
Landersdorfer CB, He YL, Jusko WJ. Mechanism-based population modelling of the effects of vildagliptin on GLP-1, glucose and insulin in patients with type 2 diabetes. Br J Clin Pharmacol. 2012;73(3):373–90.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
Landersdorfer CB, He YL, Jusko WJ. Mechanism-based population pharmacokinetic modelling in diabetes: vildagliptin as a tight binding inhibitor and substrate of dipeptidyl peptidase IV. Br J Clin Pharmacol. 2012;73(3):391–401.
de Winter W, Polidori D, Hoeben E, Devineni D, Neyens M, Vermeulen A. Population PK/PD analysis linking the direct acute effects of canagliflozin on renal glucose reabsorption to the overall effects of canagliflozin on long-term glucose control using HbA1c as the response marker from clinical studies; PAGE. Abstracts of the Annual Meeting of the Population Approach Group in Europe; ISSN 1871-6032 2013. PAGE 22 (2013) Abstr 2872. http://www.page-meeting.org/?abstract=2872.
van der Walt JS, Hong Y, Zhang L, Pfister M, Boulton DW, Karlsson MO. A nonlinear mixed effects pharmacokinetic model for dapagliflozin and dapagliflozin 3-O-glucuronide in renal or hepatic impairment. CPT Pharmacometr Syst Pharmacol. 2013;2:e42. doi:10.1038/psp.2013.20.
Demin OJ, Yakovleva T, Kolobkov D, Demin O. Analysis of the efficacy of SGLT2 inhibitors using semi-mechanistic model. Front Pharmacol. 2014;5:218. doi:10.3389/fphar.2014.00218.
Gao W, Jusko WJ. Modeling disease progression and rosiglitazone intervention in type 2 diabetic Goto-Kakizaki rats. J Pharmacol Exp Ther. 2012;341(3):617–25. doi:10.1124/jpet.112.192419.
Jose T, Inzucchi SE. Cardiovascular effects of the DPP-4 inhibitors. Diabetes Vasc Dis Res. 2012;9(2):109–16. doi:10.1177/1479164111436236.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–26. doi:10.1056/NEJMoa1307684.
Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and heart failure: a meta-analysis of randomized clinical trials. Nutr Metabol Cardiovasc Dis. 2014;24(7):689–97. doi:10.1016/j.numecd.2014.01.017.
Eddy DM, Schlessinger L. Archimedes: a trial-validated model of diabetes. Diabetes Care. 2003;26(11):3093–101.
Hayes AJ, Leal J, Gray AM, Holman RR, Clarke PM. UKPDS outcomes model 2: a new version of a model to simulate lifetime health outcomes of patients with type 2 diabetes mellitus using data from the 30 year United Kingdom Prospective Diabetes Study: UKPDS 82. Diabetologia. 2013;56(9):1925–33. doi:10.1007/s00125-013-2940-y.
Willis M, Asseburg C, He J. Validation of economic and health outcomes simulation model of type 2 diabetes mellitus (ECHO-T2DM). J Med Econ. 2013;16(8):1007–21. doi:10.3111/13696998.2013.809352.
Pacanowski MA, Hopley CW, Aquilante CL. Interindividual variability in oral antidiabetic drug disposition and response: the role of drug transporter polymorphisms. Expert Opin Drug Metabol Toxicol. 2008;4(5):529–44. doi:10.1517/17425255.4.5.529.
Food and Drug Administration Safety and Innovation Act [S.3187, 112th Congress of the United States of America, 2nd session]; 2012. http://www.gpo.gov/fdsys/pkg/BILLS-112s3187enr/pdf/BILLS-112s3187enr.pdf.
Samant TS, Mangal N, Lukacova V, Schmidt S. Quantitative clinical pharmacology for size and age scaling in pediatric drug development: a systematic review. J Clin Pharmacol. 2015;55(11):1207–17. doi:10.1002/jcph.555.
Zisowsky J, Krause A, Dingemanse J. Drug development for pediatric populations: regulatory aspects. Pharmaceutics. 2010;2(4):364.
Tamborlane WV, Klingensmith G. Crisis in care: limited treatment options for type 2 diabetes in adolescents and youth. Diabetes Care. 2013;36(6):1777–8. doi:10.2337/dc13-0743.
Christensen ML, Franklin BE, Momper JD, Reed MD. Pediatric drug development programs for type 2 diabetes: a review. J Clin Pharmacol. 2015;55(7):731–8. doi:10.1002/jcph.497.
Nesina® [package insert]. Deerfield: Takeda Pharmaceuticals America, Inc.; January 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022271s000lbl.pdf.
Tradjenta® [package insert]. Ridgefield: Boehringer Ingelheim Pharmaceuticals, Inc.; May 2014. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/201280s009lbl.pdf.
Onglyza® [package insert]. Princeton: Bristol-Myers Squibb Company; May 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022350s011lbl.pdf.
Januvia® [package insert]. Whitehouse Station: Merck & Co., Inc.; March 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021995s032lbl.pdf.
DeFronzo RA, Fleck PR, Wilson CA, Mekki Q. Alogliptin Study Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin in patients with type 2 diabetes and inadequate glycemic control: a randomized, double-blind, placebo-controlled study. Diabetes Care. 2008;31(12):2315–7. doi:10.2337/dc08-1035.
Covington P, Christopher R, Davenport M, Fleck P, Mekki QA, Wann ER, et al. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-blind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther. 2008;30(3):499–512. doi:10.1016/j.clinthera.2008.03.004.
Tanzeum® [package insert]. Wilmington: GlaxoSmithKline LLC; April 2014. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125431s000lbl.pdf.
Trulicity® [package insert]. Indianapolis: Eli Lilly and Company; March 2015. http://pi.lilly.com/us/trulicity-uspi.pdf.
Byetta® [package insert]. Wilmington: AstraZeneca Pharmaceuticals LP; February 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021773s040lbl.pdf.
Victoza® [package insert]. Bagsvaerd: Novo Nordisk A/S; March 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022341s023lbl.pdf.
Grunberger G, Chang A, GarciaSoria G, Botros FT, Bsharat R, Milicevic Z. Monotherapy with the once-weekly GLP-1 analogue dulaglutide for 12 weeks in patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomized, double-blind, placebo-controlled study. Diabet Med. 2012;29(10):1260–7. doi:10.1111/j.1464-5491.2012.03745.x.
Rosenstock J, Reusch J, Bush M, Yang F, Stewart M. Albiglutide Study Group. Potential of albiglutide, a long-acting GLP-1 receptor agonist, in type 2 diabetes: a randomized controlled trial exploring weekly, biweekly, and monthly dosing. Diabetes Care. 2009;32(10):1880–6. doi:10.2337/dc09-0366.
Matthews JE, Stewart MW, De Boever EH, Dobbins RL, Hodge RJ, Walker SE, et al. Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in patients with type 2 diabetes. J Clin Endocrinol Metab. 2008;93(12):4810–7. doi:10.1210/jc.2008-1518.
Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379(9824):1403–11. doi:10.1016/S0140-6736(11)61879-5.
Leifke E, Naik H, Wu J, Viswanathan P, Demanno D, Kipnes M, et al. A multiple-ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clin Pharmacol Ther. 2012;92(1):29–39. doi:10.1038/clpt.2012.43.
Nunez DJ, Bush MA, Collins DA, McMullen SL, Gillmor D, Apseloff G, et al. Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: results from two randomized studies. PLoS One. 2014;9(4):e92494. doi:10.1371/journal.pone.0092494.
Ansarullah, Lu Y, Holstein M, DeRuyter B, Rabinovitch A, Guo Z. Stimulating beta-cell regeneration by combining a GPR119 agonist with a DPP-IV inhibitor. PloS One. 2013;8(1):e53345. doi:10.1371/journal.pone.0053345.
Gao J, Tian L, Weng G, O’Brien TD, Luo J, Guo Z. Stimulating beta-cell replication and improving islet graft function by AR231453, a GPR119 agonist. Transpl Proc. 2011;43(9):3217–20. doi:10.1016/j.transproceed.2011.10.021.
Mu J, Qureshi SA, Brady EJ, Muise ES, Candelore MR, Jiang G, et al. Anti-diabetic efficacy and impact on amino acid metabolism of GRA1, a novel small-molecule glucagon receptor antagonist. PLoS One. 2012;7(11):e49572. doi:10.1371/journal.pone.0049572.
Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig. 2015;6(2):124–32. doi:10.1111/jdi.12294.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–6. doi:10.2337/dc11-1200.
Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95(11):5028–36. doi:10.1210/jc.2010-1041.
Invokana® [package insert]. Titusville: Janssen Pharmaceuticals, Inc.; March 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204042s005lbl.pdf.
Farxiga® [package insert]. Wilmington: AstraZeneca Pharmaceuticals LP; March 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/202293s002lbl.pdf.
Jardiance® [package insert]. Ridgefield: Boehringer Ingelheim Pharmaceuticals, Inc.; August 2014. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204629s000lbl.pdf.
Stenlof K, Cefalu WT, Kim KA, Alba M, Usiskin K, Tong C, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15(4):372–82. doi:10.1111/dom.12054.
Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721–8. doi:10.1111/dom.12081.
Veltkamp SA, van Dijk J, Collins C, van Bruijnsvoort M, Kadokura T, Smulders RA. Combination treatment with ipragliflozin and metformin: a randomized, double-blind, placebo-controlled study in patients with type 2 diabetes mellitus. Clin Ther. 2012;34(8):1761–71. doi:10.1016/j.clinthera.2012.06.027.
Rosenstock J, Aggarwal N, Polidori D, Zhao Y, Arbit D, Usiskin K, et al. Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35(6):1232–8. doi:10.2337/dc11-1926.
Bolinder J, Ljunggren O, Kullberg J, Johansson L, Wilding J, Langkilde AM, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97(3):1020–31. doi:10.1210/jc.2011-2260.
Schwartz SL, Akinlade B, Klasen S, Kowalski D, Zhang W, Wilpshaar W. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13(12):1219–27. doi:10.1089/dia.2011.0012.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32(4):650–7. doi:10.2337/dc08-1863.
Zambrowicz B, Ding ZM, Ogbaa I, Frazier K, Banks P, Turnage A et al. Effects of LX4211, a dual SGLT1/SGLT2 inhibitor, plus sitagliptin on postprandial active GLP-1 and glycemic control in type 2 diabetes. Clin Thera. 2013;35(3):273–85 e7. doi:10.1016/j.clinthera.2013.01.010.
Zambrowicz B, Freiman J, Brown PM, Frazier KS, Turnage A, Bronner J, et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin Pharmacol Ther. 2012;92(2):158–69. doi:10.1038/clpt.2012.58.
Zhou L, Cryan EV, D’Andrea MR, Belkowski S, Conway BR, Demarest KT. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem. 2003;90(2):339–46. doi:10.1002/jcb.10631.
Wright EMI. Glucose galactose malabsorption. Am J Physiol. 1998;275(5 Pt 1):G879–82.
Welchol® [package insert]. Parsippany: Daiichi Sankyo, Inc.; 2013. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021176s037,022362s020lbl.pdf.
Fonseca VA, Handelsman Y, Staels B. Colesevelam lowers glucose and lipid levels in type 2 diabetes: the clinical evidence. Diabetes Obes Metab. 2010;12(5):384–92. doi:10.1111/j.1463-1326.2009.01181.x.
Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med. 2008;168(14):1531–40. doi:10.1001/archinte.168.14.1531.
Bays HE, Goldberg RB, Truitt KE, Jones MR. Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: glucose and lipid effects. Arch Intern Med. 2008;168(18):1975–83. doi:10.1001/archinte.168.18.1975.
Myalept® [package insert]. Wilmington: AstraZeneca Pharmaceuticals LP; June 2014. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125390s004lbl.pdf.
Moon HS, Matarese G, Brennan AM, Chamberland JP, Liu X, Fiorenza CG, et al. Efficacy of metreleptin in obese patients with type 2 diabetes: cellular and molecular pathways underlying leptin tolerance. Diabetes. 2011;60(6):1647–56. doi:10.2337/db10-1791.
Gimeno RE, Moller DE. FGF21-based pharmacotherapy—potential utility for metabolic disorders. Trends Endocrinol Metab. 2014;25(6):303–11. doi:10.1016/j.tem.2014.03.001.
Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, Wroblewski VJ, et al. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS One. 2013;8(3):e58575. doi:10.1371/journal.pone.0058575.
Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32(8):1542–6. doi:10.2337/dc09-0684.
Fouqueray P, Leverve X, Fontaine E, Baquié M, Wollheim C. Imeglimin—a new oral anti-diabetic that targets the three key defects of type 2 diabetes. J Diabetes Metabol. 2011;02(04). doi:10.4172/2155-6156.1000126.
Pirags V, Lebovitz H, Fouqueray P. Imeglimin, a novel glimin oral antidiabetic, exhibits a good efficacy and safety profile in type 2 diabetic patients. Diabetes Obes Metab. 2012;14(9):852–8. doi:10.1111/j.1463-1326.2012.01611.x.
Fouqueray P, Pirags V, Inzucchi SE, Bailey CJ, Schernthaner G, Diamant M, et al. The efficacy and safety of imeglimin as add-on therapy in patients with type 2 diabetes inadequately controlled with metformin monotherapy. Diabetes Care. 2013;36(3):565–8. doi:10.2337/dc12-0453.
Bergenstal RM, Rosenstock J, Arakaki RF, Prince MJ, Qu Y, Sinha VP, et al. A randomized, controlled study of once-daily LY2605541, a novel long-acting basal insulin, versus insulin glargine in basal insulin-treated patients with type 2 diabetes. Diabetes Care. 2012;35(11):2140–7. doi:10.2337/dc12-0060.
Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, et al. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33(7):1516–22. doi:10.2337/dc09-2315.
Acknowledgments
The authors would like to thank Roman Götz for his critical input into the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Puneet Gaitonde was supported by a postdoctoral Grant by Eli Lilly and Company, and is currently an employee of Pfizer Inc. Parag Garhyan and Jenny Chien are employees of Eli Lilly and Company, and own stock in Eli Lilly and Company. Catharina Link, Mirjam N. Trame and Stephan Schmidt have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
About this article

Cite this article
Gaitonde, P., Garhyan, P., Link, C. et al. A Comprehensive Review of Novel Drug–Disease Models in Diabetes Drug Development. Clin Pharmacokinet 55, 769–788 (2016). https://doi.org/10.1007/s40262-015-0359-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0359-y