
Abstract
Selecting dosing regimens for phase 2 studies for a novel glucokinase activator LY2599506 is challenging due to the difficulty in modeling and assessing hypoglycemia risk. A semi-mechanistic integrated glucose-insulin-glucagon (GIG) model was developed in NONMEM based on pharmacokinetic, glucose, insulin, glucagon, and meal data obtained from a multiple ascending dose study in patients with Type 2 diabetes mellitus treated with LY2599506 for up to 26 days. The series of differential equations from the NONMEM model was translated into an R script to prospectively predict 24-h glucose profiles following LY2599506 treatment for 3 months for a variety of doses and dosing regimens. The reduction in hemoglobin A1c (HbA1c) at the end of the 3-month treatment was estimated using a transit compartment model based on the simulated fasting glucose values. Two randomized phase 2 studies, one with fixed dosing and the other employing conditional dose titration were conducted. The simulation suggested that (1) Comparable HbA1c lowering with lower hypoglycemia risk occurs with titration compared to fixed-dosing; and (2) A dose range of 50–400 mg BID provides either greater efficacy or lower hypoglycemia incidence or both than glyburide. The predictions were in reasonable agreement with the observed clinical data. The model predicted HbA1c reduction and hypoglycemia risk provided the basis for the decision to focus on the dose-titration trial and for the selection of doses for the demonstration of superiority of LY2599506 to glyburide. The integrated GIG model represented a valuable tool for the evaluation of hypoglycemia incidence.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glucokinase (GK) is one of a family of isoenzymes that catalyze the first step in glucose metabolism and acts as a glucose sensor in pancreatic β-cells [1, 2]. It also controls the conversion of glucose to glycogen in the liver and regulates hepatic glucose production [3]. GK activation is being evaluated for the treatment of Type 2 diabetes mellitus (T2DM) [4–6].
LY2599506 is a novel, small molecule GK activator (GKA). Robust and dose-dependent glucose lowering effect in fasting plasma glucose (FPG) and postprandial glucose (PPG) has been observed in an earlier proof of concept study [7]. There were no significant safety concerns and hypoglycemia appeared to be the primary dose-limiting factor. The key objective for phase 2 development of LY2599506 was to identify doses and dosing regimens that provide greater decrease in the hemoglobin A1c (HbA1c) and/or lower hypoglycemia incidence compared to standard of care, sulfonylureas (SUs).
Modeling and simulation (M&S) based on pharmacokinetic and pharmacodynamic (PKPD) models is a well-established approach and has provided opportunities to accelerate the evaluation of new drugs in humans [8–13]. Mechanistic models of diabetes that integrate complex glucose-insulin feedback mechanisms following standardized meals are increasingly used in the development of novel anti-hyperglycemic agents [14]. Although PKPD models are readily available for describing the dose/exposure response for the glucose-lowering effect for LY2599506, selecting doses for phase 2 studies based on phase 1 data is challenging due to the difficulty in assessing and predicting hypoglycemia risk. A semi-mechanistic integrated glucose-insulin model was developed previously that, in contrast to empiric models, offers the ability to describe the 24-h glucodynamic profiles with drug treatment, from which hypoglycemia incidence can be evaluated [15–17]. The model was updated with the addition of a glucagon component for the better understanding of the hypoglycemia risk. On the basis of the semi-mechanistic integrated glucose-insulin-glucagon (GIG) model, the phase 2 outcomes were simulated for various doses and dosing regimens in a prospective manner.
Two randomized, double-blinded phase 2 studies, one with parallel design with fixed dosing and placebo control, and the other employing dose titration with an active comparator, glyburide, were designed based on the simulations and conducted to further evaluate efficacy and safety of LY2599506 in subjects with T2DM.
The main objectives of this work was to optimize dose selection for phase 2 studies using simulations based on the semi-mechanistic integrated GIG model, specifically, to select optimal doses and dosing regimens to maximize the HbA1c lowering effect with minimal hypoglycemia risk of LY2599506 against an active comparator; and to investigate whether the simulation can reasonably predict the outcome of the actual trials.
Methods
Input output models
Pharmacokinetic model
The population PK (PopPK) model was a one compartment model with first-order elimination developed based on data from a multiple ascending dose (MAD) study. A non-linear mixed effects model was developed using NONMEM (version 7.2, ICON Development Solutions, Ellicott City, MD). The concentration of LY2599506 at the site of action was estimated via a biophase compartment. Inter-subject variability was included on absorption rate constant (ka), apparent clearance (CL/F), and apparent volume of distribution (V/F). Residual error was characterized with a proportional error model. The PK model was used to predict the time course of drug concentrations.
The integrated GIG model
A semi-mechanistic integrated GIG model that incorporates the feedback mechanism between glucose, insulin, and glucagon was developed and updated for LY2599506 based on PK, glucose, insulin, glucagon, and meal data obtained from the MAD study in patients with T2DM treated with LY2599506 for up to 26 days, using NONMEM (version 7.2). Drug effect of LY2599506 was modeled as a dual mechanism of action with a stimulating effect on insulin secretion and an inhibitory effect on hepatic glucose production. The semi-mechanistic GIG model was used to relate drug exposure to glucose and insulin response over time [18].
Relationship between glucose and HbA1C
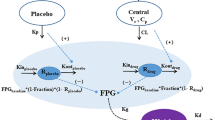
As the output from the GIG model is glucose, to further predict HbA1c from the time course data of FPG, a modification of a transit compartment model [19] was applied. The transit compartment model in a mechanistic manner, describes the dependence of HbA1c on circulating glucose concentrations [19]. Briefly, the model consists of four in-series-coupled transit compartments that describe red blood cell (RBC) aging, starting with a zero-order release of RBCs into the circulation (Kin RBC), as depicted in Fig. 1. A first-order rate constant (Ktr) defines the RBC transitions from one age stage to the next until the cell dies with Ktr = 4/RBCLS, where RBCLS is RBC life span. At any given age stage, the RBCs can become glycosylated to HbA1c as a function of FPG, described by a power function (Kglucose × FPGλ). The changes in the hemoglobin levels in compartment 1–4 are described by Eqs. 1 and 2, and the changes in the HbA1c levels in compartment 5–8 are described by Eqs. 3 and 4.
where A’s are amounts in each of the transit compartments.

Schematic representation of the mechanism-based transit compartment model for the FPG–HbA1c relationship (adapted from Hamren et al. [19]). FPG fasting plasma glucose, HbA1c glycosylated hemoglobin, K glucose glycosylation rate constant of RBCs to HbA1c, K in RBC zero-order release constant of RBCs into the circulation, K tr first-order transit rate constant, RBCs red blood cells
The model was fit using an internal data set combining data from 19 studies, a total of 1,181 patients with T2DM who were being administered SUs for at least 3 months. SUs were chosen among available oral antihyperglycemic medications because they possess a similar mechanism of action to GKA, namely stimulation of insulin secretion, and display a similar treatment effect on the time course of glucose. The information in the data set includes time matched FPG and HbA1c with sampling time and patient factors such as age, gender, and weight. Hemoglobin data was not available and therefore not included in the modeling. RBC life span, Kglucose and the FPG power function (λ) were estimated. Inter-subject variability was evaluated on λ with a log-normal distribution. Covariate effects were also evaluated on λ.
The FPG time course data simulated using the GIG model was introduced into the transit compartment model for the estimation of HbA1c at the end of 3 months for various dosing regimens in a sequential manner.
Placebo effect model
Placebo effect (disease progression) was based on data of 12 days of dosing from the MAD study.
Covariate distribution model
Age was included as a covariate on CL/F. Weight was included as a covariate on total daily calorie intake, and volume of distribution of glucose, insulin, and glucagon.
Trial execution model
Subject inclusion and exclusion criteria
As in the actual trial protocol, at trial entry, patients with T2DM with HbA1c from 7 to 10 % were included. Accordingly, a baseline distribution for glucose and HbA1c was defined for the simulations for both phase 2 trials as shown in Table 2. Men or women between the ages of 18–70 years, inclusive, and with a BMI between 20 and 40 kg/m2, inclusive, were included.
Compliance
Full adherence with the dosage regimens was assumed.
Dosing regimen
Simulations were performed for a range of dosing regimens including fixed dose and dose titration. The simulated dosing scenarios are listed in Table 1. For the conditional dose titration, the dose titration criteria were set per the following glucose targets:
-
1.
Morning doses to be adjusted to obtain evening pre-meal glucose of less than 110 mg/dL.
-
2.
Evening doses to be adjusted to obtain morning fasting glucose of less than 110 mg/dL.
The hypoglycemia threshold was predefined as < 70 mg/dL.
Simulation of phase 2 trials
The series of differential equations for the PK, GIG, and transit compartment model was translated from NONMEM to R (R deSolve package for ordinary differential equation, v. 2.9.2). Two hundred and fifty patients were simulated for each dosing scenario using a Monte Carlo simulation. Variability but no parameter uncertainty was accounted for in the simulations.
Study diet was standardized and assumed a total daily calorie intake of 30 kcal/kg, consisting of 55 % carbohydrate, 30 % fat, and 15 % protein. The total daily food intake was split by 20, 30, 40 and 10 % for breakfast, lunch, dinner, and evening snack, respectively. Variability was introduced in total daily carbohydrate intake (mean = 55 % of the total daily caloric intake, SD = 20 %) to practically mimic a subjects’ diet.
From the simulated 24-h glucose profiles and HbA1c at the end of a 3-month treatment of LY2599506, values for change from baseline in the FPG, HbA1c, and hypoglycemia incidence were summarized for each simulation scenario. Hypoglycemia incidence was summarized as percent of patients with at least one hypoglycemia event (defined as glucose <70 mg/dL) during the 3-month treatment for each simulation scenario.
Clinical studies
Study design
Based on the simulation results, two randomized, multi-center, double-blind phase 2 studies with 3-month treatment duration were planned in patients with T2DM. One study employed fixed dosing of LY2599506 and placebo control and the other compared conditional dose titration of LY2599506 and an active comparator, glyburide. The study protocols were approved by the ethics committees of the centers in which the studies were carried out and written informed consent to participate was obtained from each patient prior to enrollment. Both clinical studies enrolled patients with inadequate glycemic control, based on Hb1Ac values ranging from 7.0 to 10.0 %, inclusive, which are typical for phase 2 diabetes studies.
In the fixed-dose study, patients were randomized to placebo or 1 of 4 oral treatments of LY2599506 (50 mg BID, 100 mg BID, 200 mg BID and 200 mg QD) based on the simulation results. During the first 4 weeks, dose reduction was allowed in the event of hypoglycemia. Patients continued their treatment for an additional 8 weeks without further dose adjustments, unless necessary for management of patient safety.
In the dose-titration study, randomized patients were treated with either LY2599506 or glyburide, BID. The doses for LY2599506 were selected based on the simulations (i.e., 50–100 mg to 200–400 mg BID depending on whether glucose target was met). All patients in the glyburide treatment group started at 2.5 mg BID, and were titrated up to 5, 7.5, or 10 mg BID sequentially at a one-week interval during the first 4 weeks.
In the clinical studies, hypoglycemia was defined as any time a patient feels (s)he was experiencing a sign or symptom associated with hypoglycemia or has a blood glucose <70 mg/dL even if it was not associated with signs or symptoms of hypoglycemia.
PKPD assessments
A sparse sampling approach was utilized to collect blood samples in both phase 2 studies. Two blood samples were taken for PK analysis at the end of each of weeks 1, 2, 3, and 12. Each designated visit had 2 blood sample collections (predose and 1 of 2 randomly assigned time intervals [either 1–3 or 4–12 h post-dose]) for each patient.
The determination of LY2599506 in plasma was performed using a validated LC/MS/MS assay. The column used was a Gemini C18 (2.1 × 50 mm, 5-μm particle size) with a flow rate of 500 μL/min. Mobile phase A was water at pH of 8.5 and B was acetonitrile/methanol (50:50). The lower limit of quantification for LY2599506 was 0.1 ng/mL, and the upper limit of quantification was 250 ng/mL. Samples above the limit of quantification were diluted and re-analysed to yield results within the appropriate calibrated range. The inter-assay accuracy (% relative error) ranged from −2.78 to −1.37 % and the inter-assay precision (% relative standard deviation) was ≤14.96 %.
The primary efficacy endpoints in the phase 2 studies were HbA1c and FPG values at the end of the study. FPG and 7-point self-monitored blood glucose (SMBG) were measured at baseline, weeks 1, 2, 3, 4, 6, 8, 10, 12, and 16. The 7-point SMBG was measured before and 2 h after each meal and at bedtime. HbA1c was measured at screening, baseline, weeks 4, 8, 10, 12, and 16. Plasma glucose concentrations were analyzed using a glucose oxidase method.
Results
Model fitting with the transit compartment model
The parameter estimates of the transit compartment model for the estimation of HbA1c from the time-course of FPG after treatment with SUs are presented in Table 2. Due to the unavailability of the hemoglobin data from the 19 internal studies, the parameter Kin RBC could not be reliably estimated and was fixed to the literature value of 1.11 g/L/day for male, and 1.02 g/L/day for female [19]. The typical value was estimated to be 133 days for RBC life span, 0.000371/day/FPGλ mM, for Kglucose and 1.21 for λ. Between-subject variability was estimated to be approximately 9 % for λ and could not be reliably determined for the other parameters. None of the patient factors such as age, gender or weight had an effect on λ. Diagnostic plots for the evaluation of the quality of the model fit illustrate that the model described the data adequately (Figs. 2, 3).

Diagnostic plots of HbA1c for the transit compartment model with the sulfonylurea data. Upper population predicted versus observed and weighted residuals versus population predicted values. Lower individual predicted values versus observed and individual weighted residuals versus individual predicted values

Visual predictive check plots of HbA1c for the transit compartment model with the sulfonylurea data. a visual-predictive check plot for all patients. Symbols are observed data. Dashed lines are observed median profile and corresponding 90 % CI. Solid lines are predicted median profile and corresponding 90 % PI. b, c, and d are for representative individuals. Symbols are observed data and solid lines are individual predicted profiles
Predicted FPG, HbA1c, and incidence of hypoglycemia
Demographic characteristics of virtual patients in the simulations are presented in Table 3. The subjects simulated were moderately diabetic with mean baseline fasting glucose of 171 mg/dL. The patient characteristics are generally comparable between the simulations and the clinical studies. The predicted mean change from baseline values for FPG, HbA1c, and corresponding hypoglycemia incidence at the end of the 3-month treatment are summarized in Table 4. The simulation results suggest that there would be a mean change in FPG of ~80 mg/dL with a total daily dose of 800 mg with fixed dosing. The advantage of a BID over a QD regimen was modest. As expected, the incidence of hypoglycemia increased at higher doses. In comparison, a titration schedule resulted in a robust HbA1c reduction of approximately 1 % with a modest incidence of hypoglycemia over 3 months.
The predicted mean glucose profiles at baseline and after 3 months of treatment following a 50, 100, 200, and 400 mg BID or 100, 200, 400, and 800 mg QD dose under fixed-dose regimen are illustrated in Fig. 4, along with the predicted mean PK profiles of LY2599506. Glucose nadirs were generally predicted to be around pre-dinner or a night pre-snack time. At the higher doses of 400 mg BID (or 800 mg QD), approximately 50 % of the patients would develop hypoglycemia (the lower half of the 90 % prediction interval (PI) band fall < 70 mg/dL). Based on these simulations, doses of 50, 100, and 200 mg BID and 200 mg QD were selected for the fixed-dose phase 2 study. In the dose-titration study, all patients in the LY2599506 treatment group started at 50 mg BID and were titrated up to 100, 200 and 400 mg BID sequentially at a one-week interval during the first 4 weeks, depending on whether or not glucose control is achieved.

Predicted 24-h glucose and LY2599506 profiles for various doses with fixed-dosing regimens. Dash lines are predicted median glucose at baseline. Solid black lines and the grey bands are the predicted median glucose and corresponding 90 % prediction intervals at steady-state after treatment, Red solid lines are predicted median LY2599506 concentrations. The horizontal solid line indicates the hypoglycemia threshold of 70 mg/dL. The predictions for glucose and plasma LY2599506 concentrations are following 50, 100, 200, 400, or 800 mg administered BID (left) or QD (right). The predicted glucose corresponds to the left y-axis. The predicted LY2599506 concentration corresponds to the right y-axis (Color figure online)
Clinical data and comparisons with the model predictions
Since both phase 2 studies were terminated prior to completion of the studies due to unanticipated nonclinical findings, only limited clinical data was collected. In addition, this work focuses on simulations using the GIG model, therefore only relevant clinical results from the phase 2 clinical studies are presented in this paper. For the clinical studies, a total of 78 patients were randomized in the fixed-dose study with 6 patients completing the 3-month treatment upon termination of the study. A total of 38 patients, 16 in the LY2599506 treatment group and 22 in the glyburide treatment group were randomized in the titration study with 1 completer (glyburide) (Table 3).
The observed plasma exposure of LY2599506 at steady state was compared to the predictions performed using the PK model and the actual dosing regimens for both phase 2 studies. The predicted and observed concentrations were generally in agreement for both studies. However the observed values in the 200 mg dose group (BID and QD) in the fixed-dose study appeared lower to some extent than the predictions (Fig. 5, right column).

Predicted and observed 7-point SMBG and PK profiles at week 4 in the fixed-dose phase 2 study. The lines show the predictions for population median following 50, 100, 200 mg BID and 200 mg QD at steady-state for glucose (left) and LY2599506 concentrations (right). The shaded area represents the corresponding 90th prediction interval. Black symbols are observed data at week 4 of dosing in the fixed dosing phase 2 study
The simulations of glucose responses were performed with the actual dosing regimen, baseline glucose and insulin levels, and patient factors (such as age and weight) for patients enrolled in the phase 2 studies. The comparison of observed self monitored 7-point glucose-time course after 4 weeks of dosing of either placebo or LY2599506 and the predicted profiles is shown in Fig. 5 for the fixed-dose study. The predicted median profiles and corresponding 90 % PI shown in Fig. 5 are from simulations without incorporating data from the phase 2 studies. While data from all subjects in each cohort are not available, overall, the model predicts the central tendency and distribution well. The model predicted a slightly higher glucose lowering effect for the 200 mg BID and QD cohorts than the observed; the observations were well within the distributions.
The comparison between predicted and observed HbA1c reductions and hypoglycemia incidence at the end of 3 months is illustrated in Fig. 6 for the fixed-dose study. While the predicted HbA1c reductions at the end of 3 months are generally greater than the observed values as expected, the model well predicted the observed effect size of HbA1c after the actual treatment durations were accounted for. There also appears to be reasonable agreement between predictions and observations in the hypoglycemia incidence (Fig. 6b).

Predicted versus observed reduction in HbA1c (a) and incidence of hypoglycemia (b) after treatment with fixed-dosing of placebo (PBO) or 50, 100, 200 mg BID, and 200 mg QD LY2599506. Predictions were made for treatment for 3 months (white box) and treatment with actual duration as in the fixed-dose phase 2 study (yellow box). In a filled circles are observed change in HbA1c for completers and open circles are for non-completers. In b open squares are predicted hypoglycemia incidence and filled squares are observed hypoglycemia incidence (Color figure online)
In the titration study, every subject started at 50 mg BID and was titrated up to 400 mg BID depending on whether glucose control was achieved. Figure 7 compares the predicted and observed percentage of patients with FPG < 110 mg/dL and the corresponding hypoglycemia incidence at the end of each of the 4 weeks, with the pre-specified titration criteria. Overall, FPG levels of approximately 60 % of the patients met the target of less than 110 mg/dL at the end of week 4. The overall hypoglycemia incidence was predicted to be approximately 15 % at the top titrated doses, similar to the observed incidence of 18.8 % (Fig. 7). A mean reduction in HbA1c of 0.96 % (SD = 0.99) was predicted for the 3-month treatment. After correction for the actual treatment duration, the predicted HbA1c decrease is approximately 0.41 % (SD = 0.90), which is consistent with the observed value of 0.56 % (SD = 0.53). In comparison to glyburide, LY2599506 provided a greater HbA1c lowering effect (−0.56 % (SD = 0.53) and −0.27 % (SD = 0.70) for LY2599506 and glyburide, respectively) and lower hypoglycemia incidence (18.8 and 40.9 % for LY2599506 and glyburide, respectively) from the titration study. The observed changes in HbA1c and hypoglycemia incidence for glyburide in this study are consistent with the reported values in the literature [20–24].

Predicted versus observed percentage of patients with FPG < 110 mg/dL and incidence of hypoglycemia during the first 4 weeks of treatment in the dose titration phase 2 study. Shaded and open bars are for predicted and observed FPG (n = 16, 14, 13, and 11 for weeks 1, 2, 3, and 4), respectively. Open triangles are for predicted and filled triangles are for observed hypoglycemia incidence
To explore potential patient factors that contribute to whether the glucose of a certain patient may reach target or not at the top titrated dose of 400 mg BID in the titration study, the distribution of baseline fasting insulin (FSI) and FPG was investigated and is shown in Fig. 8. It appears the patients who did not meet glucose target had relatively high baseline FPG.

Distribution of baseline insulin and glucose levels in the dose titration simulation for patients whose glucose were normalized and not normalized. Filled symbols and open are patients who met and did not meet glucose target at completion of titration (both FPG and pre-dinner glucose are less than 110 mg/dL). Horizontal and vertical dashed lines indicate the mean values for baseline fasting insulin and baseline FPG, respectively
Discussion
The application of clinical trial simulations has been suggested to potentially revolutionize the conduct of clinical trials in drug development. The benefits of conducting clinical trial simulation are to better incorporate prior knowledge [25, 26], account of sources of variability [12, 27] and explore the impact of key variables [12, 28]. However, there are few and rare examples [29–33] regarding the prospective application of clinical trial simulation with impact and value in drug development.
The clinical development of drugs for the treatment of T2DM involves many uncertainties, mainly attributable to the complexity of the disease and its progression. Extrinsic factors such as diet and variable response to treatment add to the high inter-patient variability [34]. In this paper, a prospectively conducted clinical trial simulation drove decisions regarding the clinical development of the novel GKA LY2599506 where the GIG model developed and updated with data from a proof of concept study was used to simulate and test “what if” scenarios related to doses and dosing regimens for phase 2 studies. Data collected from the phase 2 studies consequently confirmed the learning from the early development, thus efficiently completing one “learn and confirm” cycle [8].
Before embarking on phase 2 clinical trials to further evaluate the efficacy and safety properties of LY2599506, the program elected to evaluate planned doses and assess the likelihood of success of the clinical trials using clinical trial simulations using the GIG model [35]. This model was a practical representation of a complex glucose-insulin homeostasis system where the dynamic control mechanisms and time aspects of the underlying physiology were taken into account. Specifically, the approach has the potential to account for the enhancing effects of glucose on insulin secretion and of insulin on glucose elimination as well as the inhibiting effect of plasma glucose on glucose production by the liver in a semi-mechanistic manner [15]; and the ability to account for glucose absorption [17] and thereby predict the 24-h glucose and insulin concentration–time profiles, which provided the basis for predicting hypoglycemia incidences.
The clinical trial simulation was informative on several aspects. First, it confirmed that dose titration was needed to maximize efficacy while minimizing the risk of hypoglycemia. Consequently, the phase 2 program focused on a dose titration study with the incorporation of an active comparator. This was later confirmed with results from the titration phase 2 study. Secondly, the population simulations identified the modest advantage of BID dosing (versus QD dosing). Comparison of the distributions of the glucose profiles between BID and QD suggested that QD dosing only minimally compromised the effects on glucose (and HbA1c) reduction with slightly higher hypoglycemia incidence (Table 4). This led to an addition of a QD arm in the fixed-dose phase 2 study for further evaluation. The simulations also illustrated that higher doses would be needed in the more severe diabetics (higher baseline FPG). As a result, patients in both phase 2 studies were stratified based on their baseline glucose.
Overall, the model reasonably predicted the clinical outcomes for both studies (Figs. 5, 6, 7). Several causes were identified for the minor discrepancy with further investigation of the model and the clinical data. The baseline glucose levels in the clinical studies were slightly lower than that in the simulations (Table 2). This may cause smaller reduction in glucose given that greater effect size would be expected in patients with higher baselines. In addition, early termination for both studies led to that majority of the patients did not complete the 3-month treatment with mean duration on treatment varying approximately between 4 and 8 weeks across treatment groups (Table 2). The new steady state for HbA1c after treatment were therefore not fully attained in the non-completers as 3 months were considered the minimum duration for HbA1c to reach new plateau given the turnover rate of hemoglobin [36]. After taking into consideration the actual treatment duration in the phase 2 studies, the changes in HbA1c were accurately predicted for all dose groups (Fig. 6a). Furthermore, PK exposures in the fixed-dose study, although fell largely within the 90 % PIs, lie mainly in the lower half portion of the prediction interval (Fig. 5), more evident in the 200 mg BID and QD dose groups. A simulation with updated baseline glucose and PK exposure based on the actual data from the clinical studies led to significant improvement in the GIG model prediction (data not shown).
Although several mechanism-based models reasonably describing the feedback mechanisms between glucose and insulin have been developed, including the well-known minimal model and the integrated glucose insulin model [15, 37], few have incorporated the key counter-regulatory hormone, glucagon, that is also central in the regulation of glucose homeostasis [38–40]. The addition of a glucagon component in the integrated GIG model is especially valuable for the GKA class to better predicting the incidence of hypoglycemia. The good agreement between the predicted and observed hypoglycemia incidence for LY2599506 in both phase 2 studies suggested the key influences of glucagon regulation are reasonably captured in the model. The clinical study results also confirmed that the use of a titration design did reduce the risk of hypoglycemia, as predicted by the GIG model.
The between-subject variability (BSV) was not included in the glucagon- and drug effect-related parameters in the GIG model since it could not be reliably determined due to insufficient data. This could be a potential limitation for the clinical trial simulation. However, since between-subject variability was estimated or included for most other key parameters such as CLGI, the variability in the parameters for which the BSV was not estimated were shifted to the parameters for which the BSV was determined, therefore the overall variability in the glucose response should be reasonably represented in the simulation. As shown in the VPC plots in the companion paper and Figs. 5 and 6 in this paper, the observed glucose response range is realistically covered.
There have been many published reports of converting steady state glucose (fasting, average etc.) into corresponding HbA1c values [41, 42]. These methods are not suitable for explaining the dynamic nature of FPG and HbA1c and their interplay as these relationships were devised based on steady state glucose and HbA1c data. The transit compartment model [19] used in the current work describes the dependence of HbA1c on circulating glucose concentrations in a mechanistically plausible manner, incorporates the nature of RBC aging and the chemical process of glycosylation, and was validated using in-house patient level clinical trial data for three classes of oral anti-diabetic drugs (metformin, SUs and pioglitazone). Since hemoglobin values were not available in these datasets, a literature reported value was used in the transit compartment model. Fixing this parameter is acceptable for therapies which do not have an effect on hemoglobin. The estimated RBC life span of 133 days is within the known RBC life span of 120–140 days [36]. Common demographics such as weight, gender, and age did not appear to affect the relationship between glucose and HbA1c. This is consistent with previous report [41].
In conclusion, the mechanism-based drug-disease modeling approach predicting treatment outcome according to the mechanism of action of a drug is promising. The predicted glucose/HbA1c reduction and hypoglycemia risk provided the basis for the decision to focus on the dose titration trial and for the selection of doses for the demonstration of superiority of LY2599506 compared to glyburide; and were largely confirmed by the clinical trial results. The GIG model represents a unique tool for the evaluation of hypoglycemia incidence, and was used to guide dose selection for the clinical studies. The simulation exercise yielded valuable insight into how PKPD-based simulation of clinical trials makes an impact on decision making in drug development.
Abbreviations
- ka:
-
Absorption rate constant
- CL/F:
-
Apparent clearance
- V/F:
-
Apparent volume of distribution
- BMI:
-
Body mass index
- CFB:
-
Change from baseline
- FPG:
-
Fasting plasma glucose
- GIG:
-
Glucose-insulin-glucagon
- GK:
-
Glucokinase
- GKA:
-
Glucokinase activator
- HbA1c:
-
Hemoglobin A1c
- LOCF:
-
Last observation carried forward
- LC–MS/MS:
-
Liquid chromatography/tandem mass spectrometry
- MAD:
-
Multiple ascending dose
- M&S:
-
Modeling and simulation
- PopPK:
-
Population PK
- PPG:
-
Postprandial glucose
- RBC:
-
Red blood cell
- SMBG:
-
Self-monitored blood glucose
- SUs:
-
Sulfonylureas
- T2DM:
-
Type 2 diabetes mellitus
References
Matschinsky FM (2005) Glucokinase, glucose homeostasis, and diabetes mellitus. Curr Diab Rep 5(3):171–176
Wilson JE (2003) Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 206(Pt 12):2049–2057
Iynedjian PB (2009) Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66(1):27–42
Kevin R, Guertin JG (2006) Small molecule glucokinase activators as glucose lowering agents: a new paradigm for diabetes therapy. Curr Med Chem 13(15):1839–1843
Grimsby J, Berthel SJ, Sarabu R (2008) Glucokinase activators for the potential treatment of type 2 diabetes. Curr Top Med Chem 8(17):1524–1532
Matschinsky FM, Porte D (2010) Glucokinase activators (GKAs) promise a new pharmacotherapy for diabetics. F1000 Med Rep 2:43
Bue-Valleskey JM, Schneck KB, Sinha VP, Wondmagegnehu ET, Kapitza C, Miller JW (2011) LY2599506, a novel glucokinase activator (GKA), improves fasting and postprandial glucose in patients with type 2 diabetes mellitus (T2DM). Diabetes 60(Suppl 1):A272
Sheiner LB, Steimer JL (2000) Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol 40:67–95
Karlsson MO, Anehall T, Friberg LE, Henningsson A, Kloft C, Sandstrom M, Xie R (2005) Pharmacokinetic/pharmacodynamic modelling in oncological drug development. Basic Clin Pharmacol Toxicol 96(3):206–211
Allerheiligen SR (2010) Next-generation model-based drug discovery and development: quantitative and systems pharmacology. Clin Pharmacol Ther 88(1):135–137
Zhang L, Pfister M, Meibohm B (2008) Concepts and challenges in quantitative pharmacology and model-based drug development. AAPS J 10(4):552–559
Holford N, Ma SC, Ploeger BA (2010) Clinical trial simulation: a review. Clin Pharmacol Ther 88(2):166–182
Chien JY, Friedrich S, Heathman MA, de Alwis DP, Sinha V (2005) Pharmacokinetics/pharmacodynamics and the stages of drug development: role of modeling and simulation. AAPS J 7(3):E544–E559
Dahl SG, Aarons L, Gundert-Remy U, Karlsson MO, Schneider YJ, Steimer JL, Troconiz IF (2010) Incorporating physiological and biochemical mechanisms into pharmacokinetic-pharmacodynamic models: a conceptual framework. Basic Clin Pharmacol Toxicol 106(1):2–12
Jauslin PM, Silber HE, Frey N, Gieschke R, Simonsson US, Jorga K, Karlsson MO (2007) An integrated glucose-insulin model to describe oral glucose tolerance test data in type 2 diabetics. J Clin Pharmacol 47(10):1244–1255
Silber HE, Jauslin PM, Frey N, Karlsson MO (2010) An integrated model for the glucose-insulin system. Basic Clin Pharmacol Toxicol 106(3):189–194
Jauslin PM, Frey N, Karlsson MO (2011) Modeling of 24-hour glucose and insulin profiles of patients with type 2 diabetes. J Clin Pharmacol 51(2):153–164
Schneck KB, Zhang X, Bauer R, Karlsson MO, Sinha VP (2012) Assessment of glycemic response to an oral glucokinase activator in a proof of concept study: application of a semi-mechanistic, integrated glucose-insulin-glucagon model. J Pharmacokinet Pharmacodyn. doi:10.1007/s10928-012-9287-8
Hamren B, Bjork E, Sunzel M, Karlsson M (2008) Models for plasma glucose, HbA1c, and hemoglobin interrelationships in patients with type 2 diabetes following tesaglitazar treatment. Clin Pharmacol Ther 84(2):228–235
Garber AJ, Donovan DS Jr, Dandona P, Bruce S, Park JS (2003) Efficacy of glyburide/metformin tablets compared with initial monotherapy in type 2 diabetes. J Clin Endocrinol Metab 88(8):3598–3604
Marbury T, Huang WC, Strange P, Lebovitz H (1999) Repaglinide versus glyburide: a one-year comparison trial. Diabetes Res Clin Pract 43(3):155–166
Wolffenbuttel BH, Landgraf R (1999) A 1-year multicenter randomized double-blind comparison of repaglinide and glyburide for the treatment of type 2 diabetes. Dutch and German Repaglinide Study Group. Diabetes Care 22(3):463–467
Dills DG, Schneider J (1996) Clinical evaluation of glimepiride versus glyburide in NIDDM in a double-blind comparative study. Glimepiride/Glyburide Research Group. Horm Metab Res 28(9):426–429. doi:10.1055/s-2007-979831
UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352(9131):837–853
Lesko LJ, Rowland M, Peck CC, Blaschke TF (2000) Optimizing the science of drug development: opportunities for better candidate selection and accelerated evaluation in humans. Pharm Res 17(11):1335–1344
Galluppi GR, Rogge MC, Roskos LK, Lesko LJ, Green MD, Feigal DW Jr, Peck CC (2001) Integration of pharmacokinetic and pharmacodynamic studies in the discovery, development, and review of protein therapeutic agents: a conference report. Clin Pharmacol Ther 69(6):387–399
Bonate PL (2000) Clinical trial simulation in drug development. Pharm Res 17(3):252–256
Kimko HC, Peck CC (eds) (2011) Clinical trial simulation: application and trends. AAPS advances in the pharmaceutical sciences series, vol 1. Springer-Verlag, New York
Bruno R, Hille D, Riva A, Vivier N, ten Bokkel Huinnink WW, van Oosterom AT, Kaye SB, Verweij J, Fossella FV, Valero V, Rigas JR, Seidman AD, Chevallier B, Fumoleau P, Burris HA, Ravdin PM, Sheiner LB (1998) Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol 16(1):187–196
Hasegawa M, Imai Y, Hiraoka M, Ito K, Roy A (2011) Model-based determination of abatacept exposure in support of the recommended dose for Japanese rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn 38(6):803–832
Marchand M, Fuseau E, Critchley DJ (2010) Supporting the recommended paediatric dosing regimen for rufinamide in Lennox-Gastaut syndrome using clinical trial simulation. J Pharmacokinet Pharmacodyn 37(1):99–118
de Greef R, Zandvliet AS, de Haan AF, Ijzerman-Boon PC, Marintcheva-Petrova M, Mannaerts BM (2010) Dose selection of corifollitropin alfa by modeling and simulation in controlled ovarian stimulation. Clin Pharmacol Ther 88(1):79–87
Kimko HC, Reele SS, Holford NH, Peck CC (2000) Prediction of the outcome of a phase 3 clinical trial of an antischizophrenic agent (quetiapine fumarate) by simulation with a population pharmacokinetic and pharmacodynamic model. Clin Pharmacol Ther 68(5):568–577
Matschinsky FM, Zelent B, Doliba N, Li C, Vanderkooi JM, Naji A, Sarabu R, Grimsby J (2011) Glucokinase activators for diabetes therapy: May 2010 status report. Diabetes Care 34(Suppl 2):S236–S243
Kimko H, Duffull SB (2003) Simulation for designing clinical trials: a pharmacokinetic-pharmacodynamic modeling perspective drugs and the pharmaceutical sciences. Marcel Dekker, New York
Hoffman R (ed) (2005) Hematology: basic principles and practice. Elsevier Churchill Livingston, Philadelphia
Bergman RN, Ider YZ, Bowden CR, Cobelli C (1979) Quantitative estimation of insulin sensitivity. Am J Physiol 236(6):E667–E677
Cherrington AD (1999) Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes 48(5):1198–1214
Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD (2011) Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab 13(Suppl 1):118–125
Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P (2011) Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 13(Suppl 1):95–105
Nathan DM, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine RJ (2008) Translating the A1C assay into estimated average glucose values. Diabetes Care 31:1473–1478
Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE (2002) Defining the relationship between plasma glucose and HbA1c. Diabetes Care 25:275–278
Acknowledgments
The authors thank the entire Lilly GKA team for making the clinical data available.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, X., Schneck, K., Bue-Valleskey, J. et al. Dose selection using a semi-mechanistic integrated glucose-insulin-glucagon model: designing phase 2 trials for a novel oral glucokinase activator. J Pharmacokinet Pharmacodyn 40, 53–65 (2013). https://doi.org/10.1007/s10928-012-9286-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-012-9286-9