
Abstract
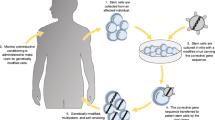
Gene therapy using autologous haematopoietic stem cells offers a valuable treatment option for patients with primary immunodeficiencies who do not have access to an HLA-matched donor, although such treatments have not been without their problems. This review details gene therapy trials for X-linked and adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome (WAS) and chronic granulomatous disease (CGD). X-linked SCID was chosen for gene therapy because of previous ‘natural’ genetic correction through a reversion event in a single lymphoid precursor, demonstrating limited thymopoiesis and restricted T-lymphocyte receptor repertoire, showing selective advantage of progenitors possessing the wild-type gene. In early studies, patients were treated with long terminal repeats-intact gamma-retroviral vectors, without additional chemotherapy. Early results demonstrated gene-transduced cells, sustained thymopoiesis, and a diverse T-lymphocyte repertoire with normal function. Serious adverse effects were subsequently reported in 5 of 20 patients, with T-lymphocyte leukaemia developing, secondary to the viral vector integrating adjacent to a known oncogene. New trials using self-inactivating gamma-retroviral vectors are progressing. Trials for ADA-SCID using gamma-retroviral vectors have been successful, with no similar serious adverse effects reported; trials using lentiviral vectors are in progress. Patients with WAS and CGD treated with early gamma-retroviral vectors have developed similar lymphoproliferative adverse effects to those seen in X-SCID—current trials are using new-generation vectors. Targeted gene insertion using homologous recombination of corrected gene sequences by cellular DNA repair pathways following targeted DNA breakage will improve efficacy and safety of gene therapy. A number of new techniques are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since the first report of a patient with primary immunodeficiency (PID) in 1956, in a boy with X-linked agammaglobulinemia, over 200 primary immunodeficiencies have been described [1].
Whilst many PID are life-threatening, haematopoietic stem cell transplantation (HSCT) can be curative. Over the last 40 years, the rate of successful treatment has dramatically improved, and for patients in optimum physical condition and with a well-matched family donor, successful HSCT can be achieved in about 90 % of cases [2, 3]. Despite these improvements in success rates, there remain significant differences in outcome related to the molecular defect. The outcome of HSCT is best for patients with subtypes of severe combined immunodeficiency (SCID), and worse for other T-lymphocyte immunodeficiencies, in particular haemophagocytic lymphohistiocytosis [2, 31]. In part, this may be due to the delay in transplanting patients with non-SCID PID, for many of whom conservative prophylactic treatments are available, in contrast to patients with SCID who generally undergo HSCT shortly after diagnosis. For patients who present with pre-existing co-morbidities, or for whom a well-matched donor cannot be found, serious adverse effects remain a challenge. The most serious of these remain steroid-resistant, or steroid-dependent graft-versus-host disease, and overwhelming infection during the period of immunocompromise following transplantation before full immune reconstitution is achieved. Significant sequalae may also follow HSCT, particularly if conditioning chemotherapy is used to facilitate stem cell engraftment. As primary immunodeficiencies demonstrate monogenic mendelian inheritance, addition of a normal complementary DNA (cDNA) copy within a progenitor cell can result in normal gene and protein expression within subsequent differentiated cells, and has the potential to cure certain diseases. Many are fatal diseases of early childhood, and as current treatment carries significant risk and sequelae, inherited immune disorders are ideal models for ex vivo gene therapy strategies, particularly those in which the results of HSCT are less favourable. This review will describe previous and current results of gene therapy trials for PID, and discuss new indications and new developments in gene editing.
2 Gene Therapy for Severe Combined Immunodeficiency
Supportive evidence for the potential of gene therapy was demonstrated in rare atypical patients who presented with attenuated SCID due to a reversion to wild-type of a mutated gene, the common gamma chain gene [4, 5]. These in vivo reversions demonstrated that ‘natural’ genetic correction through a reversion event, in even a single lymphoid precursor, can support limited thymopoiesis and provide a restricted T-lymphocyte receptor repertoire leading to attenuation of the severe disease phenotype usually associated with such conditions. Furthermore, they showed that progenitors possessing the wild-type gene had a selective advantage over those carrying the mutated gene.
In trials performed in Paris and London at the end of the 1990s and early 2000s, X-linked SCID was chosen to establish gene therapy as an effective treatment in PID [8, 9]. Infusion of haematopoietic stem cells (HSCs) from a geno-identical sibling leads to restoration of T-lymphocyte function and long-term thymopoiesis in the majority of patients [10, 11]. However, the results of treatment from a T-lymphocyte-depleted haploidentical donor are less positive, and therefore 20 patients lacking a geno-identical donor and suffering significant pre-existing co-morbidities were initially enrolled into the trials. The studies used comparable long terminal repeats (LTR)–intact gamma-retroviral vectors, with different pseudotypes but identical common gamma chain transgenes. All received ex vivo-transduced CD34+ cells infused without any additional chemotherapy (Table 1).
Early results were very encouraging, with evidence of gene-transduced cells and thymopoiesis. Seventeen patients had evidence of sustained thymopoiesis, a diverse T-lymphocyte repertoire and normal T-lymphocyte function, which led to initial clearance of life-threatening infection such as vaccine-induced BCG infection. Furthermore, long-lasting protection has been demonstrated against subsequent viral infection up to 12 years following the procedure. The results of gene therapy treatment for patients with hypomorphic mutations causing atypical SCID, or in older subjects with poor immune reconstitution following conventional HSCT, have been more disappointing, with limited or absent improvement in T-lymphocyte function, probably due to a prior loss of thymic function. Long-term natural killer cell reconstitution has not occurred to the same extent, a situation analagous to that following conventional non-conditioned HSCT. Low levels of B lymphocyte marking were found following the procedure, typically accounting for <1 % of circulating B lymphocytes. Despite this, many of the patients were independent of immunoglobulin infusions, with normal levels of immunoglobulin isotypes and evidence of IgG responses against vaccine antigens. However, not all patients achieved immunoglobulin independence.
Serious adverse effects were subsequently reported in 5 of the 20 patients, with T-lymphocyte leukaemia developing. Analysis of integration sites has shown that in the clones that became leukaemic, the viral vector had integrated adjacent to a known oncogene, most commonly LMO2, in four of five cases that developed leukeamia. The LTR enhancer activity of the retroviral vector was able to deregulate proto-oncogene expression, leading to clonal proliferation. Remarkably, the four survivors of subsequent leukaemia therapy continue to demonstrate thymopoiesis with a diverse T-lymphocyte repertoire after chemotherapy treatment, indicating that self-replicating HSCs had been transduced. Overall, 17 patients have good long-term immune recovery and are living broadly unrestricted lifestyles more than 10 years after therapy [12, 13].
Once the mutagenic potential of LTR intact vectors was uncovered, strenuous efforts have been underway to reduce the risk of adverse effects. A new generation of self-inactivating gamma-retroviral vectors, with enhancer-deleted U3 regions, have entered testing in multicentre phase I studies for SCID-X1 [14]. As in the first study, patients without HLA-matched donors are eligible for treatment with ex vivo-transduced autologous CD34+ stem cells. Early indications suggest successful reconstitution had been achieved in some of the first patients treated, although longer-term follow-up will be required to determine the risk of insertional mutagenesis.
Adenosine deaminase (ADA)-deficient SCID is an autosomal recessive disorder, and one of the most common forms of SCID. Whilst leading to a T-B-NK-SCID phenotype, the defect in the purine-pyramidine salvage pathway is systemic, and therefore patients also present with extra-immunological features due to the accumulation of toxic purine metabolites. These non-immunological manifestations include neurological abnormalities, bone malformation and lung disease. For patients with a matched sibling or matched family donor, HSCT leads to very good results, even without conditioning [15]. However, the outcome for patients with a matched unrelated donor or mismatched family donor is poor. Uniquely amongst SCID diseases, ADA-SCID can be treated with polyethylene glycosylated-ADA (PEG-ADA) given as weekly infusions and restoring at least some immune function. Treatment with PEG-ADA facilitates some immune reconstitution, with suboptimal lymphocyte numbers and function, but enough immunity for patients to amount an immune response to PEG-ADA after long-term treatment, rendering the treatment less effective.
The first gene therapy trials for PID were attempted in the early 1990s for ADA-SCID, and although these only demonstrated limited efficacy, transduced cells were detected in patients for a number of years [6, 7]. It is possible that because in these early trials, patients had received PEG-ADA for some time before gene therapy treatment, and continued treatment with PEG-ADA, gene-transduced progenitors had little competitive advantage over those cells corrected by PEG-ADA, and therefore efficacy was blunted.
At around the time that the initial common gamma chain gene therapy trials were initiated, similar gamma-retroviral ADA vectors were used in trials conducted in London, Milan and Los Angeles. Following lessons learnt from the very early ADA gene therapy trials in the early 1990s, enzyme replacement therapy was discontinued prior to gene therapy to diminish the selective advantage of the PEG-ADA-corrected cells. To date, over 40 children have been treated, most following low-dose non-myeloablative chemotherapy using either reduced-dose busulphan or melphalan. All are alive, with no reports of insertional mutagenesis, and around 70 % of these children no longer receive enzyme replacement therapy [15–17]. The success of these trials, and in particular the published experience from Milan, has led to pharmaceutical interest, with the UK-based GlaxoSmithKline entering into licencing agreements to commercialise the retroviral platform for ADA SCID. Whilst none of the subjects have developed leukaemic transformation, a number have evidence of clonal expansions, with vector integrants close to known proto-oncogenes. Alternative self-inactivating lentiviral vectors, which are less likely to insert near transcriptional start sites and have reduced potential for insertional transactivation, have recently entered phase I testing in London.
3 Gene Therapy for Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome (WAS) is an X-linked combined immunodeficiency characterised by eczema, thrombocytopenia with reduced platelet volume, which may present with haemorrhage, and recurrent infection. Infection with Epstein–Barr virus predisposes to the development of lymphoma, and there is an increased incidence of autoimmunity. HSCT is curative, with good results for patients who receive a matched sibling or well-matched unrelated donor transplant [18]. However, results following mismatched HSCT use are not so favourable. The first trial of gene therapy for WAS was performed in Hannover, Germany, using an LTR-intact gamma-retroviral vector [19]. Ten patients were treated on this protocol in which they first received reduced-dose busulphan conditioning, sufficient to cause myelosuppression and facilitate engraftment of transduced stem cells. Early reports showed restoration in myeloid and lymphoid cells of WAS protein, with improvement in clinical condition, and a rise in the platelet count, although not to normal levels. However, seven patients in this trial developed leukaemia (six T-cell acute lymphoblastic leukaemia [ALL], three acute myeloid leukemia [two secondary to ALL]). As in the SCID-X1 trial, the strong LTR enhancer associated with the gamma-retroviral vector appears to have integrated near to, and activated, proto-oncogenes such as LMO2. Alternative self-inactivating lentiviral vectors encoding an endogenous WASP promoter have been used to treat patients in Milan, with a recent publication providing the first proof-of-principle data, with good T-lymphocyte recovery and stabilisation, although not complete resolution of thrombocytopenia [20]. Additional lentiviral-based studies are underway in Paris, Boston and London with the first patients treated.
3.1 Gene Therapy for Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is a rare disorder of phagocytic cells, due to a defect in the NADPH enzyme complex responsible for generating the superoxide respiratory burst, which kills intracellular pathogens. Patients are particularly susceptible to infection with catalase-positive organisms, including Staphylococcus aureus or Aspergillus fumigatis. Inflammatory complications manifest, predominantly as inflammatory pneumonitis, inflammatory bowel disease mimicking Crohns disease, and inflammation of the genitourinary tract. The defect may affect any of the five subunits of NADPH, but the most common defect affects the CYBB gene on the X-chromosome, encoding gp91phox. Whilst conventional treatment with prophylactic antibiotics and antifungals prevents the majority of infections, inflammatory sequelae remain serious complications. Outcomes following HSCT are very good when a matched sibling or well-matched unrelated donor is available [3], although pre-existing inflammation or fungal infection is associated with a worse prognosis [21]. HSCT using a T-lymphocyte-depleted graft is particularly challenging [22]. However, gene therapy for CGD also has particular challenges. Gene-transduced neutrophils have no survival advantage over faulty neutrophils. Furthermore, neutrophils have a lifespan of only a few days, and therefore to achieve stable, long-term production of gene-transduced neutrophils, engraftment of relatively high numbers of gene-transduced haematopoietic stems cells is required, necessitating the administration of myeloablative chemotherapy prior to infusion of transduced cells. A gamma-retroviral gene therapy trial for CGD was performed in Frankfurt and Zurich [23]. Significant clinical benefit was seen in the early phases following treatment, with clearance of life-threatening fungal infection. However, the clinical benefits were not sustained as functional neutrophils disappeared over time. Methylation of the LTR promoter elements led to silencing of gene expression, whilst intact enhancer activity drove transactivation of proto-oncogenes, including MDS/EVI, leading to myelodysplasia in three patients. Two of these subjects died, although the third was successfully treated by HSCT. A fourth patient in Zurich developed clonal expansion, although not associated with chromosomal abnormalities, and has successfully undergone HSCT. An additional study in London also found evidence of transient benefit from autologous gene-modified CD34+ stem cells, but long-term persistence of corrected neutrophils was not demonstrated. A new multicentre trial sponsored by Genothon has recently opened using a self-inactivating lentiviral vector encoding a chimeric myeloid promoter.
4 Additional Candidate Immune Deficiencies
The success of gene therapy approaches for SCID-X1 and ADA SCID has encouraged development of strategies for more complex SCID disorders. For example, defects of antigen receptor VDJ recombination arising due to mutations in recombinase activating genes (RAG1 or RAG2) are being targeted for lentiviral correction. However, complex compartment-specific and time-limited regulation of these genes is required to mimic the normal gene expression required during thymopoiesis and during B-cell maturation and to prevent mutagenic transformation and immune dysregulation [32, 33]. CD40 ligand deficiency (Hyper-IgM syndrome) has similarly complex gene regulation and early attempts to use retroviral vectors to correct HSC uncovered an unacceptably high rate of T-lymphocyte lymphoproliferation [24]. Inclusion of CD40 ligand regulatory sequences in vector configurations, or targeted gene insertion to the CD40L locus may address these issues. Amongst the neutrophil disorders, leukocyte adhesion deficiency (LAD), where expression of the b-integrin CD18 is defective, leading to defective neutrophil migration, is an attractive candidate disorder. Correction of LAD dog models has been achieved using foamy virus vectors [25], and, of note, even low levels of neutrophil recovery can maintain health. Another group of candidate disorders for correction by autologous ex vivo gene delivery to HSC are the haemophagocytic lymphohistiocytosis syndromes. Here, defective cytotoxic pathways can arise due to single gene disorders, most notably within the perforin and MUNC genes. Lentiviral-mediated correction of these pathways is under development and could offer an alternative to allogeneic HSCT where donor matching is poor.
5 Emerging Technologies and Future Perspectives
To date, clinical gene therapy approaches have relied on the harvest, ex vivo manipulation and re-infusion of engineered HSC. The approaches have relied on the stable integration of an additional copy (or copies) of cDNA, delivered by viral vectors. A more elegant approach would be to correct or edit defect genes at their genomic locus, thereby maintaining appropriate regulatory control of gene expression and not risking genotoxicity through ectopic vector insertion. Technologies that allow highly specific double-stranded DNA cleavage are beginning to be used for targeted gene insertion. This can be achieved by homologous recombination of corrected gene sequences by cellular DNA repair pathways following targeted DNA breakage. Zinc finger nucleases (ZFNs), meganucleases (MN), transcription activator-like effector nucleases (TALENS) and, more recently, clustered, regularly interspaced, short palindromic repeat (CRISPR) nucleases are all being developed to create highly specific gene targeting [26–29]. The efficiency of gene editing using these techniques has improved notably in cell lines and certain primary cell lineages, although remains limited in primary HSC. Nonetheless, for disorders such as SCID-X1, where corrected cells acquire a notable survival advantage, gene repair by homologous recombination holds promise as the efficiency of nuclease reagents improves [30]. Whilst the most notable successes in the field of gene therapy for PID have been for those patients with SCID disorders, the impact of successful therapy should be notable for patients with non-SCID PID because these constitute a greater proportion of the PID population and, historically, results of HSCT are generally less positive. Furthermore, HSCT for SCID in the newborn period offers excellent results [34], and the advent of newborn screening is likely to mean that many patients with SCID will be diagnosed before symptoms appear [35]. Conversely, the adoption of a safe therapy with minimal adverse effects, including toxic short- and long-term effects of cytoreductive chemotherapy delivered to pre-symptomatic newborn SCID patients [36, 37] may advocate the continuation of gene therapy in this population. There are certainly a number of challenges ahead before gene-based therapies become more widely applicable. Production of sufficient vector stocks, in particular when self-inactivating configurations are employed, is problematic. Furthermore, the expertise and infrastructure required to harvest autologous stem cells, culture and transduce them is only available in a limited number of centres. Most of the current approaches have collected, engineered and infused stem cells without cryopreservation steps, adding to the logistic complexity of the procedures. Nonetheless, as vectors evolve and these practical hurdles are addressed, wider applicability should follow.
6 Concluding Remarks
Gene therapy has made significant progress over the last 20 years. Previous trials have demonstrated efficacy. The mechanism of unexpected adverse events is now understood, and has driven the search for safer viral vectors. New technologies to target gene insertion should enhance safety further. As data from the current clinical trials become available, it is likely that gene therapy will move from an experimental approach to become part of the mainstream therapeutic cellular armament, although long-term follow-up of treated patients will be required to monitor potential long-term adverse events. The adoption of vector manufacture by mainstream pharmaceutical companies is likely to accelerate the adoption of gene therapy as a standard tool of cellular therapies. Further details of trials currently in progress can be found at the following websites:
References
Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Fischer A, Franco JL, Geha RS, Hammarström L, Nonoyama S, Notarangelo LD, Ochs HD, Puck JM, Roifman CM, Seger R, Tang ML. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54.
Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, Amrolia PJ, Gaspar HB, Davies EG, Friedrich W, Hoenig M, Notarangelo LD, Mazzolari E, Porta F, Bredius RG, Lankester AC, Wulffraat NM, Seger R, Güngör T, Fasth A, Sedlacek P, Neven B, Blanche S, Fischer A, Cavazzana-Calvo M, Landais P, Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation, European Society for Immunodeficiency. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126:602-10.e1–11.
Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, Vermont C, Ahmad I, Shaw PJ, da Cunha JM, Schlegel PG, Hough R, Fasth A, Kentouche K, Gruhn B, Fernandes JF, Lachance S, Bredius R, Resnick IB, Belohradsky BH, Gennery A, Fischer A, Gaspar HB, Schanz U, Seger R, Rentsch K, Veys P, Haddad E, Albert MH, Hassan M, on behalf of the Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383:436–48.
Bousso P, Wahn V, Douagi I, Horneff G, Pannetier C, Le Deist F, et al. Diversity, functionality, and stability of the T cell repertoire derived in vivo from a single human T cell precursor. Proc Natl Acad Sci USA. 2000;97(1):274–8.
Hirschhorn R, Yang DR, Puck JM, Huie ML, Jiang CK, Kurlandsky LE. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet. 1996;13(3):290–5.
Aiuti A, Vai S, Mortellaro A, Casorati G, Ficara F, Andolfi G, et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8(5):423–5.
Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–3.
Cavazzana-Calvo M, Hacein-Bey S, Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–72.
Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364(9452):2181–7.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3(97):97ra79.
Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363(4):355–64.
Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42.
Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–50.
Thornhill SI, Schambach A, Howe SJ, Ulaganathan M, Grassman E, Williams D, et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008;16(3):590–8.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3(97):97ra80.
Aiuti A, Cassani B, Andolfi G, Mirolo M, Biasco L, Recchia A, et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest. 2007;117(8):2233–40.
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–58.
Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, Cant AJ, Thrasher AJ, Cowan MJ, Albert MH, Small T, Pai SY, Haddad E, Lisa A, Hambleton S, Slatter M, Cavazzana-Calvo M, Mahlaoui N, Picard C, Torgerson TR, Burroughs L, Koliski A, Neto JZ, Porta F, Qasim W, Veys P, Kavanau K, Hönig M, Schulz A, Friedrich W, Notarangelo LD. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott–Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011;118:1675–84.
Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, et al. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N Engl J Med. 2010;363(20):1918–27.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science. 2013;341(6148):1233151.
Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100:4344–50.
Horwitz ME, Barrett J, Brown MR, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. 2001;344:881–8.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–9.
Brown MP, Topham DJ, Sangster MY, Zhao J, Flynn KJ, Surman SL, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998;4(11):1253–60.
Bauer TR Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM, et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med. 2008;14(1):93–7.
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435(7042):646–51.
Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther. 2007;7(1):49–66.
Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333(6040):307.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308.
Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–306.
Ouachée-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, Neven B, Casanova JL, Tardieu M, Cavazzana-Calvo M, Blanche S, Fischer A. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–50.
Lagresle-Peyrou C, Yates F, Malassis-Séris M, Hue C, Morillon E, Garrigue A, Liu A, Hajdari P, Stockholm D, Danos O, Lemercier B, Gougeon ML, Rieux-Laucat F, de Villartay JP, Fischer A, Cavazzana-Calvo M. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. 2006;107:63–72.
van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G, Malshetty V, Cortes P, Jollet A, Danos O, Cassani B, Zhang F, Thrasher AJ, Fontana E, Poliani PL, Cavazzana M, Verstegen MM, Villa A, Wagemaker G. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol. 2014;133:1116–23.
Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, Veys P, Gennery AR, Gaspar HB. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117:3243–6.
Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, Lewis DB, McGhee SA, Moore TB, Stiehm ER, Porteus M, Aznar CP, Currier R, Lorey F, Puck JM. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132:140–50.
Horn B, Cowan MJ. Unresolved issues in hematopoietic stem cell transplantation for severe combined immunodeficiency: need for safer conditioning and reduced late effects. J Allergy Clin Immunol. 2013;131:1306–11.
Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, Schwarz K, Schulz AS, Hoenig M, Sparber-Sauer M, Gatz SA, Denzer C, Blanche S, Moshous D, Picard C, Horn BN, de Villartay JP, Cavazzana M, Debatin KM, Friedrich W, Fischer A, Cowan MJ. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014;123:281–9.
Acknowledgments
Waseem Qasim is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
Waseem Qasim and Andrew R. Gennery declare no relevant conflicts of interest. No sources of funding were used to support the writing of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Qasim, W., Gennery, A.R. Gene Therapy for Primary Immunodeficiencies: Current Status and Future Prospects. Drugs 74, 963–969 (2014). https://doi.org/10.1007/s40265-014-0223-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0223-7