
Abstract
Allele identification for agro-morphological traits and stress resistance is a major concern across the globe for improving productivity of finger millet. Here, we used 46 genomic and 58 genic simple sequence repeats (SSRs) markers in a set of 66 accessions used to constitute a global mini-core collection for analysing their genetic structure as a population and establishing association among markers and twenty morphological traits including resistance to finger blast. Phenotypic data revealed a wide range of variation for all traits except flag leaf width and flag leaf sheath width. We got amplification of 81 alleles by the 31 genomic SSRs at an average of 2.61 alleles per locus. Polymorphism information content (PIC) values varied from 0.21 to 0.75 and average gene diversity was 0.49. Structure analysis of the population using the genomic SSR data divided the accessions into two clusters where Indian and exotic accessions were grouped in separate clusters. Genic SSRs which were associated with blast resistance genes, amplified 36 alleles at an average of 2 alleles per locus. PIC values ranged from 0.32 to 0.37 and average gene diversity was 0.45. Population structure analysis using data from these SSRs grouped the accessions into three clusters, which broadly correspond to their reaction to blast disease. Twenty-two significant associations were found using the GLM approach for 20 agro-morphological traits both in 2012 and 2014, while, 7 and 5 significant marker-trait associations were identified using MLM in 2012 and 2014 respectively. The SSR markers FMBLEST35 and FMBLEST36 designed from the Pi21 gene sequence of rice were found to be associated with blast disease resistance in finger millet indicating that the gene homologues play a significant role in an important role for neck blast resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Finger millet is a potential nutraceutical food grain crop, widely grown in East Africa and India. It is highly self-fertilized allotetraploid (2n = 4x = 36) annual plant, belongs to Poaceae family. It has a wide range of seasonal adaptation, and is grown at sea level i.e. Andhra Pradesh and Tamil Nadu to high hills (Uttarakhand, Sikkim and Arunachal Pradesh) in India. Its seeds are consumed in variety of forms, such as unleavened bread (roti), thin or thick porridge, fermented porridge, and also used in brewing. Karnataka state is the major producer of finger millet in India, where it is mostly consumed in the form of finger millet balls (locally known as ragi mudde). The crop has high nutritional value, storage quality and also provides food security because of its climate resilience (Dida et al. 2007). Grains are rich source of calcium, dietary fibre and essential amino acids particularly tryptophan and methionine. Thus, as a crop, finger millet is important for identification of climate resilience and storage-related traits. Morphologically finger millet exhibits higher variations, which need to be captured at a molecular level for identification of QTLs responsible for various traits. The two most commonly used methods for marker trait associations are linkage and association mapping (Bodmer 2011). The latter offers few advantages over linkage mapping like it gives high resolution mapping of the QTLs, uses natural germplasm; reference population is broader; more alleles are considered, and it takes less time (Mott et al. 2000). Molecular marker studies in finger millet have been gaining importance and recently few reports have been published using microsatellite or SSR markers (Babu et al. 2014a; Ramakrishnan et al. 2016a). However, in finger millet very few genomic SSRs are available in the public domain (Dida et al. 2007). The genic SSRs are the alternate way for the genomic applications in finger millet, which were developed and used in several studies (Babu et al. 2014a, b) The SSR or EST-SSR markers are more widely used by molecular breeders and geneticists because of its high amount of polymorphism, accuracy, rapidity, simplicity, low cost, repeatability throughout the genome and small DNA quantity requirement for different applications like linking phenotypic and genotypic variation. In finger millet, yield is influenced by several characters, including disease resistance; ear head size, number of fingers, flag leaf etc. The finger millet blast disease caused by Magnaporthe grisea, affects all growth stages and causes yield loss of 28–36% (Nagaraja et al. 2007), and sometimes yield loss could be as high as 80–90% (Rao 1990). The full genome sequence available in rice is an added advantage for identification of linked loci for blast disease resistance in finger millet through comparative genomics approach.
The first report on the population structure analysis among African and Asian finger millet germplasm was by Dida et al. (2008) using forty-five SSR markers. The three groups identified by them were africana, African coracana and Asian coracana lines. Later, after a long gap of 6-year Babu et al. (2014a, b, c) did a population structure and association mapping analysis of large collection of global finger millet accessions (190 accessions) from different countries for different traits like blast resistance, opaque2 modifiers, and other agro-morphological characters. Recently, Ramakrishnan et al. (2016b) also studied the genetic diversity and structure of 128 finger millet genotypes using RAPD markers and grouped them into four groups. Kumar et al. (2015) studied the genetics of high calcium accumulation in finger millet grains. The association analyses revealed 9 markers significantly associated with the calcium trait in their study. However, all the above reports were with 1 year/location data, and the reliability of QTLs mainly depends upon multi-seasons/environments phenotyping. Hence, in the present study, data from 104 genic and genomic SSR markers across a set of 66 mini-core germplasm of finger millet was used to determine the population structure and phenotypic data over 2 years collected from one location was used for association mapping and identification of linked markers for 20 important morphological traits, including blast disease resistance.
Materials and methods
Plant materials and experiments
A total of sixty-six homozygous finger millet accessions belonging to a mini-core developed from a collection of global germplasm, obtained from ICRISAT, Hyderabad were used in the study. It also included one local check variety, VL Mandua 149. These accessions are from different geographical regions, i.e., Zimbabwe, Uganda, India, Zambia, Senegal, Kenya, Maldives, Malawi and Nepal. The list of the accessions used in the present study along with their places of collection is given in Table S1.
Experimental design and data collection
The experiments were conducted for 2 years in the sub-mountainous Himalayan region at ICAR-VPKAS research farm, Almora, Uttarakhand, India (29.36°N, 79.30°E, altitude—1250 m above msl) in Augmented design during the year 2012 and Alpha Lattice design with two replications in the year 2014. The experiments were conducted during the rainy season i.e. June to October. The plot size was single row and three rows of 3 m each in 2012 and 2014, respectively, with 22.5 cm spacing between rows and 10 cm between plants within rows. Fertilizers were applied at the rate of 50:40:25 (N:P:K) kg/ha as per the recommended dose for the crop. Half of the nitrogen along with entire amount of phosphorous and potassium were given at the time of field preparation as a basal dose. Top dressing was given after 45 days of sowing with the remaining half of the nitrogen. Manual weeding was practiced twice (20 and 40 days after sowing) to control weeds. Data were recorded on 20 quantitative traits in 2012 and 23 traits in 2014 on five representative plants from each plot selected randomly (Table S2). The data on morphological traits was recorded as per descriptors of finger millet (IBPGR 1985). Field screening for blast disease (caused by Magnaporthe grisea) severity was done according to Nagaraja et al. (2007) under natural epiphytotic conditions. The disease incidence for neck blast was calculated by counting the number of infected peduncles in a plot and calculated as per cent incidence. Finger blast severity was recorded in each ear heads in a plot and how much areas of each finger were infected was converted as per cent severity. Finger and neck blast data was subjected to square root transformation for analysis.
Analysis of variance (ANOVA) for augmented and alpha lattice design was computed in Indian NARS Statistical Computing Portal of ICAR-IASRI, New Delhi using SAS software (SAS 9.3) for each design separately. The adjusted mean values of all agro-morphological traits for both the locations were used for phenotypic correlation analysis using SPSS 20.0.
DNA extraction and quantification
The isolation of DNA was carried out from freshly collected leaves by using the standard protocol of Murray and Thomson (1980). Quantification of genomic DNA was done by electrophorasing 1 µL of DNA samples of all accessions in 0.8% agarose gels with standards of uncut lamda DNA. The normalized DNA samples were used for SSR genotyping.
Microsatellite amplification
A total of 46 genomic and 58 genic SSRs were used in the study. The list of genic SSRs used in the study is given in Table S3. The genomic SSR sequences were retrieved from earlier studies (Dida et al. 2007); however, genic SSR marker sequences were obtained from Babu et al. (2014a). The genic SSRs were selected based upon the BLASTx analysis of EST sequences of finger millet NBS-LRR region available in the NCBI website and broad-spectrum rice blast resistant genes. The PCR were performed with 25 ng of template DNA in a 20 µL reaction volume as described in Babu et al. (2014a, c). The amplified products were size fractionated on 3.0% super fine resolution (SFR) agarose gel at 100 volts for 3 h. Ethidium bromide staining was done to visualize the bands using Alpha Imager System (Alpha Innotech, USA). Scoring of the PCR amplified alleles was done based on allelic size and were estimated using standard 100 bp DNA ladder. The SSR loci data were used for estimating the polymorphism information content (PIC), gene diversity, allele frequency, most frequent and rare alleles using Power Marker V3.0 (Liu and Muse 2005). Out-crossing rate (t) was calculated as F = (He − Ho)/He; t = (1 − F)/(1 + F), where F is fixation index, He is expected heterozygosity; Ho is observed heterozygosity. The same allelic size scoring was used as the input format for statistical analysis in STRUCTURE v2.3.4 and TASSEL 3.0.
Population structure analysis
Structure version 2.3.4 (Pritchard and Wen 2003) was used for population structure analysis of mini core finger millet accessions. It was presumed that there exists number of sub-populations within the sample to be analyzed. To determine the groups in the population i.e. K value, the programme was run at varying K values from 1 to 10 with 4 independent runs for each K value at a burn-in period of 100,000 with a model allowing for admixture. Final K values (K = 2 and K = 3 were found optimum for total SSRs and genic SSRs, respectively) were determined using the ‘Structure Harvester’ programme (http://taylor0.biology.ucla.edu) based on both LnP(D) and Evanno’s ΔK (Evanno et al. 2005). Later, simulations were done at K = 2/3 with the same burn-in period and the membership coefficient with the lowest likelihood value was used to assign each genotype into its relevant K (Roy et al. 2015). The structure results were displayed graphically using ‘DISTRUCT’ programme (Rosenberg 2004).
Association analysis
The association mapping was carried out by using data from blast scoring and 17 agro-morphological traits recorded on finger millet accessions, genotypic SSR data of total polymorphic markers and data of population structure (Q matrix) by using software TASSEL (Bradbury et al. 2007). The two approaches of marker trait association i.e. General linear model (GLM) and mixed linear model (MLM) were used. For MLM analysis, the kinship matrix was also used in addition to genotypic, phenotypic and Q matrix data. The significant threshold (P) was taken at 0.01 for the association analysis.
Results
Phenotypic variation
The variation for agronomic traits has been presented in Table S4 for both the years. Days to flowering, plant height, peduncle length, ear exsertion, ear length, length of longest finger, finger number, grain yield and finger blast showed similar distributions in both the years (Fig. S1). Wide range of variation was observed for all the traits except flag leaf and sheath width. Grain yield varied from 3.0 to 39.0 g in the year 2012 and 3.0 to 35.0 g in 2014. Ear length, finger number and basal tillers, which are considered as major component traits of grain yield showed variation ranging from 3.6 to 16.6, 5 to 12 and 1 to 14 in the year 2012 and 3.6 to 15.1, 4 to 9 and 1 to 4.5 in 2014, respectively (Table S4).
Correlation analysis of phenotypic traits
Thirty-one highly significant (P, 0.01) and twenty-nine significant positive correlations were observed among different agro-morphological traits including grain yield correlation with ear width (P, 0.01), width of longest finger (P, 0.01), fodder weight (P, 0.01), culm thickness (P, 0.05) and flag leaf sheath length (P, 0.05) (Table 1). Highly significant negative association was observed for days to flowering and days to maturity with neck blast. There was almost perfect correlation between days to flowering and days to maturity, ear length and length of longest finger, peduncle length and ear exsertion, and flag leaf sheath length and peduncle length (Table 1).
SSR polymorphism analysis
A total of 104 SSR markers (46 genomic and 58 genic SSRs for blast resistance) were used in the study, of which 31 genomic (67%) and 18 genic (31%) SSRs were found to be polymorphic across the 66 finger millet accessions. The 31 polymorphic genomic SSRs yielded 81 alleles, ranging from 2 to 5 alleles with an average of 2.61 alleles per locus (Table 2). The genomic SSR marker UGEP24 produced five alleles followed by four alleles each produced by UGEP60, UGEP65 and RM5963. Eighteen genic SSRs associated with blast resistance genes amplified 36 alleles with average 2 alleles per locus (Table 2). The genomic SSRs showed PIC values ranging from 0.21 to 0.75, while for EST SSRs it was 0.32 to 0.37. The mean of PIC values was 0.41 and 0.35 for genomic and genic SSRs, respectively. The gene diversity was found to be highest with genomic SSR marker UGEP24 (0.79) followed by UGEP65 (0.74) and UGEP60 (0.72). The gene diversity was in the range of 0.24–0.79 for genomic and 0.20–0.50 for genic SSRs. The average gene diversity value was 0.49 for genomic and 0.45 for genic SSRs, respectively. The heterozygosity varied from 0.00 to 1.00 at an average of 0.26 for genomic and 0.22–0.82 at an average of 0.18 for genic SSRs.
Analysis of population wise genetic diversity and out-crossing showed high observed heterozygosity (Ho) for all the groups (Table 3). Indian population showed low Ho in comparison to all other populations. The out-crossing rate among different populations based on their origin country was also very high ranging from 67 to 100%. The populations from Zimbabwe, Malawi and Nepal showed 100% out-crossing (Table 3).
Population structure analysis
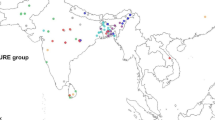
Sixty-six diverse finger millet accessions were used for population structure analysis using two sets of SSR markers. One set included all polymorphic genic as well as genomic SSRs spread across all the chromosomes, whereas the other set included only polymorphic genic SSR markers belonging to blast resistance genes or region of the genome governing blast resistance. The population structure (Structure I) depicted the place of collection of the accessions when data from all the polymorphic SSR markers were used and at K = 2 (Fig. 1), maximum peak was observed. However, when data of only the genic SSR markers were used, K was found to be maximum at peak of 3 (K = 3) (Fig. 2), indicating that the mini-core represents three populations (structure II) for their reaction to blast disease. The overall proportion of membership of the sample in each of the two populations of structure I was 0.469, and 0.531 (Table S5). The higher Fst value identified between PI and PII showed high diversity existed between these two populations. The average distances i.e. expected heterozygosity between individuals within each population were 0.4610, and 0.4662 respectively. Similarly, in structure II the proportion of membership was 0.314, 0.356 and 0.330 for three populations. The Fst estimates among the three populations of finger millet accessions are given in Table S6. The average distances (expected heterozygosity) between individuals within each population were 0.44, 0.38 and 0.45 respectively.

Identification of appropriate sub-population number (K): sub-population number K against delta K and the maximum K value observed at K = 2

Identification of appropriate sub-population number (K): sub-population number K against delta K and the maximum K value observed at K = 3
Trait-marker associations
The GLM model identified twenty-two significant (< 0.01%) marker-trait associations for 20 agro-morphological traits both in 2012 and 2014 (Table 4). However, seven and five significant (< 0.01%) associations were found using MLM approach in 2012 and 2014 respectively (Table 5). The weaker associations having P value of > 0.01were removed to avoid wrong interpretation of results. Only two marker-trait associations were common in both the years using GLM approach, however, using MLM approach only neck blast-FMBLEST35 marker association was common in both the years.
In the year 2012 using MLM approach, the agronomic traits viz., basal tiller number and flag leaf width, were found to be linked with RM5963 and FMBLEST2 markers respectively. The SSR marker FMBLEST36 was found to be associated with three traits viz., length of longest finger, ear length and neck blast (Table 5). The neck blast disease resistance was found to be linked to FMBLEST36 at an R2 of 10.5%. In the year 2014, the MLM approach resulted in identification of five trait-marker associations. Here, the neck blast disease resistance was found to be linked to FMBLEST35 at an R2 of 5.6%. The same marker was found to be associated to rice blast Pi21 gene. The agro-morphological traits leaf sheath length; lengths of longest finger and leaf blade length were found to be linked to FMBLEST10, UGEP10 and RM5963 markers respectively. In the years 2012 and 2014, three traits viz, longest finger length, ear length and neck blast were common. However, only neck blast disease was found to be linked by same gene (Pi21) markers by both the approaches (GLM and MLM) in both the years, whereas remaining two traits were linked by different markers.
Discussion
Phenotypic variation and correlation analysis
We observed wide range of phenotypic variation for all agronomic traits suggesting greater scope of improvement through introgression and development of better genotypes with high-yield potential and disease resistance. Phenotypic correlation results revealed that ear width, width of longest finger, fodder weight, culm thickness and flag leaf sheath length were major positive contributors for grain yield. These component traits could be used in formulating a selection index for yield improvement in finger millet. Further, high positive correlation between peduncle length and ear exsertion and ear length and length of the longest finger suggested that laborious observations like ear exsertion, length of the longest finger and ear width could be avoided by taking observations on its correlated traits such as peduncle and ear length as described earlier also by Upadhyaya et al. (2006).
SSR polymorphism analysis
The informativeness of the SSR loci is demonstrated by polymorphism information content (PIC), which detects the genotypic difference between individuals. The PIC for each marker was calculated to make distinctions, assess the quality of markers and compare the primer efficiency in displaying the genetic information. Genomic SSRs were more polymorphic than genic SSRs in our study, and the findings were in close agreement to Ramakrishnan et al. (2016a), who reported similar PIC ranging from 0.32 to 0.64 in 128 finger millet accessions. Nirgude et al. (2014) developed 30 SSRs belonging to o2 modifiers of rice, maize and sorghum and found 15 (50%) markers to be polymorphic in finger millet. Arya et al. (2009) analysed genetic diversity using 31 EST SSRs in a set of 11 elite finger millet genotypes and found 17 polymorphic markers. Similar observations were also reported in an analogous study (Kumar et al. 2012). Ramakrishnan et al. (2016a) found 2.13 alleles per primer with a set of SSR markers among 128 finger millet accessions, which is almost identical to our observation. In an earlier study, Babu et al. (2014c) also found similar results; however, Reddy et al. (2012) found higher PIC values.
The expected heterozygosity (He) also known as ‘Gene diversity’ is the probability that an individual will be heterozygous at a given locus or loci. EST-SSRs represent the functional regions of the genome and the polymorphism analyzed with these markers show ‘‘true gene diversity’’, within or flanking regions of the genes (Thiel et al. 2003). Nirgude et al. (2014) found much lower gene diversity (0.024–0.327) than our results for o2 modifiers EST-SSRs. The reason for the same could be less number of markers and the study on a specific trait. Our results however, were similar to Bharathi (2011), who also found the range of values from 0.20 to 0.85 for He. The results of gene diversity clearly indicated that the markers were more polymorphic in our study in comparison to earlier studies. The highest heterozygosity was found in the UGEP65 SSR marker (1.00), followed by UGEP78 and RM 262 (0.98 each). The probable reason for high heterozygosity in finger millet could be due to its allo-tetraploid nature with two different genomes. Dida et al. (2007) also reported high heterozygosity for UGEP3 locus due to two alleles as a result of its mapping in both the genomes of finger millet. The use of diverse germplasm collection may also be the reason of high heterozygosity in the present study due to some residual heterozygosity at least at some of the loci. Another probable reason could be high bias for and rate of mutation at SSR loci could (Udupa and Baum 2001). Out-crossing rate of different populations was very high and three of the populations showing 100% out-crossing revealed that there is very high admixture of alleles among different populations used in the present study.
Population structure analysis
All the SSR markers presented one or more alleles with a frequency of more than 5%. Generally, an allele with a frequency of less than 5% is considered as a rare allele. The maximum allele frequency was observed with the SSR marker RM21 for the 90 bp allele (86%). The selection of diverse accessions results in a normalizing effect which minimizes the population structure, thereby making a suitable condition for association mapping. In structure I, population I (PI) consisted of mostly Indian, whereas Population II (PII) comprised of mostly exotic accessions (Fig. 3). Among the Indian accessions, seven accessions were found to have less percentage of admixtures while remaining all accessions were having a good amount of admixture. The accessions IE2312 and IE6082 were found to be pure lines. In the PII, nine exotic accessions had less percent of admixture. All these less admixture accessions were mostly of Zimbabwe origin. The results showed that there were not many movements of finger millet germplasm between Zimbabwe and other countries. The results were in close agreement with earlier reports, where Babu et al. (2014c) found two populations largely corresponding to their geographic origin. Similarly, Bharathi (2011) grouped a large finger millet germplasm collection into four subpopulations where mixture of East Africa and South Asian accessions formed subpopulation 1 and subpopulation 2, maximum number of South Asian accessions were in subpopulation 3 and subpopulation 4 had mixture of accessions from all other geographical origins. The results of structure analysis using all SSRs showing two populations were in agreement with high out-crossing rate observed from the SSR data among different populations based on the origin country. Taking 70% purity as a criterion for accession to be pure, 10 out of 19 accessions from Zimbabwe, 3 out of 7 from Kenya, 9 out of 17 from India, 2 out of 7 from Uganda, 1 out of 2 from Malawi and 4 out of 9 from Nepal were pure. It was observed that all the populations had admixtures from other depicting high gene flow due to exchange of germplasm between countries.

The population structure of 66 mini-core finger millet accessions using the SSR loci revealed by STRUCTURE software
In structure II, three populations were found viz, population I (HR-R), population II (HS-S-MS/MR), and population III (MR/MS-R) with few exceptions (Fig. 4). Among these populations, the PII consisted of fewer admixtures of accessions; whereas remaining two populations consisted of high admixture. The results showed that alleles for R and MR against blast disease were spread in two populations (PI and PIII). Babu et al. (2014a) also found similar grouping pattern where the finger millet accessions were grouped into four populations based on the disease resistance. Highly susceptible and moderately resistant accessions were placed in population 1 (P1), and population 4 (P4) while, moderately resistant and resistant accessions were in population 2 (P2). The population 3 (P3) had resistant and highly resistant accessions. Here it may be noted that the population structure is based on reactions to prevalent pathotype at the location under study.

The population structure of 66 mini-core finger millet accessions using the blast specific SSR loci revealed by STRUCTURE software
Trait-marker associations
Significance of trait-marker associations was more accurate by using MLM approach rather than GLM approach, since it reduces the type I errors. Hence, in the present study, the trait-marker associations obtained by MLM approach were only used for assessing the QTL discussion. In the year 2012, seven trait-marker associations were found by MLM approach. The finger blast resistance was found to be linked with RM3330 and UGEP6 SSR markers at an R2 value of 10.2 and 9.65% respectively. The rice microsatellite RM3330 is also linked to the Pi40t blast resistant gene of rice. EST SSR, FMBLEST36 was found to be linked with neck blast resistance at R2 of 10.5%. The same SSR marker has been found to be linked to rice blast Pi21 gene. In a similar study, Babu et al. (2014a) identified four and one linked markers for finger blast and neck blast resistance, respectively. The SSR markers, RM262 and FMBLEST32 were found to be linked to finger blast disease resistance and explained 10 and 8% phenotypic variance (R2), respectively. In our study too the same SSR markers i.e. FMBLEST35 and FMBLEST36 were associated with blast disease resistance. These markers were designed from the Pi21 gene sequence of rice. This showed that Pi21 rice gene orthologs might be responsible for blast resistance in finger millet too. The same gene marker was also associated with neck blast resistance in the year 2014. So, this can be a potential QTL which needs further validation for its use in the marker assisted programmes. If the genic marker linked to a trait is present within the target gene, there is a greater chance of marker being linked to that particular trait and may be responsible for expression of target trait. In the present study also the identified genic markers, FMBLEST35 and FMBLEST36 represent high phenotypic variation for blast resistance. The genetics of blast disease resistance in rice has been studied in detail and many genes for resistance (nearly 40 genes) have been identified. Among the genes identified for blast resistance in rice, pi21 is recessive while most others are dominant (Fukuoka and Okuno 2001). In rice, the region between two markers RM0527 and RM3330 (3.5 cM) in chromosome 6 explained 58.9–66.5% phenotypic variation, and thus showed strong association with blast resistance (Jeung et al. 2007). The pi21gene in rice has been introduced from an upland Japanese rice, and shows strong resistance to blast by restricting the development of lesions. The results obtained in our study revealed that major blast resistant genes might be present on the long arm of chromosome 4 and 6 in finger millet genome, which has been projected in our earlier reports also (Babu et al. 2014a).
There were few reports on the association mapping for different morphological traits in finger millet. The genomic marker UGEP81 was strongly linked to basal tiller number (Babu et al. 2014c) which explained the phenotypic variance of 10.8%. Another marker FM9 was linked to flag leaf width and plant height, which explained 14.1 and 11.2% phenotypic variance (Babu et al. 2014c). In another study involving large germplasm collection of finger millet, Bharathi (2011) reported association of UGEP8 loci with basal tiller number, which is located on chromosome 3B. She also reported multi-marker single trait associations. These include association of four genomic SSR markers UGEP8, UGEP 56, UGEP77 and UGEP90 to days to 50% flowering. In another study, Babu et al. (2014b) found two QTLs for tryptophan content and one QTL for protein content by using 74 genic and 46 genomic SSR loci. In their study, the genic marker OM5 showed strong association with tryptophan content. In all the above reports and present study, it was found that basal tiller number, flag leaf width, days to flowering and plant height were the common traits linked to different molecular markers.
Allelic distribution of linked SSR markers for blast resistance
Association analysis with the data from combined or genic markers and phenotypic data collected over 2 years indicated that in the year 2012 neck blast was linked to the genic marker FMBLEST36 while in the year 2014 it was linked to FMBLEST35. The marker FMBLEST36 produced two alleles, 220 and 170 bp. A total of 38 HR, R and MR accessions were heterozygous and contained 220/170 bp allele while the S and HS accessions were homozygous for the 170 bp allele. The SSR marker FMBLEST35 amplified two alleles, 420 and 430 bp, in the accessions studied. It was found that 35 R and HR accessions were homozygous for the 430 bp allele. Thus, it appears that the 430 bp allele is linked to neck blast resistance. As regards finger blast, analysis with phenotypic data of 2012 and genotypic data showed that RM3330 and UGEP6 were linked with the disease. RM3330 marker produced 360 and 350 bp alleles while UGEP6 produced 230 and 200 bp alleles. Other alleles like finger length and flag leaf width also showed an association with markers. Marker UGEP10 produced three alleles of 190, 200 and 210 bp, respectively. Analysis with phenotypic data of 2014 showed that most of the long finger accessions contained the 210 bp allele. Similarly, the marker FMBLEST2 showed linkage with flag leaf width (2012 data). The marker produced two alleles, 300 and 310 bp. Accessions homozygous for the 310 bp allele had flag leaf width of more than 1.3 mm. The identified markers can be successfully deployed for screening of germplasm accessions of finger millet for neck and finger blast resistance as well as for important yield component traits and hasten the process of varietal development. Further, by developing bi-parental mapping populations through hybridization between identified contrasting individuals, the distance between marker and gene can be worked out. Later, fine mapping of these alleles could give further authentication of the observed associations.
The identification of QTLs linked to important agro-morphological traits is very important in the crop breeding programmes. There are limited reports on mapping of important traits in finger millet. The present investigation resulted in identification of seven and five significant marker-trait associations using MLM in years 2012 and 2014 respectively. The SSR markers FMBLEST35 and FMBLEST36 designed from the Pi21 gene sequence were found to be linked to leaf and neck blast disease resistance from earlier reports and also in the present study. This showed that Pi21 gene orthologs might be responsible for resistance to blast disease in finger millet. It was found that basal tiller number, flag leaf width, days to flowering and plant height were the common morphological traits linked to different molecular markers. The results clearly revealed that major blast resistant genes might be present on chromosome 4th and 6th in finger millet genome. These markers can be further used for fine mapping and validation studies using bi-parental mapping populations, which can be further used in cloning of full-length blast resistant genes. Marker assisted breeding programmes aid in transferring the resistance genes in less time with more authenticity in comparison to conventional breeding programmes.
References
Arya L, Verma M, Gupta VK, Karihaloo JL (2009) Development of EST SSRs in finger millet (Eleusine coracana ssp coracana) and their transferability to pearl millet (Pennisetum glaucum). J Plant Biochem Biotech 18(1):97–100
Babu BK, Agrawal PK, Pandey D, Jaiswal JP, Kumar A (2014a) Association mapping of agro-morphological characters among the global collection of finger millet genotypes using genomic SSR markers. Mol Biol Rep 41:5287–5297
Babu BK, Agrawal PK, Pandey D, Kumar A (2014b) Comparative genomics and association mapping approaches for opaque2 modifier genes in finger millet accessions using genic, genomic and candidate gene-based simple sequence repeat markers. Mol Breed 34:1261–1279
Babu BK, Pandey D, Agrawal PK, Sood S, Chandrashekara C et al (2014c) Comparative genomics and association mapping approaches for blast resistant genes in finger millet using SSRs. PLoS ONE 9(6):e99182. https://doi.org/10.1371/journal.pone.0099182
Bharathi A (2011) Phenotypic and genotypic diversity of global finger millet (Eleusine coracana (L.) Gaertn.) composite collection. Dissertation, Tamil Nadu Agriculture University, India
Bodmer WF (2011) Human genetics: the molecular challenge. Cold Spring Harb Symp Quant Biol 51:1–13
Bradbury P, Zhang Z, Kroon D, Casstevens T, Ramdoss Y, Buckler E (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Dida MM, Srinivasachary Ramakrishnan S, Bennetzen JL, Gale MD, Devos KM (2007) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114:321–332
Dida MM, Wanyera N, Harrison Dunn MLN, Bennetzen JL, Devos KM (2008) Population structure and diversity in finger millet (Eleusine coracana) germplasm. Trop Plant Biol 1:131–141
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fukuoka S, Okuno K (2001) QTL analysis and mapping of pi21, a recessive gene for field resistance to rice blast in Japanese upland rice. Theor Appl Genet 103:185–190
IBPGR (1985) Descriptors for finger millet. International Board for Plant Genetic Resources Secretariat, Rome, Italy. http://www.bioversityinternational.org. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/417.pdf. Accessed 2 Feb 2017
Jeung JU, Kim BR, Cho YC, Han SS, Moon HP, Lee YT, Jena KK (2007) A novel gene, Pi40(t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice. Theor Appl Genet 115:1163–1177
Kumar A, Sharma N, Panwar P, Gupta AK (2012) Use of SSR, RAPD markers and protein profiles based analysis to differentiate Eleusine coracana genotypes differing in their protein content. Mol Biol Rep 39(4):4949–4960
Kumar A, Yadav S, Panwar P, Gaur VS, Sood S (2015) Identification of anchored simple sequence repeat markers associated with calcium content in finger millet (Eleusine coracana). Proc Natl Acad Sci India Sect B Biol Sci 85(1):311–317
Liu K, Muse M (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Mott R, Talbot CJ, Turri MG, Collins AC, Flint J (2000) From the cover: a method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci USA 97:12649–12654
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326
Nagaraja A, Jagadish PS, Ashok EG, Krishne Gowda KT (2007) Avoidance of finger millet blast by ideal sowing time and assessment of varietal performance under rainfed production situations in Karnataka. J Mycopathol Res 45(2):237–240
Nirgude M, KalyanaBabu B, Shambhavi Y, Singh UM, Upadhyaya HD, Kumar A (2014) Development and molecular characterization of genic molecular markers for grain protein and calcium content in finger millet (Eleusine coracana (L.) Gaertn.). Mol Biol Rep 41(3):1189–1200
Pritchard JK, Wen W (2003) Documentation for the structure software, version 2. Department of Human Genetics, University of Chicago, Chicago. http://pritchardlab.stanford.edu/software/readme_2_1/. Accessed 2 Feb 2017
Ramakrishnan M, Antony Ceasar S, Duraipandiyan V, AlDhabi NA, Ignacimuthu S (2016a) Assessment of genetic diversity, population structure and relationships in Indian and non-Indian genotypes of finger millet (Eleusine coracana (L.) Gaertn) using genomic SSR markers. SpringerPlus 5:120
Ramakrishnan M, Antony Ceasar S, Duraipandiyan V, AlDhabi NA, Ignacimuthu S (2016b) Using molecular markers to assess the genetic diversity and population structure of finger millet (Eleusine coracana (L.) Gaertn.) from various geographical regions. Genet Resour Crop Evol 63(2):361–376
Rao ANS (1990) Estimates of losses in finger millet (Eleusine coracana) due to blast disease (Pyricularia grisea). J Agric Sci 24:57–60
Reddy INBL, Narasu ML, Sivaramakrishnan S (2012) Identification and characterization of EST-SSRs in finger millet (Eleusine coracana (L.) Gaertn.). J Crop Sci Biotechnol 15(1):9–16
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Roy S, Banerjee A, Mawkhlieng B, Misra AK, Pattanayak A, Harish GD, Singh SK, Ngachan SV, Bansal KC (2015) Genetic diversity and population structure in aromatic and quality rice (Oryza sativa L.) landraces from north-eastern India. PLoS ONE 10(6):e0129607. https://doi.org/10.1371/journal.pone.0129607
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Udupa SM, Baum M (2001) High mutation rate and mutational bias at (TAA)n microsatellite loci of chickpea (Cicer arietinum L.). Mol Genet Genomics 265:1097–1103
Upadhyaya HD, Gowda CLL, Pundir RPS, Reddy VG, Singh S (2006) Development of core subset of finger millet germplasm using geographical origin and data on 14 quantitative traits. Genet Resour Crop Evol 53:679–685
Acknowledgements
The work was supported by the Institute grant received from Indian Council of Agricultural Research, New Delhi, India. Authors are thankful to Mr. G.S. Bisht, technical officer, ICAR-VPKAS, Almora for assistance in phenotypic data generation.
Funding
The Study was funded by Indian Council of Agricultural Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Babu, B.K., Sood, S., Chandrashekara, C. et al. Mapping quantitative trait loci for important agronomic traits in finger millet (Eleusine coracana) mini core collection with genomic and genic SSR markers. J. Plant Biochem. Biotechnol. 27, 401–414 (2018). https://doi.org/10.1007/s13562-018-0449-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-018-0449-7