
Abstract
Late wilt disease (LWD) caused by Harpophora maydis (Samra, Sabet and Hing) is emerging as major production constraint in maize across the world. As a prelude to develop maize hybrid resistance to LWD, genetic basis of resistance was investigated. Two F2:3 mapping populations (derived from CV156670 × 414-33 (P-1) and CV156670 × CV143587 (P-2)) were challenged with LWD at two locations (Kallinayakanahalli and Muppadighatta) during 2017 post-rainy season. A wider range of LWD scores was observed at both locations in both the populations. LWD response was influenced by significant genotype × location interaction. Six and 56 F2:3 progeny families showed resistance level better than resistant parent. A total of 150 and 199 polymorphic single nucleotide polymorphism markers were used to genotype P-1 and P-2, respectively. Inclusive composite interval mapping was performed to detect significant Quantitative Trait Loci (QTL), QTL × QTL, QTL × location interaction effects. Three major and four minor QTL controlling LWD resistance were detected on chromosome-1. The position and effect of the QTL varied with the location. Significant di-QTL interactions involving QTL (with significant and/or non-significant effects) located within and between all the ten chromosomes were detected. Five of the seven detected QTL showed significant QTL × location interaction. Though two major QTL (q-lw-1.5 and q-lw-1.6) with lower Q×L interaction effects could be considered as stable, their phenotypic variance is not large enough to deploy them in Marker Assisted Selection (MAS). However, these QTL are of paramount importance in accumulating positive alleles for LWD resistance breeding.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Maize productivity in India is constrained by several prevalent and emerging insect pests and diseases. Of these, post-flowering stalk rot (PFSR) is a complex disease caused by three pathogens, namely, Fusarium moniliforme J. (Sheld), Macrophomina phaseolina (Tassi) Goid and Harpophora maydis (Samra, Sabet and Hing) (Khokhar et al. 2014). Stalk rot caused exclusively by seed-borne and soil-borne pathogen H. maydis causes premature wilting symptoms at post-flowering stage, a condition known as late wilt disease (LWD). Harpophora maydis is similar to Harpophora anamorphic states of Gaeumannomyces species in culture. Harpophora spp. mycelia are characterized olivaceous brown with radiating hyphae, and cylindrical conidia with curved borne in slimy heads (Gams, 2000). Older hyphae are heavily pigmented, younger hyphae are nearly hyaline and phialides are intermediate in pigmentation (Gams, 2000; Saleh and Leslie, 2004). Typical symptoms of LWD include pre-mature wilting of leaves, discoloration of the stalk followed by stalk tissue disintegration and fibrousness in advanced stages (Samra et al. 1963; Ramana et al. 1997; Johal et al. 2004). Upon severe infection, pathogen colonizes in the kernels causing seed rot and early stage damping-off (El-Shafey and Claflin 1999). Although it is a weakly competitive saprophyte, the production of sclerotia in infested host debris ensures its long-term survival (Sabet et al. 1970; Johal et al. 2004). It is also known to infest cotton, lupine, green foxtail and watermelon in addition to corn (Dor and Degani, 2019). The Egyptian, Indian and Hungarian isolates have been reported to differ in their morphology, pathogenicity and route of infection (Warren 1983; Degani et al. 2019).
LWD has been reported in more than 10 maize growing countries (Chalkley, 2016), with significant economic losses in Egypt (Sabet et al. 1961), India (Payak et al. 1970; Sunitha et al. 2020), Spain and Portugal (Molinero-Ruiz et al. 2010) and Israel (Drori et al. 2013; Degani et al. 2019). It is opined that dry environments like India, Egypt and Iberian peninsula maize growing regions are more prone to late wilt infection (Ortiz-Bustos et al. 2019) in the context of increased temperatures due to climate change. Based on survey, economic losses of 40% have been reported in Egypt (Labib et al. 1975; Galal et al. 1979), up to 100% in Israel (Degani et al. 2019) and up to 51% in India (Johal et al. 2004) due to LWD. However, 3.5 to 38.4% loss of grain yield attributable to soil inoculation by H. maydis has been reported based on an empirical study using a limited number of genotypes in Egypt (El-Naggarr et al. 2015). Very recently, we have reported 5.8 to 44.2% grain yield losses attributable to LWD by creating artificial epiphytotic condition through injecting H. maydis inoculum into the stem internode (Sunitha et al. 2020) in India. It is considered as an endemic disease in maize growing areas (Degani and Cercina, 2014). Despite increasing reports from Israel and Egypt in the last 10 years, there are only a few reports on the incidence of late wilt disease in India. It was identified as threat at Hyderabad, Pantnagar and Rajasthan (Payak et al. 1970; Singh and Siradhana 1986); Johal et al. (2004) reported 70% incidence and economic losses up to 51% in India. Even though this pathogen was identified 30 years back in India, further reports on its incidence and damage were very limited. However, several maize researchers have reported its potential of causing endemic from last one decade (Shekhar et al. 2010; Biradar et al. 2020).
Genetic intervention is regarded as eco-friendly and cost-effective option to mitigate losses caused by LWD (El-Shafey et al. 1988; Zeller et al. 2000). As a prelude to genetic intervention, stable sources of resistance to H. maydis (Satyanarayana 1995; Shekhar et al. 2010; Rakesh et al. 2016b; Aruna 2017; Biradar et al. 2020; Kamara et al. 2021) have been identified. The inheritance of resistance to LWD has been reported as complex with significant genotype × environment interaction (Shekhar et al. 2010; Rakesh 2016; Aruna 2017; Biradar 2019). Hence, direct selection for LWD resistance is likely to be less effective. Nevertheless, DNA markers could be used as effective surrogates of such traits in maize for which a priori identification and validation of closely linked markers are essential. As a step towards this, Quantitative Trait Loci (QTL) conferring LWD tolerance (Rakesh 2016) on chromosomes 1, 3, 5, 7 and 10 have been detected. The objectives of our study were to (i) validate the QTL detected by Rakesh (2016), (ii) to detect and estimate size effects of new LWD resistance governing QTL, if any in two different genetic backgrounds, and (iii) detect and estimate size effects of their interaction with two spatial environments.
Material and methods
Basic genetic material and development of mapping populations
The basic genetic material consisting of one LWD-resistant inbred line, CV156670 and two LWD susceptible inbred lines, 414-33 and CV143587, developed and being maintained at Bayer Crop Science (erstwhile Monsanto India Ltd.) were used as parents to develop two connected mapping populations. For intellectual property protection reasons, the pedigree of these inbreds is not included. The two F1’s (available with Bayer Crop Science), CV156670 × 414-33 and CV156670 × CV143587 were selfed during 2017 summer season to derive F2 populations. Two F2 populations derived from connected crosses CV156670 × 414-33 and CV156670 × CV143587 were selfed during 2017 rainy season. These F2:3 populations (300 progeny families each) constituted inter-connected mapping populations to detect QTL controlling LWD resistance.
Phenotyping mapping populations for responses to LWD
Field experimentation
The mapping populations were evaluated at two locations, Kallinayakanahalli (KNH) 13.464013°N, 77.519178°E and Muppadighatta (MPG) 13.241161°N, 77.483637°E. 300 F2:3 progeny families each from two F2 populations, parental inbred lines and checks (susceptible and resistant check) were sown in single rows of 4-m length with 0.6-m spacing between rows and 0.2-m spacing between plants in two replications following randomized complete block design (RCBD) during 2017 post-rainy season. Recommended package of practices like fertilizer (N and P) application in intervals, earthing-up at knee-height stage and regular weeding were followed to establish good crop stand till artificial inoculation. The F2:3 progeny families were screened for responses to LWD using artificial inoculation. Mass multiplication and inoculum preparation has been followed as per the protocol given by Rakesh et al. (2016a).
Artificial inoculation of H. maydis inoculum
Isolation and mass multiplication of H. maydis were done as per Shekhar and Kumar (2012) modified by Rakesh et al. (2016a). Naturally infested LWD stalk specimens collected from commercial maize production fields were used for isolation of pathogens and inoculum preparation. Pure cultured isolate characteristics were compared with reported morphology and fruiting body characteristics of H. maydis (Gams, 2000). Similarity with spore characters reported by Rakesh (2016), Aruna (2017) and Biradar et al. (2020) was considered. Pathogenicity test based on Koch’s postulates were proved to confirm the pathogen used for inoculation. The inoculum concentration was assessed using hemocytometer and was adjusted to 4×106 spore ml−1. A hole was made at the second internode on the stem of plants using stem jabbers, and 2 ml of the H. maydis was injected to the holes using a syringe twice at 45 and 55 days after sowing (DAS). From each progeny family, 20 plants were inoculated. All the recommended production practices were followed except for the application of potassium fertilisers and fungicides to maintain the plants after inoculation.
Sampling and data recording
The stalks of the artificially inoculated plants were split open at 110 DAS to examine the typical symptoms of LWD. Disease symptoms were manifested by discoloration, disintegration and fibrousness at the inoculated internode. Based on the coverage of disease symptom within an internode and its spread across internodes, the data on LWD severity was recorded on all the inoculated plants using modified 1–9 scale given in Supplementary table 1 (Rakesh et al. 2016a).
Genotyping F 2:3 families
Five F2:3 seeds from each plant of two F2 populations were pooled and ground to fine powder using liquid nitrogen. DNA isolated from fine powder using CTAB method (Khairallah and Hoisington, 1994) and genotyped using Monsanto proprietary-single nucleotide polymorphism (SNP) markers (Supplementary table 2 and 3) through TaqMan assay (Semagn et al. 2015). A total of 150 and 199 SNP markers polymorphic between the parents of the two mapping populations were selected from proprietary database (unpublished) for genotyping the two mapping populations derived from CV156670 × 414-33 and CV156670 × CV143587, respectively.
Statistical analysis
LWD responses
LWD responses from 20 plants of each F2:3 progeny family were recorded (Supplementary figure 1). Data were cured based on missing data points, outliers and correlation between replicates (Mailman et al. 2007; Rakesh 2016). Following data curation, mean LWD scores of 236 and 187 F2:3 progeny families out of 300 families of P-1 and P-2, respectively were used for statistical analysis. Analysis of variance (ANOVA) of F2:3 progeny families was performed to partition the total variation in LWD response scores into those attributable to F2:3 progeny families and within families. Additionally, pooled ANOVA was performed to test for the consistency of the F2:3 progeny families for LWD response across two locations and to quantify the contribution of location and genotype × location interaction. Genotypic data of markers with >15% missing data and markers showing significant segregation distortion were not considered for statistical analysis.
Detection of QTL controlling resistance to LWD
Markers showing segregation distortion were removed (Supplementary table 4 and 5). Information on 115 out of 150 and 137 out of 199 markers were used in P-1 and P-2 respectively after data curation. The genotypic and phenotypic data of F2:3 progeny families evaluated in two locations were integrated to detect QTL controlling LWD resistance, initially using single marker analysis. Subsequently, Inclusive Composite Interval Mapping (ICIM) was performed to detect and estimate size effects of QTL and QTL × QTL interactions (Li et al. 2015) controlling LWD resistance at individual locations and across locations (pooling data from both locations). The accuracy of QTL position and significance of size effect of QTL and QTL × location interaction conferring LWD resistance was determined using data-driven estimates of threshold LOD scores obtained by 1000 permutations (Churchill and Doerge 1994). Similarly, significant QTL × QTL interactions controlling LWD resistance were detected at threshold LOD value of 5.0, and their size effects were estimated. All these statistical analyses were implemented using QTL ICiMapping software version 4.0 (Wang et al. 2016). The detected di-QTL interactions associated with resistance to LWD, dominance × dominance di-QTL interactions were interpreted based on theoretical investigations of Kearsey and Pooni (1996).
Results
LWD responses of F 2:3 mapping populations
The LWD response scores of the F2:3 progeny families derived from CV156670 × 414-33 (P-1) and CV156670 × CV143587 (P-2) (Supplementary figure 1) recorded at both KNH and MPG locations were normally distributed (Fig. 1a–d). While LWD response scores of F2:3 progeny families ranged from 3.17 to 9.00 and from 3.13 to 8.11 at KNH and MPG, respectively in P-1 (Fig. 2a), the responses ranged from 2.38 to 8.38 and from 3.12 to 8.64 at KNH and MPG, respectively in P-2 (Fig. 2b). A wider range of LWD scores at both locations in both the populations indicated sufficient LWD expression. Further, there were considerable numbers of plants towards both resistant and susceptible parents (Fig. 2). Six progeny families including two at KNH and four at MPG in P-1; 56 progeny families including 37 at KNH and 19 at MPG in P-2 showed resistance level better than resistant parent. Similarly, six progeny families including four at KNH and two at MPG in P-1; five progeny families including two at KNH and three at MPG in P-2 showed higher susceptibility than susceptible parent. The F2:3 families derived from each of the two mapping populations differed significantly across both the locations except P-1 at MPG (Table 1). Further, LWD responses of F2:3 families of both populations interacted significantly with location as evident from significance of genotype× location (G×L) interaction mean squares in pooled ANOVA (Table 2). While mean squares attributable to location was non-significant, those attributable to genotype × location were significant in P-2. This trend was reversed in P-1 (Table 2).

Frequency distribution for responses to LWD of F2:3 populations: a P-1 at KNH (Wilk’s statistic = 0.988; P = 0.087). b P-1 at MPG (Wilk’s statistic = 0.993; P = 0.563). c P-2 at KNH (Wilk’s statistic = 0.987; P = 0.177). d P-2 at MPG (Wilk’s statistic = 0.984; P = 0.069)

Patterns of expression of responses to LWD by two F2:3 populations (a P-1 and b P-2) across two locations
Detection of QTL controlling LWD resistance
QTL main effects
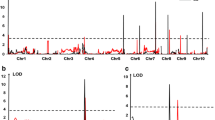
Seven QTL, i.e. two (q-lw-1.1 and q-lw-1.2) in P-1 (Fig. 3) and five (q-lw-1.3, q-lw-1.4, q-lw-1.5, q-lw-1.6 and q-lw-1.7) in P-2 detected on chromosome-1 (Fig. 4) were found associated with LWD resistance with phenotypic variation explained (PVE) ranging from 4.34 to 13.06% (Table 3). Among these, three major QTL (q-lw-1.4 and q-lw-1.6 at MPG; q-lw-1.5 at KNH) with > 10% PVE were detected in P-2. Among the remaining four minor QTL, two (q-lw-1.1 and q-lw-1.2) were from P-1, and other two (q-lw-1.3 and q-lw-1.7) were from P-2. Further, while five (q-lw-1.1, q-lw-1.3, q-lw-1.5, q-lw-1.6 and q-lw-1.7) of these seven detected QTL showed dominance effects, one (q-lw-1.4) showed additive effects in desirable direction (decreasing effects) for LWD resistance (Table 3).

Genome-wide detection of QTL and their effects controlling LWD response mapped in F2:3 of CV156670 (R) × 414-33(S) (P-1) at (a) Kallinayakanahalli and (b) Muppadighatta

Genome-wide detection of QTL controlling LWD response and corresponding additive and dominance effects mapped in F2:3 of CV156670(R) × CV143587(S) (P-2) at (a) Kallinayakanahalli and (b) Muppadighatta
QTL × location interaction
Pooled analysis across locations indicated significant QTL × location (Q×L) interaction. Of the seven detected QTL, five (q-lw-1.2, q-lw-1.4, q-lw-1.5, q-lw-1.6 and q-lw-1.7) interacted significantly with location with their size effects ranging from 0.04 to 6.80% PVE (Table 3). Of these five interacting QTL, only q-lw-1.7 was not detected when individual location-wise analysis was performed. This means that, remaining four (q-lw-1.2, q-lw-1.4, q-lw-1.5 and q-lw-1.6) of these five QTL detected using pooled analysis were detected only in either of the locations but not in both the locations. These four QTL were also stable for their position but unstable for their effects (decreased size effects) as evident from pooled analysis.
Of the five QTL which showed significant Q × L interaction, one QTL, q-lw-1.4 (6.80% PVE Q × L), interacted with locations to a greater extent than the other four QTL (Fig. 5). None of the QTL detected through location-wise analysis were common in both locations. Two QTL (q-lw-1.3 and q-lw-1.5) flanked by same pair of markers, namely, Marker-151 and Marker-152 (with inter-marker distance of 23.8 cM) were detected in P-2. While the QTL, q-lw-1.3 was mapped at 11.40 cM in KNH (in location-wise analysis), q-lw-1.5 was mapped at 9.40 cM in MPG (in location-wise as well as pooled analysis). Further, their position and effects differed (Table 3).

Genome-wide detection of QTL controlling LWD response and QTL × location interactions in F2:3 of (a) CV156670(R) × 414-33(S)/P-1 and (b) CV156670(R) × CV143587(S)/P-2
Di-QTL epistasis
A total of 48 significant di-QTL interactions with size effects (in % PVE) ranging from 4.00 to 19.03% were detected across both locations in P-1. Of these, ten were between QTL located within the same chromosome (Fig. 6a). Only nine of these 48 interactions involved QTL with significant effects. Similarly, 52 significant di-QTL epistatic interactions including ten within same chromosome were detected in P-2 with their size effects (in % PVE) ranging from 3.18 to 13.83% (Fig. 6b). Only four of these 52 significant di-QTL interactions involved QTL with significant effects. Summation of direction and effects of dominance (h) and dominance × dominance (l) effects associated with all di-QTL interaction effects indicated duplicate di-QTL epistasis in both the populations. Duplicate di-QTL epistasis was in desirable direction (dominant decreasing effects) at KNH and MPG in P-1 and P-2, respectively (Table 4).

Genome-wide detection of significant di-QTL interactions controlling LWD response mapped in F2:3 of (a) CV156670(R) × 414-33(S)/P-1 (b) CV156670(R) × CV143587(S)/P-2
Validation of reported QTL conferring resistance to LWD
Rakesh (2016) reported QTL on chromosome-1 at 103.20 cM (IMD- 10.6 cM), flanked by MONIND14101828 and MONIND14262087. This QTL explained only 7.26% of LWD resistance in mapping population derived by CV138811 × CV143587. We identified one of these markers namely, Marker-161 (MONIND14101828) flanking the QTL, q-lw-1.6 could be validated in P-2 (CV156670 × CV143587). However, this QTL flanked by Marker-160 and Marker-161 was detected at position (83.40 cM) and phenotypic variation (13.06% PVE at MPG and 4.35% PVE across locations) different from that reported by Rakesh (2016) (Table 3).
Discussion
LWD is emerging as a major and serious biotic production constraint with a potential to reduce yield and quality of both grain and fodder (Sunitha et al. 2020; El-Naggarr et al. 2015; Drori et al. 2013; Johal et al. 2004). Developing LWD-resistant cultivars is considered as an eco-friendly and sustainable approach to mitigate losses caused by LWD. Precise information on the number and mode of action of genes/QTL controlling resistance to LWD help devise suitable strategies to develop resistant cultivars. The results of our study indicate possible involvement of a large number of genes controlling resistance to LWD as evident from the normal distribution of LWD responses in F2:3 populations. Several previous work by Shehata (1976), Nawar and Salem (1985), Abdel-Snbour and Bekhit (1993) and El-Hosary and El-Fiki (2015) have also reported the involvement of a large number of genes controlling LWD resistance in maize. Further, recovery of individuals that surpass the levels of resistant and susceptible parents indicate (i) presence of alleles controlling resistance even in susceptible parent and those controlling susceptibility even in resistant parent and (ii) complementation of parental alleles controlling resistance. Thus, F2:3 populations serve as potential source for recovering inbred lines with levels of resistance to LWD better than the resistant parent.
Most of the reports (Wu et al. 2020; Zhang et al. 2012; Yang et al. 2010; Suneetha 2016) related to mapping QTL controlling resistance to stalk rot are predominantly on stalk rot caused by Fusarium sp. followed by Macrophomina phaseolina but not on the stalk rot caused exclusively by Harpophora maydis (Late wilt disease). This could be because of co-existence of Harpophora with Fusarium sp. and/or Macrophomina sp. which form a pathogen complex causing PFSR (Degani et al. 2020; Khokhar et al. 2014). Additionally, soil-borne Fusarium sp. and/or Macrophomina sp. are known to invade the H. maydis infested stalks (Drori et al. 2013). This causes difficulty in phenotyping the symptoms caused exclusively by H. maydis. To address the issue of possible confounding effects of pathogens other than H. maydis, we used Indian pure isolates of H. maydis to inoculate the spores directly into the second internode of the stem (Rakesh et al. 2016a, b) using stem jabbers (specialized injectors) twice at 45 DAS and 55DAS (Sunitha et al. 2020).
LWD resistance conferring QTL
Difficulty arising due to complexity of the disease and phenotyping could be the potential causes for sporadic reports on genetic basis of LWD resistance. The only report (Rakesh 2016) on mapping genomic regions controlling LWD resistance in two F2:3 populations (derived using CV138811 and CV143587 as donor and susceptible parents, respectively) resulted in the identification of three major QTL on chromosomes 3, 5 and 10; and six minor QTL on chromosomes 1, 2, 3, 5, 6 and 7. QTL analysis is not only intended to implement Marker Assisted Selection (MAS) for QTL but also for understanding the genetics of the quantitative trait. Hence, all the identified QTL, whether their effects are large or small and with or without environmental sensitivity are informative (Asins 2002). We could identify three major QTL and four minor QTL. In most of the studies, QTL with small effects go undetected due to bias caused by less-stringent threshold levels. To minimize such bias, we used LOD threshold derived from 1000 permutations (Churchill and Doerge 1994).
QTL×location interaction
The position and effect of the QTL associated with LWD resistance varied significantly with the spatial environments represented by two locations. If a quantitative trait exhibits significant genotype by environment interaction, it then follows that underlying QTL should also display significant interaction with either temporal or spatial environments (Bernardo 2020). F3 progenies of both the crosses displayed significant interaction with the environment represented by two locations for responses to LWD (Table 2). Hence, detected significant interaction of LWD resistance governing QTL with location environments did not surprise us. Several researchers have also reported significant QTL × location interaction associated with resistance response of maize diseases such as Fusarium ear rot (Wu et al. 2020; Robertson-Hoyt et al. 2006), northern leaf bight (Xia et al. 2020; Chen et al. 2016) and rough dwarf disease (Wang et al. 2019). The major QTL (>10% PVE) detected through location-wise mapping behaved as minor QTL (<10% PVE) when detected through pooled analysis. We could also observe shift in the position of QTL (q-lw-1.3 and q-lw-1.5) within the same flanking marker interval in different locations. However, such QTL despite showing significant G × E interaction help select genotypes adapted to specific locations (Asins 2002). As expected based on previous reports by Shekhar et al. (2010), we could also observe significant influence of location and genotype × location interaction on the expression of LWD. Significance of G × E interaction for a trait reflects that underlying genomic regions exhibit significant QTL × Location interaction (Bernardo 2020). Among the three major QTL identified by us, although two QTL (q-lw-1.5 and q-lw-1.6) showed significant Q×L interaction, the magnitude of interaction effects was relatively low. These QTL with lower Q×L interaction effects could be considered as stable and deployed to implement MAS for LWD resistance after validation. This is because, the efficiency of MAS may get reduced if detected QTL controlling target trait exhibits significant interaction with either the environment or background genotype used (Asins 2002).
QTL×QTL interaction
The chances of recovering recombinant inbred lines (RILs) with dominant di-QTL interactions are fewer with any population other than F2. However, even a large F2 mapping population may contain fewer two-QTL RILs limiting the statistical power of detecting di-QTL interactions (Bernardo, 2020; Pandey et al. 2006). Hence, there are chances that epistatic interactions go undetected. We addressed this limitation by increasing the LOD significance of >5.0 and considering phenotypic data from replicated trials from F2:3. Some of the di-QTL interactions which involve QTL with their individual effects being non-significant are also informative, provided their di-QTL interaction effects are of higher magnitude. Under such instance, deployment of QTL alone in MAS without considering its significant di-QTL interaction would be ineffective. Hence, such QTL showing significant epistatic effects of large magnitude need to be introgressed together into recipient genetic background. However, there are chances that the magnitude of the significant di-QTL interaction may get reduced or get enhanced in recipient parent genetic background. The effects of such interacting QTL in recipient genetic background will be known after introgression. The direction of cumulative dominance effects associated with duplicate di-QTL epistasis varied with location. The duplicate di-QTL epistasis with decreasing dominance effects on LWD response is desirable. However, these di-QTL interaction effects need to be confirmed through extensive multi-location evaluation.
QTL genetic background interaction
Genetic background has considerable influence on the position and effect of QTL conferring disease resistance (Asins 2002; Awata et al. 2020). The same was evident in our study, and none of the QTL was common between both populations, P-1 and P-2 (which differed for susceptible parent genome). However, we could validate only one of the reported markers, MONIND14101828 (Marker-161), flanking the reported QTL (Rakesh 2016) in P-2. While the QTL region having this marker, MONIND14101828 (Marker-161), in common is flanked by an inter-marker distance of 11.8 cM in the present study, it is flanked by an inter-marker distance of 10.6 cM in a previous report (Rakesh 2016). Thus, this region needs fine mapping to identify QTL with a narrow inter-marker distance for efficient deployment in MAS.
In summary, three major QTL, q-lw-1.4, q-lw-1.5 and q-lw-1.6, were identified from our study. However, PVE% explained by these three QTL is not large enough for use in MAS, but these are of paramount importance in accumulating positive alleles for LWD resistance breeding. Further, only one of these QTL, q-lw-1.6, could be validated. The three major QTL exhibited significant interaction with locations. Considering that the validated QTL reported by us and those reported by previous researcher exhibit significant interactions with locations, further research investigation is essential to identify stable QTL with large PVE for implementing MAS.
References
Abdel-Snbour MS, Bekhit MMM (1993) Genetic variability and diallel analysis of late wilt resistance and some other characters in maize. Zagazig J Agril Res 20(5):61–73
Aruna K (2017) Identification of High Yielding and Late Wilt Resistant Inbred Lines of Maize (Zea Mays L.) (Doctoral dissertation, UNIVERSITY OF AGRICULTURAL SCIENCES GKVK, BENGALURU)
Asins MJ (2002) Present and future of quantitative trait locus analysis in plant breeding. Plant Breed 121(4):281–291. https://doi.org/10.1046/j.1439-0523.2002.730285.x
Awata LA, Beyene Y, Gowda M, Suresh LM, Jumbo MB, Tongoona P, Danquah E, Ifie BE, Marchelo-Dragga PW, Olsen M, Ogugo V, Mugo S, Prasanna BM (2020) Genetic analysis of QTL for resistance to maize lethal necrosis in multiple mapping populations. Genes 11(1):32. https://doi.org/10.3390/genes11010032
Bernardo R (2020) Breeding for quantitative traits in plants, 3rd edn. Stemma press, Woodbury
Biradar M (2019) Identification of high yielding and late wilt (Harpophora Maydis) disease resistant hybrids of maize (Zea Mays L.) (Doctoral dissertation, UNIVERSITY OF AGRICULTURAL SCIENCES GKVK, BANGALORE).
Biradar M, Gangappa E, Ramesh S, Sowjanya PR, Sunitha N, Parveen G, Sowmya H, Suma K (2020) Association between GCA and per se performance of parents and hybrids for grain yield, its attributing traits and late wilt disease (Harpophora maydis) resistance in maize (Zea mays L.). Int J Curr Microbiol Appl Sci 9(3):2560–70
Chalkley D (2016) Invasive fungi: late wilt of maize Harpophora maydis. Systematic Mycology and Microbiology Laboratory, Agricultural Research Service, United States Department of Agriculture, pp 1 – 6
Chen G, Wang X, Long S, Jaqueth J, Li B, Yan J, Ding J (2016) Mapping of QTL conferring resistance to northern corn leaf blight using high-density SNPs in maize. Mol Breeding 36(1):4. https://doi.org/10.1007/s11032-015-0421-3
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971. https://doi.org/10.1093/genetics/138.3.963
Degani O, Dor S, Abraham D, Cohen R (2020) Interactions between Magnaporthiopsis maydis and Macrophomina phaseolina, the causes of wilt diseases in maize and cotton. Microorganisms 8(2):249. https://doi.org/10.3390/microorganisms8020249
Degani O, Dor S, Movshovitz D, Rabinovitz O (2019) Methods for studying Magnaporthiopsis maydis, the maize late wilt causal agent. Agronomy 9(4):181. https://doi.org/10.3390/agronomy9040181
Dor S, Degani O (2019) Uncovering the host range for maize pathogen Magnaporthiopsis maydis. Plants 8(8):259
Drori R, Sharon A, Goldberg D, Rabinovitz O, Levy M, Degani O (2013) Molecular diagnosis for Harpophora maydis, the cause of maize late wilt in Israel. Phytopathol Mediterr 52(1):16–29
El-Hosary AAA, El-Fiki IAI (2015) Diallel cross analysis for earliness, yield, its components and resistance to late wilt in maize. Int J Agric Sci Res 5:199–210
El-naggarr AAA, Sabryr AM, Yassin MA (2015) Impact of late wilt disease caused by Harpophora maydis on maize yield. J Biol Chem Env Sci 10(3):577–595
El-Shafey HA, Claflin LE (1999) Late Wilt. In: White DG (ed) Compendium of corn diseases (3rd). APS Press, St. Paul, pp 43–44
El-Shafey HA, El-Shorbagy FA, Khalil II, El-Assiuty EM (1988) Additional sources of resistance to the late-wilt disease of maize caused by Cephalosporium maydis. Agric Res Rev 66:221–230
Galal AA, El-Rouby MM, Gad AM (1979) Genetic analysis of resistance to late wilt (Cephalosporium maydis) in variety crosses of maize. Zeitschriftfür Planzenzüchtung 83:176–183
Gams W (2000) Phialophora and some similar morphologically little-differentiated anamorphs of divergent ascomycetes. Stud Mycol 45:87–199
Johal L, Huber DM, Martyn R (2004) Late wilt of corn (maize) pathway analysis: intentional introduction of Cephalosporium maydis. In: Pathways Analysis for the Introduction to the U.S. of Plant Pathogens of Economic Importance. U.S. Department of Agriculture, animal and plant health inspection service technical report no. 503025
Kamara MM, Ghazy NA, Mansour E, Elsharkawy MM, Kheir A, Ibrahim KM (2021) Molecular genetic diversity and line× tester analysis for resistance to late wilt disease and grain yield in maize. Agronomy 11(5):898
Kearsey MJ, Pooni HS (1996) The genetical analysis of quantitative traits. Chapman and Hall, New York
Khairallah M, Hoisington D (1994) Laboratory protocols: CIMMYT applied molecular genetics laboratory, 3rd edn. D. F. CIMMYT, Mexico
Khokhar MK, Hooda KS, Sharma SS, Singh V (2014) Post flowering stalk rot complex of maize-present status and future prospects. Maydica 59(3):226–242
Labib HA, Salem A, Abd El Rahim ME, Abd El Fattah A (1975) DC 19, a new maize hybrid seed resistant to late-wilt disease caused by Cephalosporiummaydis. Agric Res Rev 53(8):1–4
Li S, Wang J, Zhang L (2015) Inclusive composite interval mapping of QTL by environment interactions in biparental populations. PLoS ONE 10(7):e0132414. https://doi.org/10.1371/journal.pone.0132414
Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N (2007) The NCBI dbGaP database of genotypes and phenotypes. Nat Genet 39(10):1181–1186. https://doi.org/10.1038/ng1007-1181
Molinero-Ruiz ML, Melero-Vara JM, Mateos A (2010) Cephalosporium maydis, the cause of late wilt in maize, a pathogen new to Portugal and Spain. Plant Dis 94(3):379. https://doi.org/10.1094/PDIS-94-3-0379A
Nawar AA, Salem MA (1985) Diallel analysis of the inheritance of late wilt and leaf blight in maize [Egypt]. Monoufeyah J Agric Res 10(2):719–737
Ortiz-Bustos CM, López-Bernal A, Testi L, Molinero-Ruiz LJ (2019) Environmental and irrigation conditions can mask the effect of Magnaporthiopsis maydis on growth and productivity of maize. Plant Pathol 68(8):1555–1564
Pandey SK, Sarkar DE, Sharma SU (2006) Quantitative Trait Loci (QTLs) for plant disease response. Proc Indian Natl Sci Acad 72(3):179
Payak MM, Lal S, Renfro BL (1970) Cephalosporium maydis—a new threat to Maize in India. Indian Phytopathol 23(3):562–569
Rakesh B (2016) Rakesh B. (2016) Mapping Genomic Regions Controlling Late Wilt Disease Resistance Caused by Harpophora Maydis in Maize (Zea Mays L.) (Doctoral dissertation, UNIVERSITY OF AGRICULTURAL SCIENCES GKVK, BENGALURU)
Rakesh B, Gangappa E, Sonali G, Gowda RV, Swamy SD, Ramesh S, Hemareddy HB, Nagaraju N (2016a) Modified method of screening maize inbred lines to late wilt disease caused by Harpophora maydis. Mysore J Agric Sci 50:684–690
Rakesh B, Gangappa E, Sonali G, Ramesh S, Nagaraju N (2016) Identification of maize (Zea mays L.) inbred lines resistant for late wilt disease caused by Harpophora maydis. Mysore J Agric Sci 50(2):366–368
Ramana VV, Reddy VK, Aeddy SM, Reddy KJ (1997) Production of cellulases, hemicellulases, pectinases, proteinases, and lipases by Cephalosporiummaydis isolated from Zea mays stalks. In: Reddy SM, Srivastava HP, Purchit DK, Ram Reddy S (eds) Microbial Biotechnology. Scientific Publishers, Jodphur, pp 187–192
Robertson-Hoyt LA, Jines MP, Balint-Kurti PJ, Kleinschmidt CE, White DG, Payne GA, Maragos CM, Molnár TL, Holland JB (2006) QTL mapping for Fusarium ear rot and fumonisin contamination resistance in two maize populations. Crop Sci 46(4):1734–1743. https://doi.org/10.2135/cropsci2005.12-0450
Sabet KA, Samra AS, Mansour IM (1970) Saprophytic behavior of Cephalosporium maydis and C. acremonium. Annals of Applied Biology 66:265–271
Sabet KA, Samra AS, Hingorani MK, Mansour IM (1961) Stalk and root rots of Maize in the United Arab Republic. Plant Protection Bulletin, FAO 9(7):121–125
Saleh AA, Leslie JF (2004) Cephalosporium maydis is a distinct species in the Gaeumannomyces-Harpophora species complex. Mycologia 96:1294–1305
Samra AS, Sabet KA, Hingorani MK (1963) Late wilt disease of maize caused by Cephalosporium maydis. Phytopathol 53:402–406
Satyanarayana E (1995) Genetic studies of late wilt and turcicum leaf blight resistance in maize. Madras Agricultural Journal 82:608–609
Semagn K, Beyene Y, Babu R, Nair S, Gowda M, Das B, Tarekegne A, Mugo S, Mahuku G, Worku M, Warburton ML (2015) Quantitative trait loci mapping and molecular breeding for developing stress resilient maize for sub-Saharan Africa. Crop Sci 55(4):1449–1459. https://doi.org/10.2135/cropsci2014.09.0646
Shehata AH (1976) Gene action involved in the manifestation of late wilt (Cephalosporium maydis) of maize. Egyptian J Genet Cytol 5(1):42–47
Shekhar M, Kumar S (2012) Inoculation methods and disease rating scales for maize diseases. ICAR, New Delhi, pp 17–19
Shekhar M, Kumar S, Sharma RC, Singh R (2010) Sources of resistance against post-flowering stalk rots of maize. Arch Phytopathol Plant Protect 43(3):259–263. https://doi.org/10.1080/03235400701803846
Singh SD, Siradhana BS (1986) Germplasm evaluation against Cephalosporium maydis incitant of late wilt of maize. Indian J Mycol Plant Pathol 16:295
Suneetha P (2016) Mapping of gene(s) for resistance to post flowering stalk rot in maize (Zea mays L.) caused by Macrophomina phaseolina (Tassi) Goid (Doctoral dissertation, Professor Jayashankar Telangana State Agricultural University Rajendranagar, Hyderabad)
Sunitha NC, Gangappa E, Gowda RV, Ramesh S, Biradar S, Swamy D, Hemareddy HB (2020) Assessment of impact of late wilt caused by Harpophora maydis (Samra, Sabet and Hing) on grain yield and its attributing traits in maize (Zea mays L.). Mysore J Agric Sci 54(2):30–36
Wang J, Li H, Zhang L, Meng L (2016) User’s manual of QTL IciMapping ver. 4.1.’. Quantitative Genetics group, Institute of Crop Science, Chinese Academy of Agricultural Sciences (CAAS): Beijing/Genetic Resources Program, International Maize and Wheat Improvement Center (CIMMYT): Mexico City
Wang X, Yang Q, Dai Z, Wang Y, Zhang Y, Li B et al (2019) Identification of QTLs for resistance to maize rough dwarf disease using two connected RIL populations in maize. PLoS ONE 14(12):e0226700. https://doi.org/10.1371/journal.pone.0226700
Warren HL (1983) Potential disease problems-late wilt of maize. In Phytopathology (Vol. 73, No.5, pp.782-782). 3340 Pilot Knob Road, ST Paul, MN 55121: Amer Phytopathological Soc
Wu Y, Zhou Z, Dong C, Chen J, Ding J, Zhang X, Mu C, Chen Y, Li X, Li H, Han Y (2020) Linkage mapping and genome-wide association study reveals conservative QTL and candidate genes for Fusarium rot resistance in maize. BMC Genomics 21:357. https://doi.org/10.1186/s12864-020-6733-7
Xia H, Gao W, Qu J, Dai L, Gao Y, Lu S, Zhang M, Wang P, Wang T (2020) Genetic mapping of northern corn leaf blight-resistant quantitative trait loci in maize. Medicine 99(31)31):e21326. https://doi.org/10.1097/MD.0000000000021326
Yang Q, Yin G, Guo Y, Zhang D, Chen S, Xu M (2010) A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 121(4):673–687
Zeller KA, Jurgenson JE, El-Assiuty EM, Leslie JF (2000) Isozyme and amplified fragment length polymorphisms from Cephalosporium maydis in Egypt. Phytoparasitica 28(2):121–130
Zhang D, Liu Y, Guo Y, Yang Q, Ye J, Chen S, Xu M (2012) Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 124(3):585–596
Acknowledgements
Extension of field and lab facilities by Monsanto India Limited under collaborated project with University of Agricultural Sciences, Bengaluru, Karnataka, India is gratefully acknowledged.
Funding
This research was funded by Monsanto India Limited under collaborated project with University of Agricultural Sciences, Bengaluru, Karnataka, India including financial support in the form of ‘Monsanto student fellowship’.
Author information
Authors and Affiliations
Contributions
EG, VRPG and SR conceptualized the work. SNC conducted experiments and analyses. DS and SNC performed inoculation and screening for late wilt disease. SB, SR and SNC interpreted results. SNC and SR revised the manuscript. EG, SR, VRPG and HBH reviewed and edited the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by Izabela Pawłowicz.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sunitha, N.C., Gangappa, E., Gowda, R.P.V. et al. Discovery of genomic regions associated with resistance to late wilt disease caused by Harpophora maydis (Samra, Sabet and Hing) in maize (Zea mays L.). J Appl Genetics 63, 185–197 (2022). https://doi.org/10.1007/s13353-021-00672-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-021-00672-x