
Abstract
Northern corn leaf blight (NCLB) is a prevalent foliar disease in maize. Deployment of resistant cultivars is an effective way to control NCLB. In this study, 207 recombinant inbred lines derived from a K22 × By815 cross were planted in Yangling, China, in 2012 and 2013. NCLB score and lesion size were investigated after artificial inoculation. Significant phenotypic variation in NCLB resistance was observed in both years. Using a genetic map containing high-density single-nucleotide polymorphisms with average genetic distance of 0.74 cM, quantitative trait loci (QTL) for NCLB score and lesion size were analyzed. For NCLB score, four and three QTL were identified in 2012 and 2013, respectively. Two stable QTL were identified in both years. Of these, qNCLB5.04, located on chromosome 5 (bin 5.04), had the largest resistance effect, accounting for 19 and 20 % of the phenotypic variation in 2012 and 2013, respectively. For lesion size, six QTL were identified. Of these, one consensus QTL was associated with both lesion length and width, and the other five were associated only with lesion width. Among all QTL identified, only qNCLB5.04 was associated with both NCLB score and lesion size. Thus, our mapping results suggest that qNCLB5.04 could be a desirable target for marker-assisted selection for NCLB resistance in maize breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Foliar diseases are a significant limiting factor in maize production globally (Pratt and Gordon 2006). Northern corn leaf blight (NCLB), caused by the hemibiotrophic fungal pathogen Setosphaeria turcica (anamorph Exserohilum turcicum, formerly known as Helminthosporium turcicum), is one of the major foliar diseases in maize. NCLB can dramatically reduce maize yield by destroying the photosynthetically active leaf during the grain-filling period (Raymundo and Hooker 1981; Tefferi et al. 1996). Severe epidemics often occur throughout maize-producing regions wherever moderate temperatures and humidity prevail (Balint-Kurti and Johal 2009).
Deployment of resistant cultivars is an effective way to control NCLB. Resistant lines carrying Ht genes (for Helminthosporium turcicum) have been widely used in maize breeding programs (Welz and Geiger 2000). Both dominant genes (Ht1, Ht2, Ht3, Htm1, Htn1, N, and HtP) and recessive genes (ht4 and rt) conferring resistance to specific races of Setosphaeria turcica have been identified (Welz and Geiger 2000; Ogliari et al. 2005), and several Ht genes have been mapped with molecular markers. Ht1 and HtP are located on chromosome 2L and map 10 cM from each other (Bentolila et al. 1991; Ogliari et al. 2007), Ht2 and Htn1 are on chromosome 8L (Zaitlin et al. 1992; Simcox and Bennetzen 1993), and rt is located on chromosome 3L (Ogliari et al. 2007). Recently, Htn1 was cloned using a map-based cloning approach. Htn1 encodes a wall-associated receptor-like kinase that acts as an important component of the plant innate immune system by perceiving pathogen- or host-derived elicitors (Hurni et al. 2015).
Historically, both qualitative and quantitative disease resistances were important for controlling NCLB. Compared with Ht genes, quantitative trait loci (QTL) are now emphasized in maize breeding, because they show durable resistance and are less likely to be overcome by evolution of novel resistant pathogen races (Parlevliet 2002). Various studies have been conducted to map QTL for resistance to NCLB, and these QTL seem to be distributed throughout the genome (reviewed by Welz and Geiger 2000; Wisser et al. 2006; Ali and Yan 2012). Several major QTL have been further fine-mapped, including qNLB1.02 and qNLB1.06 on chromosome 1 (Chung et al. 2010a). Recently, the location of qNLB1.06 was refined to a 3.60-Mb interval, and association mapping in this region implicated candidate genes, including a putative zinc finger protein-encoding gene and pan1 (Jamann et al. 2014). Another major QTL, qNLB8.06 DK888 , has also been fine-mapped and delimited to a 0.46-Mb region on chromosome 8, and three candidate genes in this region were identified (Chung et al. 2010b). In recent years, the nested association mapping (NAM) population has been widely used for dissecting the genetic architecture of agronomic traits, including resistance to foliar diseases. For resistance to NCLB, 29 QTL have been identified. However, no major QTL has been detected, and the large variation in resistance to NCLB has been attributed to the accumulation of many loci with small additive effects (Poland et al. 2011).
Recent advances in high-throughput genotyping technologies allow construction of genetic maps with high-density single-nucleotide polymorphism (SNP) markers, which can significantly improve mapping resolution when compared to earlier QTL linkage maps constructed from low-density markers such as simple sequence repeats (Yan et al. 2010; Chen et al. 2014). In the present study, we evaluated variations in NCLB indices (disease severity and lesion size) in a recombinant inbred line (RIL) population, and conducted QTL mapping using a genetic map constructed with high-density SNPs. The objectives were to determine the number and positions of resistance QTL and to identify consensus QTL for NCLB severity and lesion size that could advance the use of marker-assisted selection in maize breeding programs.
Materials and methods
Plant materials and field design
A mapping population of 207 F6:7 RILs was produced by single-seed descent from a cross between K22 (resistant) and By815 (susceptible). The RILs and their parents were planted at the Yangling Experimental Station (N34°20′, E108°42′), Northwest Agricultural and Forestry University, Shaanxi, China, in 2012 and 2013. The soil type in the experimental station is classified as yellow–brown earth (Shao et al. 2009). The resistant parent K22 is an elite subtropical inbred line derived from the K11 × Ye478 cross and is widely used in maize breeding programs in China (Li et al. 2004). The susceptible parent By815 is a high-oil inbred line developed from a Beinongda high-oil population (Zhou et al. 2009). A randomized complete block design with two replications was used in these studies. K22 and By815 were planted in each block as the resistant and susceptible controls, respectively. Lines were grown in single 3-m rows spaced 0.67 m apart with a planting density of 45,000 plants/ha. Standard agricultural practices were followed throughout the growing season.
Inoculation and disease assessment
Setosphaeria turcica was isolated from diseased plants of susceptible lines collected in previous seasons. To obtain large amounts of inoculum for field inoculation, the spores were cultured on potato dextrose agar medium with subsequent subculturing on sorghum (Sorghum bicolor) kernels at room temperature (25–28 °C) for 2 weeks prior to collection. Experimental plots were inoculated at the V8–V10 growth stages by placing 20–30 pathogen-colonized sorghum grains into the leaf whorl (Carson 1995). Four weeks after inoculation, all plants in the plots were visually scored for NCLB severity using a scale from 1 (most resistant phenotype) to 9 (most susceptible phenotype; Supplemental Fig. S1). In addition, in the 2012 season, five representative plants from the middle of each plot were selected for measurement of NCLB lesion size. For each plant, we selected three leaves (the primary ear leaf and two leaves around the ear); from these, we measured the length and width of a total of four or five randomly selected lesions/plant.
Data analysis
The phenotypic data (NCLB score and lesion size) of the parental lines and RILs were subjected to standard analysis of variance (ANOVA) using the PROC GLM procedure of SAS (SAS Institute 1999). Components of variance were estimated using a complete random effects model, and broad-sense heritability was calculated as defined by Knapp et al. (1985).
Linkage map construction
All parental lines and RILs were genotyped using the MaizeSNP50 BeadChip, an Illumina BeadChip array of 56,110 maize SNPs developed from the B73 reference sequence (Ganal et al. 2011). SNPs showing polymorphism between the parental lines (K22 and By815) were selected to construct the genetic map. All polymorphic markers were arranged according to physical location on the maize AGPv2 reference sequence. Linkage analysis was performed using the est.map function of the R/qtl package (Broman et al. 2003). The fill.geno command and the Viterbi method were used to fill in missing genotyping data. Recombination fractions were converted into map distances in centi-Morgans (cM) using the Kosambi mapping function (Kosambi 1944).
QTL analysis
We first used composite interval mapping (CIM; Zeng 1994) in Windows QTL Cartographer 2.5 for QTL mapping. A genome-wide critical threshold value was set by 1000 random permutations (Churchill and Doerge 1994). For additive effect QTL, positive and negative signs of the estimates indicated that resistance effects were from By815 or K22, respectively. The proportion of phenotypic variation explained by a single QTL was determined by the square of the partial correlation coefficient (R2).
Taking year as an environmental factor, mixed-model composite interval mapping (MCIM) was also adopted for QTL mapping using QTLNetwork 2.0 software (Wang et al. 1999; Yang et al. 2007). MCIM is an alternative method for QTL mapping of complex traits in multiple environments that takes into account the effects of multiple QTL, epistasis, QTL-by-environment (Q × E) interactions, and epistasis-by-environment interactions in the full-QTL model. For comparisons of QTL positions detected by the CIM and MCIM methods, QTL within the same chromosome interval and sharing the same flanking markers were defined as the same QTL.
Results
Evaluations of phenotypic variation
The average performance and the descriptive statistics for NCLB score in the two parents and 207 RILs are given in Table 1. Significant differences in resistance to NCLB were observed between the parents (Fig. 1). K22 was highly resistant in both 2012 and 2013, while By815 was consistently susceptible to NCLB. The average NCLB scores across years for K22 and By815 were 1.0 and 6.82, respectively. Significant differences among RILs were also observed (Supplemental Fig. S2). Genotypic components of variance (σ 2g ) and genotype × environment interaction (σ 2ge ) were significant for the RIL population, and the heritability estimate was high (0.83) across environments, indicating that much of the phenotypic variance in the RIL population is genetically controlled.

Phenotypic variation in NCLB resistance between the parental lines. a Parental line K22 shows resistance to NCLB. b Parental line By815 shows susceptibility to NCLB
Extensive phenotypic variation was observed for lesion length and width in the RILs in 2012 (Supplemental Figs. S3 and S4). A high positive correlation (r 2 = 0.63) was observed between lesion length and width. A significant positive correlation was also observed between lesion size and NCLB score (Supplemental Table S1). However, the correlation coefficient of NCLB score with lesion width was higher (r 2 = 0.49) than that of NCLB score with lesion length (r 2 = 0.27).
Genetic linkage mapping
Of the 56,110 SNPs generated by the MaizeSNP50 BeadChip, 13,603 SNPs showing polymorphism between K22 and By815 were selected to construct the genetic map. The markers were distributed along all 10 chromosomes with no obvious gaps. Some markers were located within the same block without recombination with adjacent markers. In such cases, one marker in each block was randomly selected. This resulted in a genetic map with 2263 markers spanning a total length of 1670.4 cM, with an average distance of 0.74 cM between markers.
QTL mapping of resistance to NCLB
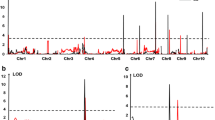
The QTL for NCLB score identified by CIM are shown in Table 2. In 2012, four additive QTL were detected after 1000 permutation tests. The most significant QTL, qNCLB5.04, was detected on chromosome 5 (bin 5.04) with a LOD score of 11.09. Other loci associated with NCLB score included qNCLB1.03, qNCLB3.08, and qNCLB7.05. Each of the four QTL identified in 2012 could explain 5–19 % of the total phenotypic variation. Combined, the four QTL could explain 41 % of the total phenotypic variation. All resistance alleles were from the resistant parent, K22, except qNCLB1.03, which was contributed by the susceptible parent, By815.
Three QTL were detected for NCLB score in 2013, including two (qNCLB5.04 and qNCLB1.03) that were identified in 2012. The third specific QTL (qNCLB9.03) detected was located on chromosome 9. Among the three QTL detected in 2013, qNCLB5.04 had the largest effect, explaining 20 % of the total phenotypic variation. Combined, the three QTL could explain 35 % of the total phenotypic variation.
Taking year as an environmental factor, epistasis and QTL-by-environment (Q × E) interactions of resistance to NCLB scores were further analyzed across environments by MCIM. Significant Q × E interactions were detected for qNCLB5.04 at the P = 0.05 level (Table 3). However, the effect of Q × E interactions could only explain 0.3 % of the total phenotypic variation. No significant effects of epistasis or epistasis-by-environment interactions were identified.
In total, six QTL related to NCLB lesion size were also identified by CIM in 2012 (Table 4). Only one of these QTL, located on chromosome 10 (bin 10.03), was associated with both lesion length and width. The other five QTL were specifically associated with lesion width and were located on chromosomes 5, 6, 7, and 8. Of the six QTL for lesion size, three (qNCLB5.04, qNCLB6.00, and qNCLB7.03) had resistance effects derived from K22, and the other three (qNCLB6.05, qNCLB8.06, and qNCLB10.03) had resistance effects derived from By815. Interestingly, qNCLB5.04, with the largest effect for NCLB score, was also associated with lesion width.
Discussion
Reliable phenotypic evaluation is a prerequisite for QTL mapping. In previous reports, disease severity has typically been used to evaluate phenotypic variation in resistance to NCLB. In the present study, both NCLB severity and lesion size were examined. The initial motivation to investigate lesion size was that it provides a continuous variation, unlike the categorical variation in NCLB score. Lesion size can serve as an alternate NCLB index and is a more objective measure of resistance variation. However, because measuring lesion size is extremely time-consuming, it was only assessed in 2012 with two replications. ANOVA showed significant positive correlations between replications and between lesion size and NCLB score (Supplemental Table S1). As expected, the consensus QTL, qNCLB5.04, was found to be associated with both lesion size and NCLB score.
In this study, QTL mapping was conducted for parameters by the CIM method. In total, five and six QTL were identified for NCLB score and lesion size, respectively, and considerable differences were observed in both the positions and effects of these QTL. The NCLB severity is a composite index determined by at least seven components, including lesion size (infection efficiency of inoculum dose, incubation period, latency period, lesion size, lesion expansion rate, lesion number, and spore sporulation rate of a given area of lesion) (Brewster et al. 1992). Furthermore, lesion size is highly correlated with inoculum dose, incubation period, latency period, and lesion expansion rate. Nonetheless, only one consensus QTL was associated with both NCLB score and lesion size in our analysis, and it showed the largest effect for NCLB score. Therefore, qNCLB5.04 can be viewed as a desirable target QTL for further fine-mapping and map-based cloning to elucidate the mechanisms of resistance to NCLB.
Of the six QTL for lesion size, only one was common for both lesion length and lesion width, and the remaining five QTL were specific for lesion width (Table 4), which implies different genetic mechanisms in controlling lesion length and width in the RIL population. We also compared the parental source of the six QTL for lesion size and found that three QTL (qNCLB5.04, qNCLB6.00, and qNCLB7.03) were from K22, while the other three QTL (qNCLB6.05, qNCLB8.06, and qNCLB10.03) were from By815. However, qNCLB5.04, which was associated with both NCLB score and lesion width, had the same resistance effects from K22.
In the present study, both CIM and MCIM were used for QTL mapping. CIM is a popular method focusing on the evaluation of additive effects in RIL populations (Zeng 1994). To examine whether there are genetic effects other than additive (i.e., additive-by-environment interactions, epistasis, and epistasis-by-environment interactions), we used MCIM, an alternative method for QTL mapping of complex traits in multiple environments (Wang et al. 1999; Yang et al. 2007). All QTL identified by CIM were also detected with MCIM. The QTL identified by both methods shared the same QTL intervals, but, in general, the QTL identified by MCIM showed lower additive effects. The reason may lie in differences in model selected for QTL mapping. More genetic effects were considered in MCIM, whereas only additive effects were analyzed in CIM. In fact, we identified Q × E interactions with MCIM. For example, qNCLB5.04 showed significant additive-by-environment interaction, although the effect on phenotypic variation was small (0.3 %, Table 3).
Numerous NCLB resistance QTL have been reported in maize, and they cover 89 % of the maize genome (Wisser et al. 2006). However, the high percentage of genome coverage partly reflects the relatively low resolution of QTL mapping. In the present study, the resolution was significantly improved by using high-density markers, and the improved resolution can facilitate comparison of our results with disease-associated QTL and genes reported previously. For example, the stable consensus QTL (qNCLB5.04) was associated with resistance to NCLB score and lesion width and was located on chromosome 5 (bin 5.04). In this region, Poland et al. (2011) detected four QTL for resistance to NCLB in the NAM population, and subsequent association mapping identified one candidate gene (GRMZM2G024612) within the QTL interval. GRMZM2G024612 is located near qNCLB5.04 (∼0.69-Mb upstream of the confidence interval of qNCLB5.04). The other important genomic region for resistance to NCLB is on chromosome 8 (bin 8.06), where Ht2 and Htn1 are located (Zaitlin et al. 1992; Simcox and Bennetzen 1993; Hurni et al. 2015). Fine-mapping delimited Ht2 to a region of ∼0.46-Mb, and candidate genes were predicted based on the B73 reference genome sequence (Chung et al. 2010b). In our study, we identified one QTL associated with lesion width on chromosome 8 (bin 8.06), and it is located ∼0.76-Mb upstream of GRMZM2G135202, a candidate gene for Ht2 (Chung et al. 2010b).
In previous studies, flowering time has also been considered for mapping NCLB resistance QTL (Poland et al. 2011; Inghelandt et al. 2012), since flowering time may confound the NCLB QTL mapping results, as late-flowering lines may appear to be more resistant. In the present study, we also collected flowering time data from parental lines and RILs. The average flowering times across years (from planting to pollen shedding) for K22 and By815 were 67 and 74 days, respectively; however, none of the identified NCLB resistance-related QTL showed overlapping association with flowering time (data not shown). Therefore, it is unlikely that the NCLB resistance QTL from K22 are false positives caused by late maturity.
In the present study, qNCLB5.04, a large and stable NCLB resistance QTL identified from K22, is a promising target QTL for improving NCLB resistance in breeding programs. Because K22 is an elite inbred line with many desirable traits, the present results should have direct application in maize breeding programs for NCLB resistance improvement.
References
Ali F, Yan JB (2012) The phenomenon of disease resistance in maize and the role of molecular breeding in defending against global threat. J Integr Plant Biol 55:134–151
Balint-Kurti PJ, Johal G (2009) Maize disease resistance. In: Bennetzen JL, Hake SC (eds) Handbook of maize: its biology. Springer, New York, pp 229–250
Bentolila S, Guitton C, Bouvet N, Sailand A, Nykaza S, Freyssinet G (1991) Identification of RFLP marker tightly linked to the Ht1 gene in maize. Theor Appl Genet 82:393–398
Brewster VA, Carson ML, Wicks ZW (1992) Mapping components of partial resistance to northern leaf blight of maize using reciprocal translocations. Phytopathology 82:225–229
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Carson ML (1995) A new gene in maize conferring the chlorotic halo reaction to infection by Exserohilum turcicum. Plant Dis 79:717–720
Chen ZL, Wang BB, Dong XM, Liu H, Ren LH, Chen J, Hauck A, Song WB, Lai JS (2014) An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genom 15:433
Chung CL, Longfellow J, Walsh EK, Kerdieh Z, Esbroeck GV, Balint-Kurti P, Nelson RJ (2010a) Resistance loci affecting distinct stages of fungal pathogenesis: use of introgression lines for QTL mapping and characterization in the maize-Setosphaeria turcica pathosystem. BMC Plant Biol 10:103
Chung CL, Jamann T, Longfellow J, Nelson R (2010b) Characterization and fine-mapping of a resistance locus for northern leaf blight in maize bin 8.06. Theor Appl Genet 121:205–227
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Ganal MW, Durstewitz G, Polley A, Bérard A, Buckler ES, Charcosset A, Clarke JD, Graner EM, Hansen M, Joets J, Paslier MCL, McMullen MD, Montalent P, Rose M, Schön CC, Sun Q, Walter H, Martin OC, Falque M (2011) A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 6:e28334
Hurni S, Scheuermann D, Krattinger SG, Kessel B, Wicker T, Herren G, Fitze MN, Breen J, Presterl T, Ouzunova M, Keller B (2015) The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc Natl Acad Sci 112:8781–8785
Inghelandt DV, Melchinger AE, Martinant J, Stich B (2012) Genome-wide association mapping of flowering time and northern corn leaf blight (Setosphaeria turcica) resistance in a vast commercial maize germplasm set. BMC Plant Biol 12:56
Jamann TM, Poland JA, Kolkman JM, Smith LG, Nelson RJ (2014) Unraveling genomic complexity at a quantitative disease resistance locus in maize. Genetics 198:333–344
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugenic 12:172–175
Li FM, Mao JC, Li XT (2004) The breeding of maize inbred line K22 and the analysis on the combine ability. J Gansu Agric Univ 39:312–315
Ogliari JB, Guimarães MA, Geraldi IO, Camargo LEA (2005) New resistance genes in the Zea mays L.-Exserohilum tucicum pathosystem. Genet Mol Biol 28:435–439
Ogliari JB, Guirnaraes MA, Aranha Carnargo LE (2007) Chromosomal locations of the maize (Zea mays L.) HtP and rt genes that confer resistance to Exserohilum turcicum. Genet Mol Biol 30:630–634
Parlevliet JE (2002) Durability of resistance against fungal, bacterial and viral pathogens; present situation. Euphytica 124:147–156
Poland JA, Bradbury PJ, Buckler ES, Nelson RJ (2011) Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci 108:6893–6898
Pratt RC, Gordon SG (2006) Breeding for resistance to maize foliar pathogens. Plant Breed Rev 26:119–173
Raymundo AD, Hooker AL (1981) Measuring the relationship between northern corn leaf blight and yield losses. Plant Dis 65:325–327
Shao Z, Chang Q, Tao W (2009) Establishment of Shaanxi soil information system based on ArcEngine. Bull Soil Water Conserv 29:125–129
Simcox KD, Bennetzen JL (1993) The use of molecular markers to study Setosphaeria turcica resistance in maize. Phytopathology 83:1326–1330
Tefferi A, Hulluka M, Welz HG (1996) Assessment of damage and grain yield loss in maize caused by northern leaf blight in western Ethiopia. J Plant Dis Protect 103:353–363
Wang DL, Zhu J, Li ZK, Paterson AH (1999) Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor Appl Genet 99:1255–1264
Welz HG, Geiger HH (2000) Genes for resistance to northern corn leaf blight in diverse maize populations. Plant Breed 119:1–14
Wisser RJ, Balint-Kurti PJ, Nelson RJ (2006) The genetic architecture of disease resistance in maize: a synthesis of published studies. Phytopathology 96:120–129
Yan JB, Yang XH, Shah T, Sánchez H, Li JS, Warburton M, Zhou Y, Crouch J, Xu YB (2010) High-throughput SNP genotyping with the Golden Gate assay in maize. Mol Breed 25:441–451
Yang J, Zhu J, Williams RW (2007) Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics 23:1527–1536
Zaitlin D, Demars SJ, Gupta M (1992) Linkage of a second gene for NCLB resistance to molecular markers in maize. Maize Genet Coop Newsl 66:69–70
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhou Y, Fu ZY, Li Q, Xu ST, Chander S, Yang XH, Li JS, Yan JB (2009) Comparative analysis of carotenoid and tocopherol compositions in high-oil and normal maize (Zea mays L.) inbreds. Acta Agronomica Sinica 35:2073–2084
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (Grant No. 31161140347) and by DuPont Pioneer and Dunhuang Pioneer Company.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, G., Wang, X., Long, S. et al. Mapping of QTL conferring resistance to northern corn leaf blight using high-density SNPs in maize. Mol Breeding 36, 4 (2016). https://doi.org/10.1007/s11032-015-0421-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0421-3