
Abstract
Despite remarkable advances in our knowledge about the function of autophagy in myocardial ischemia/reperfusion (I/R) injury, the debate continues over whether autophagy is protective or deleterious in cardiac I/R. Due to the complexity of autophagy signaling, autophagy can play a dual role in the pathological processes of myocardial I/R injury. Thus, more researches are needed to shed light on the complex roles of autophagy in cardioprotection for the future clinical development. Such researches can lead to the finding of new therapeutic strategies for improving cardiac I/R outcomes in patients. Several preclinical studies have targeted autophagy flux as a beneficial strategy against myocardial I/R injury. In this review, we aimed to discuss the complex contribution of autophagy in myocardial I/R injury, as well as the therapeutic agents that have been shown to be useful in reducing myocardial I/R injury by targeting autophagy. For this reason, we provided an updated summary of the data from in vivo, ex vivo, and in vitro experimental studies about the therapeutic agents that exert positive effects against myocardial I/R injury by modulating autophagy flux. By addressing these valuable studies, we try to provide a motivation for the promising hypothesis of “autophagy modulation as a therapeutic strategy against cardiac I/R” in the future clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic heart diseases (IHD) are one of the leading causes of mortality worldwide. In myocardial ischemia condition, complex pathological changes occur as a result of myocardial blood flow interruption, leading to the progressive damage and eventually cell death. Although reperfusion of ischemic myocardium is necessary to save cardiomyocytes, but unfortunately it leads to further damages. Myocardial ischemia/reperfusion (I/R) injury leads to further complications such as decreased cardiac contractile function, endothelial dysfunction, arrhythmias, tissue edema, electrophysiological dysfunction and myocardial infarction (MI). Therefore, it is clinically important to find new strategies in order to reduce the complications of myocardial reperfusion injury [4, 23, 53].
One of the processes involved in the pathology of myocardial I/R injury is autophagy. Under normal conditions, autophagy occurs at a basal level in the heart, which is involved in cellular homeostasis maintenance by clearing aged organelles and excessive or long-lived proteins. But elimination of autophagy can lead to the adverse effects on the myocardium. In addition, changes in autophagy flux have been detected in cardiovascular diseases such as IHD, cardiac hypertrophy and heart failure [56, 84]. Also, it has been shown that autophagy is associated with myocardial I/R injury. Therefore, interference with autophagy may be a potential therapeutic option for the treatment or prevention of myocardial I/R injury. But it should be considered that the mechanism of autophagy in the pathogenesis of I/R is either deleterious, or as an adaptive response. In other words, the adaptive or maladaptive role of autophagy in determining the fate of ischemic-reperfused myocardium is still under discussion. Since there are still many questions about the dual role of autophagy in myocardial I/R injury, more preclinical and clinical investigations in order to elucidate the molecular mechanisms of how and under what conditions autophagy contributes to the cell survival or death in cardiac I/R are needed. Also, by identifying the factors that can induce protective autophagy or prevent harmful autophagy under myocardial I/R condition, it would be possible to achieve useful and effective cardioprotective drugs and thus improve the outcomes of I/R injury in patients [45, 54].
In this review, after demonstrating the regulation of autophagy and its contribution in cardiac I/R injury, we provided an updated summary of the evidences showing positive effects of therapeutic agents against myocardial I/R via targeting autophagy flux. We aimed to draw attention to the therapeutic potential of modulating autophagy process in experimental studies for future clinical development. This review is expected to lead to the better identification of potential therapeutic targets for improving outcomes in patients with cardiac I/R in the future.
Physiological roles of autophagy in the heart
The heart contains long-lived cardiomyocytes with low regenerative capacity. Under normal conditions, the process of autophagy in the myocardium occurs at a basal level, which is involved in cellular homeostasis through the clearance of excessive or long-lived proteins, as well as aged organelles. This basal level of autophagy is essential for the structure and function of the heart, because it participates in controlling the quality of organelles and proteins [45]. In autophagy process, proteins and organelles are encircled in the autophagosomes and delivered to the lysosomes to be hydrolyzed to amino acids, fatty acids and substrates for generation of adenosine triphosphate (ATP), which can be recycled to synthesize and produce high-energy phosphates, new proteins, and other cellular elements. Despite the complexity of autophagy, it is performed in 4 separate but consecutive stages as follows: 1) sensing the activating factor of autophagy and induction of autophagy; 2) formation and accumulation of autophagosomes; 3) docking and fusion of autophagosome to the lysosomal membrane; and 4) fragmentation of autophagic bodies [5] (Fig. 1).

Positive and negative regulatory signaling pathways of autophagy. Please see the text for more details
There are three main pathways of autophagy including macroautophagy, microautophagy, and chaperone-mediated autophagy, which differ in how unnecessary elements are delivered to lysosomes for final degradation [71]. Macroautophagy is the most common form of autophagy and is usually called autophagy. As shown in Fig. 1, in autophagy, an isolation membrane called phagophore appears in the cytoplasm and forms bi-membrane vesicle called autophagosome. This type of autophagy process involves multiple proteins encoded by autophagy-related genes (ATGs). These proteins are involved in the expansion of isolation membrane and the formation of autophagosome. The cytosolic components are sequestered in the autophagosome, and then the outer membrane of the autophagosome is fused to the lysosome, leading to the autolysosome formation. The cytosolic elements and the inner membrane of the autophagosome are then degraded to amino acids and lipids by lysosomal hydrolases. Finally, degraded elements are released into the cytosol to contribute in energy homeostasis and macromolecules biosynthesis. In mitochondrial autophagy (mitophagy), which is known as a protective mechanism during myocardial I/R injury, selective clearance of damaged mitochondria occurs [66].
In microautophagy, however, lysosomal membrane invagination causes direct uptake of the substrate. In mammals, microautophagy includes the direct engulfment of cytoplasm by the lysosome, and in plant and fungi, it refers to the direct engulfment of cytoplasm by the vacuole. After invagination of lysosomal/vacuolar membrane, it is differentiated into the autophagic tube in order to encircle portions of the cytosol. At the top of the tube, the vesicles forming, homotypical fusing, and budding into the lumen are occurred [50, 66]. It should be noted that microautophagy would be either selective or non-selective. The characteristic of selective microautophagy, which is often induced in yeasts, is sequesteration of specific organelles with arm-like protrusions. It has been demonstrated that selective microautophagy has three main forms including micromitophagy, micropexophagy, and piecemeal microautophagy of the nucleus (PMN). However, the characteristic of non-selective microautophagy, which is regularly induced in mammalian cells and usually called microautophagy, is engulfment of soluble intracellular substrates via the tubular invaginations [37].
Chaperone-mediated autophagy (CMA), which is unique to mammalian cells, is a selective degradation process of those cytosolic proteins harboring the amino acid motif KFERQ, which are identified by specific chaperone complexes led by heat shock cognate 70 (HSC70), to facilitate their transport to lysosomes via lysosome-associated membrane protein (LAMP) 2A [50]. LAMP2A proteins undergo oligomerization, and lead to the formation of a translocon complex in order to internalize and degrade chaperone delivered cargo. An important difference between CMA and macroautophagy is that CMA delivers single proteins for lysosomal degradation one at a time. However, in macroautophagy, autophagosomes engulf and deliver mainly larger structures for bulk degradation of cargo. The highly selective nature of recognization and degradation of single cytosolic proteins is the characteristic of CMA as compared to macroautophagy. Important roles of microautophagy and CMA in cardiovascular diseases need further investigations [12]. It should be noted that new types of autophagy, such as RNautophagy and DNautophagy have been identified in which RNA or DNA is directly degraded by lysosomes in an ATP-dependent process, which are mainly mediated by LAMP2C. But the physiological function of these two types of autophagy is not completely determined [10, 11].
In general, autophagy is involved in cellular homeostasis maintenance in aging, starvation, inflammation, cardiac remodeling and reverse remodeling conditions [58]. Studies have shown that loss of essential genes for autophagy can lead to the cardiac dysfunction and disorders. It has been suggested that the main mediators of autophagy are LAMP2 and Atg5, so that deficiencies in these genes cause impaired autophagy, accumulation of abnormal substrates and ultimately cardiac dysfunction. LAMP2 mutations have been shown to lead to Danon disease, which is a severe and progressive myopathy. In this disease, abnormal accumulation of autophagosomes occurs due to the defect in the autophagosome-lysosome fusion [47]. Deficiency of Atg5 in mice has also been reported to lead to the accumulation of damaged proteins and organelles, which leads to ventricular dilatation and cardiac dysfunction [57].
Signaling pathways regulating autophagy flux
Negative regulation of autophagy
Class I PI3K/Akt/mTOR pathway.
Mammalian target of rapamycin (mTOR) is a protein kinase that plays an important role in regulating metabolism and cell growth in response to growth factors, nutrients and energy stress. mTOR is considered as the most important regulator of autophagy. There are two types of mTOR complexes that differ in their subunit composition and sensitivity to rapamycin. mTOR complex 1 (mTORC1) consists of mTOR, regulatory associated protein of mTOR (Raptor) and proline-rich AKT1 substrate 40 (PRAS40). mTORC1 is sensitive to rapamycin, and is involved in controlling cell growth by regulating transcription, translation and autophagy. One of the known regulators of mTORC1 activity is Ras homolog enriched in brain (Rheb), which promotes mTORC1 activity through direct association with mTOR. Another regulator of mTORC1 activity is tuberous sclerosis complex (TSC)1/TSC2, which inhibits Rheb via GTPase activator protein (GAP) activity of TSC2. mTOR complex 2 (mTORC2) consists of mTOR, rapamycin-insensitive companion of mTOR (Rictor) and G protein β subunit-like (GβS). However, this complex is not inhibited by rapamycin and also modulates cell growth by regulating cytoskeleton [3]. But long-term treatment with rapamycin has been shown to inhibit mTORC2 [63, 64].
Nutrient-rich conditions in the cell that are modulated by phosphatidylinositol 3 kinase (PI3K) signals, activate the class I PI3K/Akt/mTOR pathway. Akt can activate mTORC1 in two ways. Akt induces phosphorylation of TSC2 and thus inhibits its GAP domain which leads to the activation of Rheb and mTORC1. Likewise, in the presence of insulin and growth factor, Akt/PKB phosphorylates PRAS40 and eliminates its inhibitory effect on mTORC1. mTORC1 also inhibits the activity of the Atg1/unc-51 like autophagy activating kinase 1 (ULK1) complex through direct phosphorylation of ULK1 in serine 757. It can be said that it negatively regulates autophagy by disrupting the interaction between ULK1 and AMP-activated protein kinase (AMPK), and thus reducing the activity of ULK1. The Atg1/ULK1 complex consists of Atg13, Atg101 and focal adhesion kinase family interacting protein of 200 kDa (FIP200) [42, 58, 74] (Fig. 1).
Acetylation/deacetylation.
It has been revealed that acetylation of Atg proteins plays important role in inhibiting or promoting their function in autophagy. In nutrient-rich conditions, acetylation of Atg5, Atg7, microtubule-associated protein 1 light chain 3 (LC3) and Atg12 by p300 acetyltransferase leads to autophagy inhibition [34].
Positive regulation of autophagy
AMPK pathway.
AMPK is a serine/threonine kinase that regulates energy homeostasis by sensing the state of cellular energy. The role of AMPK in regulation of many cellular processes such as growth, apoptosis, and autophagy has been proven. An increase in cellular AMP/ATP ratio occurs under conditions such as exercise, glucose deprivation, ischemia, and oxidative stress, which is sensed and modulated by Ca2+/calmodulin-dependent kinase kinase beta (CaMKKβ), transforming growth factor β activated kinase-1 (TAK1), and liver kinase B1 (LKB1), resulting in AMPK phosphorylation and activation [29]. Activated AMPK can initiate autophagy by stimulation of p27 gene, and on the other hand inhibits mTOR by two mechanisms. One is that it stimulates GAP activity through phosphorylation of TSC2 in serine 1345, and the other is that it causes phosphorylation and inhibition of Raptor in mTORC1 and finally inhibition of mTORC1 and thus activation of ULK1 and Beclin1-class III PI3K complexes [69]. It has been demonstrated that AMPK can inhibit the effect of Akt on mTORC1 activation, while Akt activity can negatively regulate AMPK phosphorylation. It has been revealed that AMPK leads to the activation of autophagy via binding to ULK1 and its phosphorylation [33]. In other words, under glucose starvation condition, AMPK can promote autophagy through direct phosphorylation of ULK1 in serine 317 and serine 777 [68]. AMPK can also stimulate autophagy by activating vacuolar protein sorting (Vps) 34 complex by phosphorylation of Beclin1 in serine 91/serine 94 [32] (Fig. 1).
Hypoxia inducible factor (Hif-1)/protein kinase C delta (PK-Cδ)/c-Jun N-terminal kinase 1 (JNK1) pathway.
In hypoxic condition, this pathway promotes Beclin1 and class III PI3K interaction, which is an essential step in phagophore induction. It should be noted that this interaction is also promoted or induced by the loss of nutrients especially amino acids, as well as by reactive oxygen species (ROS) and calcium [39].
Acetylation/deacetylation.
Under starvation conditions, sirtuin 1 deacetylase (SIRT1) causes deacetylation of Atg proteins and ultimately upregulates autophagy flux [34]. In response to the reduction of growth factors, glycogen synthase kinase-3β (GSK-3β) causes phosphorylation of Tat-interactive protein 60-kDa (TIP60), which leads to the acetylation of ULK1 and activation of ULK1 complex, resulting activation of autophagy [41]. Under autophagy-inducing conditions, TIP60 and Esa1 also increase Atg3 acetylation, and thus increase the interaction between Atg3 and Atg8, and ultimately promote autophagy [87].
Numerous therapeutic agents can modulate the activity of autophagy through affecting its molecular cascades. In the following chapters, a summary of the data from several experimental studies using numerous therapeutic agents has been provided, suggesting that adaptive induction of controlled autophagy, or inhibition of inappropriate or excessive autophagy through modulation of signaling pathways involved in autophagy regulation, may exert cardioprotective effects against myocardial I/R injury [72]. The related signaling pathways that have been targeted with these therapeutic agents in order to induce the appropriate level of autophagy flux are mentioned in Tables 1 and 2.
Complex contribution of autophagy in myocardial I/R
Due to very limited regenerative capacity of postmitotic myocytes, the mechanism of autophagy seems to be essential for the preservation of cardiomyocytes function. This is why investigations on the complex roles of autophagy in myocardial I/R condition are of particular importance [79]. It has been revealed that autophagy is involved in both phases of myocardial ischemia and reperfusion (Fig. 2). During ischemia/hypoxia in the heart, rapid depletion of cellular ATP level leads to the activation of autophagy to preserve cellular viability and energy homeostasis. In other words, nutrient depletion during ischemia/hypoxia leads to autophagy for replenishment of metabolic substrates, as well as removing damaged organelles. Therefore, it can be said that autophagy in this phase is an adaptive response with cardioprotective effects [49]. Activation of AMPK, an energy-deprivation sensor, plays an important role in mediating metabolic adaptation during ischemia/hypoxia. Since inhibition of AMPK significantly inhibits autophagy in cardiac ischemia, it can be concluded that activation of AMPK is necessary to induce autophagy in this phase [49]. Studies have shown that AMPK activation not only inhibits mTOR through TSC2 phosphorylation, but also induces phosphorylation of eukaryotic elongation factor 2 (eEF2). mTOR inhibition and eEF2 phosphorylation cause inhibition of protein synthesis and stimulation of autophagy, resulting hypoxic adaptation. But the importance of mTOR inhibition and eEF2 phosphorylation in AMPK-mediated autophagy is unclear [48]. It has been shown that dephosphorylation/activation of GSK-3β during ischemia can phosphorylate TSC1/TSC2 and inhibit mTORC1, resulting in autophagy stimulation [67]. Besides mTOR signaling mechanism, autophagy stimulation in ischemia/hypoxia may be mediated by HIF-1 upregulation. However, the roles of mTOR inhibition and HIF-1 upregulation in autophagy induction in myocardial ischemia/hypoxia is not well defined [39, 67].

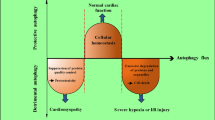
Induction of autophagy with protective and damaging roles in myocardial ischemia and reperfusion conditions. Autophagy is involved in both phases of myocardial ischemia and reperfusion in the cardiomyocytes. In ischemia phase, the mechanism of autophagy seems to be different from those in the reperfusion phase. The main stimuli for autophagy under ischemia are decreased O2 and nutrient supplies, and reduced ATP levels in the heart, which cause AMPK activation. These stimuli lead to autophagy, which may be protective. Under reperfusion phase, the energy crisis is partially resolved, and upregulation of Beclin 1 expression, accumulation of ROS, increased ER stress and oxidative stress may occur. These stimuli lead to detrimental autophagy (Please see the text for more details). ATP: adenosine triphosphate; AMPK: adenosine monophosphate-activated protein kinase; ER: endoplasmic reticulum; mTOR: mammalian target of rapamycin; Bnip3: Bcl-2/adenovirus E1B-19 KD interacting protein 3; NIX: Nip3-like protein X; ROS: reactive oxygen species
Numerous studies support the notion that autophagy generally has protective role during myocardial ischemia and exerts its protective effects against cell injury via three proven mechanisms. The first mechanism is that autophagy may provide an energy source through the autophagosome-lysosome pathway to increase ATP production. In other words, autophagy may compensate the energy loss by regeneration of amino acids and fatty acids, which then are used to synthesize ATP through the tricarboxylic acid (TCA) cycle. It should be demonstrated that in severe or prolonged ischemia, ATP cannot be produced through the TCA cycle. Because a number of processes in autophagy, such as autophagosome formation, are energy-dependent. Thus, autophagy and subsequently ATP production cannot be performed when ischemia is complete [55]. The second mechanism is that autophagy activation may be involved in the process of removing damaged proteins that are harmful for cell. So autophagy scavenges protein aggregates that have been accumulated during ischemia [36]. The third mechanism is that autophagy in cardiomyocytes plays an important role in reducing ROS, as well as removal of damaged organelles such as mitochondria that may release proapoptotic factors such as cytochrome c [18].
The mechanisms responsible for autophagy in reperfusion phase seem to be different from the mechanisms responsible for autophagy in ischemia. Because the energy crisis in the heart which is the main stimulus for autophagy in ischemia, is partially resolved in reperfusion phase, because oxygen and nutrients are made available in cardiomyocytes. Instead, ROS accumulation which is a major promoter of autophagy in reperfusion phase, causes mitochondrial damage, mitochondrial permeability transition pore (MPTP) opening, as well as fragmentation of mitochondria [13]. Studies have shown that ROS oxidizes and inhibits cysteine protease activity of Atg4, leading to LC3 lipidation and autophagy [65]. Also, ROS has been shown recently to play an important role in mediating Beclin1 upregulation in rat heart during I/R [22].
It has been revealed that increased autophagy flux in reperfusion phase may be beneficial or detrimental. This dual role of autophagy can be attributed to the dual role of Beclin1 during I/R injury [16]. Activation of Beclin1 is necessary to initiate autophagy in the early phase of ischemia, but its continuous activity during the reperfusion phase leads to excessive catabolic activity and cell death [46]. Since Beclin1 is inhibited by anti-apoptotic protein B-cell lymphoma (Bcl)-2, it can be suggested that this interaction may play a role in modulating the ratio of cell viability to cell death. Likewise, Bcl-2/adenovirus E1B-19 KD interacting protein 3 (Bnip3) which is involved in regulating MPTP opening, activates autophagy in I/R. In addition, increased intracellular calcium via depletion of sarcoplasmic reticulum stores increases autophagy in cardiomyocytes [59].
It has been shown that along with elevated Beclin1 expression under myocardial I/R condition, an increase in autophagosome formation occurs. Therefore, due to the accumulation of autophagosomes in cardiomyocytes during the reperfusion phase, it seems that autophagy has been increased during this phase. But recently, a new interpretation has been emerged that autophagy flux during the reperfusion is partly impaired rather than further activated. In this regard, researchers have concluded that the clearance of autophagosome in cardiomyocytes is reduced significantly along with reperfusion, which is damaging to the survival of cardiomyocytes during reperfusion [71]. Therefore, it can be said that the increase in cell death during I/R is due to the defective clearance of autophagosomes. This indicates the importance of autophagosome clearance by LAMP2. Thus, the increase in autophagosomes can be caused by two distinct phenomenon; autophagy upregulation or autophagy degradation blockade [79]. Therefore, inhibition of autophagy at autophagosome formation level or autophagosome-lysosome fusion level may exert different effects on I/R injury [71].
Because the molecular mechanisms mediating the different effects of autophagy in ischemia and reperfusion are not fully understood, further investigations about why autophagy can play protective or detrimental role under numerous experimental conditions are required. It has been shown that there is a direct relationship between the severity of ischemia and the extent of autophagy in myocardial I/R. This means that mild to moderate ischemia/hypoxia leads to the modest levels of autophagy. In this state, autophagy may play a protective role by preventing apoptosis. But severe ischemia/hypoxia or I/R leads to higher levels of autophagy, in which autophagy may lead to self-digestion and subsequently cell death. In other words, due to the partially nonspecific feature of autophagy degradation, increased autophagy activity may lead to the digestion of proteins and organelles which could protect cardiomyocytes from reperfusion-induced cell death. For example, autophagy degrades catalase, an enzyme essential for reducing H2O2, thereby increasing oxidative stress and apoptosis. Thus, it can be concluded that early stage of I/R induces protective autophagy, whereas at reperfusion phase in the later stage of I/R, autophagy is damaging [19]. Due to disagreements and debates about the beneficial or harmful functions of autophagy in myocardial I/R, the attention of many scientists has been attracted to elucidate the complex contribution of autophagy in cardioprotection, that is very helpful in determining new therapeutic strategies for reducing I/R injury. Numerous preclinical studies have been performed in order to evaluate pharmacological modulation of autophagy flux as an effective strategy against myocardial I/R damage, and have got very interesting results in this regard. By addressing these valuable studies, we aimed to elucidate the way for further researches on autophagy modulation to improve outcomes in cardiac I/R patients in the future.
Therapeutic targeting of beneficial autophagy for reducing myocardial I/R injury in preclinical studies
Although the role of autophagy in cardiac I/R pathophysiology is complex and not well understood, several previous experimental studies suggested that adaptive induction of autophagy may exert cardioprotective effects against myocardial I/R injury, which we mentioned some of them in Table 1.
MicroRNA (miR)-490-3p antagomir.
MicroRNAs are single-stranded and non-coding RNAs that play a role in a number of biological processes and can regulate cell growth, proliferation, differentiation and apoptosis. A study showed that the inhibition of miR-490-3p inhibited apoptosis, promoted LC3-II expression, inhibited p62 expression, increased the autolysosomes, and decreased the infarct size under cardiac I/R injury. So inhibition of miR-490-3p promoted autophagy, and decreased apoptosis in myocardial cell in order to alleviate myocardial I/R damage via ATG4B upregulation. This finding revealed new insights for the protective mechanism of autophagy in I/R [80].
6-Gingerol (6-G).
Long noncoding RNAs (LncRNAs), containing over 200 nucleotides, can combine with biological molecules and effect on important proteins or signaling pathways in different pathophysiological processes. A study reported that 6-G could regulate lncRNA H19/miR-143/ATG7 signaling axis-mediated autophagy and relieve cardiac I/R damage by enhancing autophagy. In details, 6-G enhanced the expression of cellular H19 under hypoxia/reoxygenation (H/R) condition. In addition, 6-G decreased cleaved caspase 3 and caspase 9 proteins, and elevated the expression of Bcl-2. This study showed that H19 has direct interaction with miR-143 and acts as a molecular sponge to lower its cellular abundance, and subsequently increase the expression of ATG7 and promote the autophagy process [44].
Alliin.
A study by Zhao et al. showed that alliin (S-allyl cysteine sulfoxide) exerts its cardioprotective effects in I/R or H/R condition via increasing autophagy flux. They showed that Atg9b gene, which plays an important role in the assembly of autophagosomes, was upregulated in I/R condition and was further increased following alliin treatment. Other autophagy-related genes such as becn1, Rgs16 and Irgm1 were similarly upregulated by alliin treatment. It seems that promoting autophagy flux via upregulation of these autophagy-related genes, Beclin1, LC3-II/LC3-I and Atg9b, as well as reducing apoptosis, play important role in mediating the cardioprotective effects of alliin against I/R or H/R damage [92].
Visnagin.
In a study investigating the effects of visnagin (4-methoxy-7-methyl-5H-furo [3,2-g] [1] benzopyran-5-one) on myocardial I/R in rats, it was found that encapsulation of visnagin in N-isopropylacrylamide (NIPAAm)-methacrylic acid (MAA) nanoparticles (NP), and its intravenous injection during reperfusion protected the heart against I/R injury, which was evidenced by reducing fibrosis and infarct size, and improving cardiac function. This protective mechanism of NP-visnagin in myocardial I/R was mediated by apoptosis inhibition and autophagy induction in the ischemic region of the heart [9].
Coenzyme Q10 (CQ10).
CQ10 is a fat-soluble quinone antioxidant with a structure similar to vitamin K and vitamin E. Liang et al. showed that CQ10 preconditioning increased antioxidants and decreased oxidants, and led to the balance between them in cardiac I/R in rats. It also increased ATP concentration and inhibited apoptosis. Bcl-2 antagonist/killer (Bak) and Bcl-2 associated x protein (Bax), which can increase mitochondrial outer membrane permeability (MOMP) and cell death, were decreased by CQ10. As well as, CQ10 preconditioning decreased p53 and active caspase 3. Elevation of Beclin1, LC3-II/LC3-I and Atg5 were the evidences for positive effect of CQ10 on autophagy. CQ10 also decreased total LC3 and p62. In general, CQ10 could decrease myocardial infarct size, fibrosis and necrosis, and improve myocardial function following I/R injury due to its abilities such as antioxidant function, energy survival, reduction of myocardial apoptosis and enhancement of autophagy activity [40].
Mesenchymal stem cells-derived exosome.
Confirming the protective role of autophagy in I/R condition, the results of a study revealed that after 3 h exposure to H2O2, the autophagy flux was blocked in H9c2 cells. After 6 and 12 h exposure to H2O2, autophagy was decreased with time-dependent decrease in Bcl-2/Bax ratio. Interestingly, treatment with mesenchymal stem cells (MSC)-derived exosome protected H9c2 cells against H2O2-induced damage, and also increased cell viability by reducing cell apoptosis and ROS production. On the other hand, it reduced apoptosis and infarct size, and improved heart function in I/R model in vivo. It has been proven that these positive effects of MSC-derived exosome against myocardial H2O2 or I/R injury were due to increased Beclin1 and LC3-II, and decreased p62. In addition, MSC-derived exosome increased p-AMPK and decreased p-Akt and p-mTOR in H9c2 cells after 12 h exposure to H2O2. So these beneficial effects of MSC-derived exosome were partly mediated by modulation of AMPK/mTOR and Akt/mTOR pathways, and the subsequent increase in autophagy activity in H9c2 cells [43].
Trimetazidine (TMZ).
Study on TMZ [1- (2,3,4-trimethoxybenzyl)-piperazine dihydrochloride] by Zhong et al. showed that TMZ had protective effects against H/R damage, probably due to its positive effect on autophagy flux. In other words, TMZ could improve cell survival by promoting autophagy. Therefore, it can be said that cytoprotection by TMZ was associated with a moderate increase in autophagy activity. TMZ also caused an increase in LC3-II/LC3-I, as well as autophagosomes, which may indicate induction of autophagy or defective autophagy flux. Further investigation showed that H/R led to impair and maladaptive autophagy, which was confirmed by the accumulation of autophagosomes and the reduction of autolysosomes, and thus led to detrimental effect on cardiomyocytes. But TMZ pretreatment caused restoration of autophagy flux via increasing the clearance of autophagosomes. So the protective mechanism of TMZ against H/R injury was repairing autophagy flux. Because TMZ pretreatment reduced apoptotic cells, this study identified that the crosstalk between autophagy and apoptosis is an important factor in how TMZ regulated apoptosis. Therefore, it was concluded that TMZ pretreatment reduced apoptosis and induced cardioprotection against H/R injury through autophagy induction. TMZ also increased p-AMPK and decreased p-mTOR in H/R condition. TMZ restored autophagy flux by activating AMPK/mTOR pathway, resulting in cardioprotection against H/R injury [95].
Cellular repressor of E1A-stimulated genes (CREG).
CREG is a secreted glycoprotein that is involved in regulating cell and tissue homeostasis. Song et al. revealed that prepumping recombinant CREG protein in mice before myocardial I/R reduced cardiomyocyte apoptosis and infarct size, and improved heart function. It has been shown that these protective effects of CREG in myocardial I/R condition were due to the activation of lysosomal autophagy. In vivo and in vitro experiments indicated that overexpression of CREG promoted myocardial autophagy flux after reperfusion, but low expression of CREG led to the accumulation of autophagosomes in cells. It indicates that CREG regulated autophagy after autophagosome formation. Therefore, CREG played a protective role by reducing cardiomyocyte apoptosis, as well as regulating late fusion of lysosomes with autophagosomes. It was revealed that overexpression of CREG increased the acidic pH of lysosomes and the expression of lysosome-related proteins in the lysosome component, possibly by modulating the binding efficiency between mannose-6-phosphate receptors (M6P/IGF2R) and lysosome-related proteins. It means that CREG was involved in modulating lysosome function and improving cellular autophagy. CREG regulated cardiomyocytes autophagy by altering lysosomal related protein transport into lysosome mediated by M6P/IGF2R. Generally, CREG was downregulated in myocardial I/R damage, and interestingly, treatment with recombinant CREG protein could protect the heart against myocardial I/R damage by activating autophagy [70].
Pramipexole (PPX).
A study confirming the beneficial effects of autophagy in cardiac I/R and H/R conditions showed that pretreatment with PPX at a protective dose had cardioprotective effects against I/R in mice by reducing serum creatine kinase (CK) and lactate dehydrogenase (LDH) levels, and reducing infarct size. It also protected against H/R damage in cultured H9c2 cells by reducing LDH and ROS, as well as restoring mitochondrial membrane potential (∆Ψm) and increasing cell viability. These protective effects of PPX against cardiac I/R or H/R were associated with increased autophagy flux through AMPK activation. Consequently, PPX can be proposed as a clinically useful agent against reperfusion injury due to its cardioprotective effects by increasing autophagy activity through AMPK-dependent mechanism [52].
Spermine.
Polyamines such as spermine interact with proteins and nucleic acids, and act as modulators for cell growth and differentiation, as well as the synthesis of proteins, RNA, and DNA. It was found that spermine pretreatment in simulated I/R (sI/R) model in neonatal rat cardiomyocytes (NRCMs) reduced cell damage and sI/R-induced apoptosis, as well as increased cell viability and proliferation. This useful effect of spermine was shown to be mediated by its positive effect on autophagy flux, which was proved by increased autolysosomes, increased autophagy-related proteins such as Beclin1, LC3-II, and LC3-I, and decreased p62. These effects of spermine on autophagy induction in cardiomyocytes were found to be due to its inhibitory effect on mTOR pathway, as it decreased phosphorylations of mTOR (serine 2448, serine 2481) and 70-kDa ribosomal protein S6 kinase (p70S6K). Thus, spermine may be a potential therapeutic target for the treatment of myocardial injury in children due to its pre-protective effect on immature cardiomyocytes by inducing autophagy in sI/R condition [6].
Rapamycin.
Wang et al. demonstrated that treatment with rapamycin following anoxia/reoxygenation (A/R) inhibited cardiomyocyte apoptosis through downregulation of proapoptotic proteins such as Bcl-2 interacting mediator of cell death (Bim) and caspase 3. Rapamycin protected against A/R-induced apoptosis by further increasing autophagy. It was revealed that rapamycin probably exerted its beneficial effects against A/R injury by increasing autophagy via activating PI3K/Akt signaling pathway. It can be said that rapamycin activated other kinases in order to further inhibit apoptosis via inhibiting mTOR expression. It should be noted that the pharmacological effects of rapamycin in the heart are closely related to autophagy activity. Because when autophagy has a protective role in cardiac I/R, rapamycin as an autophagy inducer also has a positive effect on cardiomyocytes. But when autophagy plays a damaging role in cardiac I/R, rapamycin has a negative role on cardiomyocytes by activating autophagy [77].
Suberoylanilide hydroxamic acid (SAHA).
Another study showed that SAHA (a histone deacetylase (HDAC) inhibitor) reduced LDH, myocardial apoptosis, and infarct size, and also improved cardiac function. SAHA exerted cardioprotective effect by inducing autophagy flux. SAHA treatment increased autophagy, especially in cardiac border zone following I/R. Increased autophagy led to the protective effects against I/R-induced cell death in the in vivo model and sI/R-induced cell death in the in vitro model. Accordingly, reactivation of I/R-induced downregulated autophagy was the main mechanism of SAHA for cardioprotection [83].
Acetylcholine (ACh).
The results of a study on the effects of ACh against H/R damage showed that ACh administration activated cytoprotective autophagy in H9c2 cells under H/R condition. This inducible effect of ACh on autophagy occured through the muscarinic receptor. ACh was also shown to stimulate autophagy following H/R by AMPK activation and mTOR inhibition. So ACh protected H9c2 cells against H/R damage by inducing autophagy through the muscarinic receptor and activation of AMPK/mTOR signaling pathway [91].
Chloramphenicol succinate (CAPS).
The protective effect of autophagy against I/R injury has also been observed with administration of CAPS. CAPS pretreatment (10 min before I/R) decreased infarct size and increased expression of Beclin1 and LC3-II, which indicated a link between cardioprotection and preischemic induction of autophagy. However, it seems unlikely that increased expressions of Beclin1 and LC3-II within 10 min after CAPS treatment were associated with de novo synthesis of proteins. Because the increase in LC3-II was probably due to the increase in the conversion of LC3-I to LC3-II by C-terminal cleavage and conjugation to phosphatidylethanolamine. The increase in Beclin1 could probably be attributed to a rapid decrease in degradation rather than an increase in synthesis, which, of course, requires further investigation. Delayed administration of CAPS during ischemia also decreased infarct size and increased LC3-II expression, but had no significant effect on Beclin1 expression. Delayed administration of CAPS showed less protective effect as compared to its administration before I/R. In general, CAPS administration increased porcine heart resistance against I/R injury by induction of autophagy [62].
Resveratrol.
Although it has been previously suggested that autophagy is involved in non-apoptotic form of programmed cell death, several studies have shown that autophagy can led to the cell survival under certain conditions. In this regard, a study showed that treatment with low doses of resveratrol was involved in reducing apoptosis and improving heart function in I/R injury, as well as reducing death and increasing viability in H9c2 cells in H/R injury, partly by inducing autophagy [17]. It was proved that low doses of resveratrol increased autophagy and survival ex vivo and in vitro. However, high doses of resveratrol reduced autophagy and increased cell death ex vivo. High doses of resveratrol in H9c2 cells also reduced autophagy, while did not change cell survival. This differences in results are probably due to differences in ex vivo and in vitro conditions. This study showed that low dose of resveratrol differently regulated mTOR to induce autophagy, as it inhibited mTOR phosphorylation in serine 2448, while increased mTOR phosphorylation in serine 2481. Also, low dose of resveratrol activated AMPK and thus inhibited the effect of Akt on mTORC1 activation. So low dose of resveratrol inhibited mTORC1 during induction of autophagy. However, low dose of resveratrol induced Rictor, which is a component of mTORC2. Since Rictor is responsible for the phosphorylation of Akt in serine 473, so it activated Akt, which leads to the phosphorylation of substrates involved in the regulation of cell survival, growth, proliferation and metabolism. Consequently, it was concluded that there was a positive relationship between induction of Rictor, autophagy and cell survival by low dose of resveratrol, and therefore mTORC2 was involved in autophagy induction. In other words, resveratrol induced a survival signal in the myocardium and H9c2 cells by inducing autophagy. However, inhibition of autophagy was found to reduce the cardioprotective effect of resveratrol against I/R and H/R damages. Overall, low dose of resveratrol-induced cell survival was mediated by induction of autophagy in the myocardium and cardiac myoblast cells via Beclin1-dependent mechanism, which also depends on the activation of Rictor-mediated mTORC2 pathway [17].
Sulfaphenazole (SUL).
Since autophagy may be a process to increase the heart resistance against ischemia, a study showed that administration of SUL induced autophagy and cardioprotection in intact mouse hearts, isolated perfused rat hearts, and adult rat cardiomyocytes (ARCMs). This study showed that an increase in autophagy occurred following cardiac I/R in rats, possibly due to the impaired clearance of autophagosomes rather than an increase in autophagosomes formation. Interestingly, SUL caused cardioprotection by inducing autophagy via PKC signaling-dependent mechanism. Accordingly, SUL caused effective autophagy via the phosphorylation of regulatory subunit of vacuolar protein ATPase (VPATPase) by PKC signaling-dependent mechanism. Consequently, SUL caused sufficient fusion of autophagosomes with functional lysosomes via lysosomal acidification by VPATPase [24].
γ-tocotrienol and resveratrol.
The results of a study showed that cardioprotection by γ-tocotrienol and resveratrol was related to their ability to induce autophagy. It was found that both compounds could act synergistically to create a high level of cardioprotection by increasing autophagy flux. It was revealed that treatment with both γ-tocotrienol and resveratrol at low concentrations further increased the levels of Beclin1 and LC3-II/LC3-I compared to single treatment, indicating the synergistic effects of these compounds to induce autophagy. The induction effect of γ-tocotrienol on autophagy was more dependent to mTOR-dependent signaling pathway. But the induction effect of resveratrol on autophagy was more independent to mTOR pathway. It was also found that both compounds caused more activation of survival proteins such as Akt and Bcl-2 compared to single treatment, indicating their additive effect on inducing survival pathways. γ-tocotrienol and resveratrol were able to protect the myocardium from I/R injury, which was confirmed by normal morphology of the heart with very few degenerative changes in cardiomyocytes, the presence of autophagosomes at different stages of maturation, reduction of infarct size, and increased heart function. Hence, the positive and synergistic effects of these two compounds against myocardial I/R injury were mediated by induction of autophagy and Akt/Bcl-2 survival pathway. But the precise mechanisms by which these compounds induced autophagy require further investigations [35].
A1 adenosine receptor agonist 2-chloro-N (6)-cyclopentyl-adenosine (CCPA).
Autophagy acts as an important mediator of cytoprotection by CCPA. CCPA treatment increased the number of autophagosomes in the heart of mCherry-LC3 transgenic mice, NRCMs, ARCMs, and HL-1 cells. It was revealed that administration of CCPA before sI/R, upregulated autophagy in HL-1 cells within 10 min, and induced cytoprotection against sI/R in the same time. Remarkably, autophagy in reperfusion phase was lower than autophagy in untreated cells exposed to sI/R, indicating that less damage occured in preconditioned cells during ischemia, and less autophagy was needed as a repair response during reperfusion phase. In other words, treatment with CCPA increased autophagy flux before sI/R. But following sI/R, CCPA reduced the formation of autophagosomes without improving their clearance. Decreased autophagy in CCPA-treated cells after sI/R may indicate that the cells were less stressed and, in fact, less autophagy activity was required in reperfusion phase (reparative autophagy). In confirming the cytoprotective effect of CCPA by autophagy modulation, transient transfection of Atg5K130R in HL-1 cells reduced autophagy and blocked the cytoprotective effect of CCPA against sI/R. So CCPA mediated delayed preconditioning through a mechanism that requires autophagy. The effects of CCPA on autophagy and thus cytoprotection against sI/R were receptor dependent. It means that CCPA induced autophagy through phospholipase C (PLC), as well as increasing intracellular calcium. In fact, PLC signaling was an upstream for inducing autophagy by CCPA [88].
Therapeutic targeting of detrimental autophagy for reducing myocardial I/R injury in preclinical studies
There are several previous experimental studies showing that reduction of excessive autophagy flux, or improving impair autophagy flux by numerous therapeutic agents may have cardioprotective effects against myocardial I/R injury, which we mentioned some of them in Table 2.
Urolithin B (UB).
In vivo and in vitro experiments by Zheng et al. showed that UB had cardioprotective effects against I/R or H/R injury by inhibiting autophagy. UB increased antioxidant and decreased oxidants levels, decreased myocardial apoptosis and infarct size, and also improved cardiac function in rats following I/R. UB also protected cardiomyocytes against H/R injury with the evidences of decreased ROS production, decreased apoptosis, and increased cell viability. UB was found to inhibit autophagy by activating autophagy upstream signaling pathways such as mTOR/ULK1 and PI3K/Akt, thus protected the heart against oxidative stress and caspase 3-depedent apoptotic death. This inhibitory effect of UB on autophagy was essential for its cardioprotective effect. It was also shown that treatment with UB caused p62 accumulation and its interaction with kelch-like ECH-associated protein 1 (Keap1), and promoted nuclear factor erythroid 2-related factor 2 (Nrf2) nuclear translocation during I/R or H/R. It means that UB treatment reduced ROS production and inhibited apoptosis by p62/Keap1/Nrf2 signaling pathway [93].
Calreticulin (CRT).
Another study found that H/R treatment in H9c2 cells caused Beclin1 upregulation, increased LC3-II/LC3-I and autophagosomes formation. On the other hand, H/R treatment decreased LAMP2, impaired autophagosome-lysosome fusion, increased intracellular ROS, and impaired autophagosomes clearance by increasing p62 in H9c2 cells. But CRT postconditioning reduced oxidative stress and H/R-induced cell death by improving H/R-induced impair autophagy flux. CRT inhibited H/R-induced excessive formation of autophagosomes, which was proved by reduction of Beclin1 and LC3-II/LC3-I. CRT also improved impair clearance of autophagosomes by promoting autophagosome-lysosome fusion and autophagosome degradation, which was evidenced by decreased number of autophagosomes, increased number of autolysosomes, increased LAMP2, and decreased p62 levels. In vivo investigation also showed that CRT protected the heart against myocardial I/R injury in rats [76].
Tetramethylprazine (TMP).
A study found that tetramethylprazine (TMP) improved I/R or H/R-induced impair function of autophagy, which was proved by reduction of CK, LDH, and myocardial infarct size in rats, as well as, reduction of LDH and cardiomyocyte apoptosis, and improvement of cell viability in neonatal rat ventricular myocytes (NRVMs). TMP was shown to induce cardioprotection against I/R or H/R through modulation of Beclin1-dependent autophagy by PI3K/Bcl-2 cascade. Therefore, PI3K/Bcl-2 cascade was associated with the ability of TMP to inhibit autophagy dysfunction and reduce cardiac I/R injury [96].
Antithrombin III (AT).
Huang et al. demonstrated that cardiac I/R injury increased autophagy which played damaging role in myocardial I/R. However, treatment with AT reduced cardiac I/R or H/R injury by inhibiting autophagy in a dose-dependent manner. Since autophagy is mediated by Beclin1 in reperfusion phase, AT reduced autophagy and exerted its protective effects by reducing Beclin1 expression. For the reason that Beclin1 has the ability to induce apoptosis via binding to Bcl-XL, AT also reduced apoptosis by reducing Beclin1 expression. It was also found that I/R-induced high levels of p-Akt/Akt were further increased by AT. Therefore, it was concluded that AT inhibited autophagy and caused cardioprotection through PI3K/Akt signaling pathway [26].
Hesperidin.
Another study in confirming the detrimental role of excessive autophagy in myocardial I/R revealed that administration of hesperidin [3',5,7-trithydroxy-4'-methoxy-flavanone-7-(6-α-L-rhamnopyranosyl-β-D glucopyranoside)] for 3 days before ischemia prevented excessive autophagy following I/R, which was associated with decreased Beclin1 and LC3-II/LC3-I. Hesperidin also activated PI3K/Akt/mTOR pathway. Thus, hesperidin inhibited excessive autophagy and showed its cardioprotective effect in reducing myocardial I/R injury by activating PI3K/Akt/mTOR pathway [38].
Trichostatin A (TSA).
In a study using cardiac myocyte specific active histone deacetylase 4 (HDAC4) transgenic mice in order to examine the role of HDAC4 in myocardial I/R damage, it was revealed that activated HDAC4 overexpression caused increases in autophagy and apoptosis, and a decrease in superoxide dismutase (SOD)1, which exacerbated myocardial I/R damage as evidenced by an increase in infarct size and decrease in cardiac function. By using an HDAC inhibitor, TSA, HDAC4 overexpression-induced exacerbated I/R injury was reduced. This beneficial function of TSA was done through reducing autophagy and apoptosis and increasing SOD1. I/R model in pig was also used to confirm the protective effect of TSA. The results showed that TSA protected the heart against I/R damage in pigs, which was proved by reduction of infarct size [90].
Sevoflurane postconditioning (SPC).
In one study, it was found that following myocardial I/R, an increase in autophagosomes number, as well as Beclin1, LC3-II/LC3-I, and p62 levels occurred, which indicated I/R-induced autophagosomes formation. However, SPC reduced infarct size and cardiac dysfunction. This cardioprotective effect of SPC against I/R injury was associated with restoration of autophagy flux, as SPC increased LAMP2 expression and decreased Beclin1, LC3-II/LC3-I, and p62 levels, thereby reduced autophagosomes accumulation. SPC also increased nitric oxide synthase (NOS) content, endothelial NOS (eNOS) and neuronal NOS (nNOS) phosphorylations, and nitric oxide (NO) production. Therefore, SPC had protective effects against myocardial I/R injury through NO-dependent mechanism. For the reason that NO can bind to cytochrome c oxidase and prevent MPTP opening by inhibiting mitochondrial respiration, it can be said that the protective effect of SPC against myocardial I/R was probably related to mitochondrial protection, and in fact, mitochondria serves as the target of SPC-derived NO. In other words, SPC was found to increase mitochondrial nicotinamide adenine dinucleotide (NAD+) levels, decrease MPTP opening and cytochrome c release, and restore I/R-induced impair autophagy flux. Thus, eNOS/NO pathway was involved in SPC positive effects on autophagy flux. It means that SPC can be considered as a potential therapeutic target for the treatment of myocardial reperfusion injury by increasing NO and restoring I/R-indused impair autophagy flux [61].
Melatonin.
Study on the beneficial effects of melatonin against cardiac I/R or H/R revealed that melatonin treatment decreased LDH, increased cell vitality, reduced infarct size, and improved cardiac function. Melatonin was found to increase ATP/AMP ratio, thereby reducing AMPK phosphorylation and increasing mTOR phosphorylation, which means that melatonin exerted its positive effects by inhibiting autophagy through AMPK/mTOR signaling pathway [2].
Choline.
Hang et al. demonstrated that in myocardial I/R, there was an increase in LC3 expression and autophagosomes. Because this increase in LC3 may be interpreted as an increase in autophagosomes due to autophagy activation, or as an accumulation of autophagosomes due to autophagy dysfunction, they evaluated p62/SQSTM1 and found that p62 was increased in cardiac I/R condition. So this increase in LC3 was attributed to the failure in degradation of autolysosomes. But pretreatment with choline decreased LC3-II/LC3-1 and p62, which were attributed to the degradation of autolysosomes by choline. It was also found that choline decreased I/R-induced autophagy activity by activating Akt/mTOR signaling pathway, resulting in cardioprotective effects against cardiac I/R. But rapamycin reduced the protective effects of choline against I/R damage [21].
Vitexin.
One study showed that treatment with vitexin (apigenin-8-C-D-glucopyranoside) caused cardioprotective effect against I/R or sI/R damage in a dose-dependent manner. Potential mechanisms by which vitexin exerted its cardioprotective effects in vivo and in vitro were inhibition of apoptosis (decreases in Bax and cleaved caspase 3, and increase in Bcl-2) and inhibition of excessive autophagy (decreases in Beclin1 and LC3-II/LC3-I, and increase in p62). Also, vitexin protected the cells from oxidative stress damage by increasing SOD concentration, and decreased I/R damage by reducing malondialdehyde (MDA), CK, and LDH concentrations. In other words, vitexin improved I/R-induced myocardial injury by inhibiting cardiomyocytes autophagy. However, activation of autophagy eliminated the inhibitory effect of vitexin on apoptosis and myocardial injury. Therefore, it can be said that vitexin inhibited apoptosis and reduced myocardial injury by inhibiting excessive autophagy flux. It was found that treatment with vitexin resulted in upregulation of p-Akt and mTOR. Therefore, vitexin inhibited autophagy by activating PI3K/Akt/mTOR pathway [73].
Berbamine.
The results of a study showed that berbamine postconditioning reduced infarct size and improved post-ischemic heart function in a concentration-dependent manner. This positive effect was partly related to the effects of berbamine on improving cell survival and mitochondrial membrane potential in cardiomyocytes, and reducing myocytes death. It was proved that berbamine led to cardioprotection via modulation of autophagy and reduction of autophagy-induced cell death. Berbamine improved autophagosome processing in the myocardium as well as cardiomyocytes, and suppressed Beclin1-dependent autophagy by reducing I/R-induced Beclin1 upregulation. In addition, berbamine further increased Akt phosphorylation. Thus, berbamine modulated Beclin1-dependent autophagy dysfunction by activating PI3K/Akt pathway, thus protected the heart against I/R injury [94].
miR-21 precursor.
A study showed that following H/R, downregulation of endogenous miR-21 occured in H9c2 cells. This downregulation of miR-21 may be associated with H/R damage in cardiomyocytes. Upregulation of miR-21 by transfection of cells with miR-21 precursor inhibited H/R-induced excessive autophagy in H9c2 cells, thereby protected cardiac cells against H/R injury. On the other hand, miR-21 increased Bcl-2 expression through direct interaction and binding to Bcl-2 mRNA. Beclin1, which is an important factor for initiation of autophagy, can be inhibited by Bcl-2 through its BH3 domain, so there is a negative interaction between Bcl-2 and autophagy activity. Therefore, pretreatment with miR-21 precursor increased Bcl-2/Bax and decreased apoptosis in H/R condition, and also inhibited autophagy. The mechanism by which miR-21 inhibited excessive autophagy was found to be through Akt/mTOR signaling pathway. In addition, upregulation of miR-21 inhibited phosphatase and tensin homolog (PTEN) expression. PTEN, as an apoptosis-related protein, inhibited PI3K pathway and prevented Akt activation. Therefore, it can be said that miR-21 upregulation reduced H/R-induced apoptosis in one hand, and had a negative regulatory role on autophagy and inhibited excessive autophagy activity in the other hand, which were partly mediated by PTEN/Akt/mTOR signaling pathway [28].
Puerarin.
A study revealed the roles of LncRNA ANRIL and puerarin in myocardial I/R injury. Prolonged hypoxia led to the downregulation of LncRNA ANRIL expression in cardiomyocytes, and reduction of cardiomyocyte viability. On the other hand, prolonged reoxygenation increased apoptosis. However, puerarin reversed the effects of H/R damage on the expression of ANRIL, the viability of cardiomyocyte, LDH and MDA contents, apoptosis, and the expressions of autophagy-related genes. In addition, in vivo experiments showed that puerarin protected cardiac tissues by ANRIL upregulation and autophagy inhibition [20].
Salidroside (Sal).
Circular RNA (circRNA), a highly stable non-coding RNA with covalently closed loop structure, plays important role in cardiac I/R damage. A study documented that pretreatment with Sal in I/R rats led to the improvement of myocardial function, reduction of infarct size, LDH and CK-MB levels, and inhibition of anti-oxidative stress, autophagy and apoptosis. It was revealed that pretreatment with Sal attenuated autophagy via upregulation of hsa_circ_0000064 (circ-0000064) expression. So pretreatment with Sal had cardioprotective effect against myocardial I/R injury, which might be mediated by circ-0000064 upregulation and autophagy inhibition [31].
Radioprotective 105 kDa protein (RP105).
RP105, a toll-like receptor 4 (TLR4) homologue, is a specific inhibitor of TLR4. Guo et al. demonstrated that overexpression of RP105 by intramyocardial injection of adenoviral vector encoding EGFP-RP105 (Ad-EGFP-RP105) before induction of I/R caused cardioprotection by inhibiting apoptosis and autophagy, which was confirmed by decreased levels of CK, LDH, and tissue apoptosis. Accordingly, RP105 decreased the expressions of TLR4, p-nuclear factor κB (NF-κB)/p65 and interleukin 6 (IL-6)/tumor necrosis factor α (TNF-α), and finally reduced apoptosis. In other words, RP105 decreased the expressions of proapoptotic cytokines such as TNF-α and IL-6 by modulating TLR4/NF-κB signaling pathway. It also had an inhibitory effect on autophagy by inactivating TLR4/p-NF-κB/p65 signaling pathway, resulting Beclin1 and LC3-II/LC3-I downregulations. In addition, RP105 increased the expression of Bcl-2, which may play an important role in autophagy inhibition by increasing the binding between Beclin1 and Bcl-2. In general, the anti-apoptotic and anti-autophagic effects of RP105 in myocardial I/R condition were mediated by inhibition of TLR4/NF-κB pathway [15].
Danshensu (DSS).
In confirming the damaging effect of excessive autophagy in cardiac I/R or H/R condition, a study by Fan et al. showed that mTOR activity was inhibited under I/R condition, which led to the excessive autophagy flux, autophagic cell death, and consequently cardiac dysfunction. However, ischemia-induced cell death and cardiac dysfunction were reduced by DSS treatment via activation of mTOR, and phosphorylation of ribosomal protein S6 kinase beta-1 (S6K1) and S6. So DSS reduced I/R-induced autophagy and ultimately prevented cardiac injury by upregulating mTOR signaling and reducing autophagosomes. Therefore, DSS negatively regulated autophagy, which was evidenced by decreased Beclin1, LC3-II, and p62. DSS also reduced ischemia-induced apoptosis in cardiomyocytes. It was revealed that mTOR activation was associated with stimulation of Bcl-2 and downregulation of Bax and caspase 3 proteins. It means that DSS affected both autophagic and apoptotic pathways through mTOR activation. So DSS inhibited excessive autophagy and apoptosis by activating mTOR [7].
17-Methoxyl-7-hydroxy-benzene-furanchalcone (MHBFC).
Another study showed that treatment with MHBFC had beneficial effects in cardiac I/R condition by reducing Beclin1 and LC3-II/LC3-I expressions. Decreased CK-MB levels indicated membrane-stabilizing ability and protective function of MHBFC in myocardial I/R. MHBFC increased PI3K/Akt pathway activity, and increased mTOR phosphorylation as an important downstream of this pathway. So MHBFC prevented the harmful effects of excessive autophagy in myocardial I/R via PI3K/Akt/mTOR pathway. On the other hand, MHBFC postconditioning decreased TNF-α and increased p-eNOS, NO production, and p-Akt. It means that MHBFC inhibited MPTP opening and also apoptosis by increasing Akt-eNOS activation and NO production, leading to the positive effects on the heart in I/R. Thus, it can be said that the anti-apoptotic mechanism of MHBFC in response to myocardial I/R injury was probably mediated by inhibition of MPTP opening. PI3K/Akt signaling as a pro-survival pathway, played a key role in anti-apoptotic function of MHBFC in myocardial I/R. Overall, it was concluded that the cardioprotective effect of MHBFC against myocardial I/R may be associated with inhibition of excessive autophagy and apoptosis by regulation of Beclin1/Bcl-2 expression, which requires activation of PI3K/Akt/mTOR pathway [86].
Bauhinia championii flavone (BCF).
An investigation showed that BCF had beneficial effects in myocardial I/R injury in rats by preventing I/R-induced excessive autophagy and apoptosis, which was probably related to the BCF ability to activate PI3K/Akt signaling pathway. BCF was able to reduce excessive autophagy by increasing PI3K, and decreasing Beclin1 and apoptosis by modulating Beclin1/Bcl-2 expression. BCF activated p-eNOS and also increased NO production through upregulation of p-Akt expression. Because NO has anti-inflammatory and anti-apoptotic effects in I/R, so BCF was able to prevent MPTP opening and apoptosis. It means that BCF exerted its cardioprotective effect by inhibiting the mitochondrial pathway of apoptosis. TNF-α can also activate the external apoptotic pathway via binding to the TNF receptor, which was reduced by BCF, and as a result, BCF also reduced apoptosis in this way [30].
Berberine.
Myocardial I/R-induced excessive autophagy flux or autophagic cell death causes cytosol and organelles damages, which ultimately leads to the cell dysfunction and death. But regulated autophagy has a protective role against cell death. Accordingly, berberine inhibited excessive autophagy, and protected the myocardium and H9c2 cells against I/R or H/R injury by preventing AMPK and mTORC2 activation. It means that berberine induced cardioprotection by inhibiting the upstream pathways of autophagy. It was also shown that the potential biomarker of excessive autophagy in H/R is Bnip3, and berberine inhibited excessive autophagy by preventing mitochondrial expression of Bnip3. Bnip3 expression seems to be more sensitive to berberine treatment than Beclin1. Berberine also maintained ATP levels in H9c2 cells, indicating that berberine was able to protect mitochondrial function by inhibiting Bnip3. In both I/R and H/R conditions, upregulation of autophagy occurred with increased expressions of Beclin1, Bnip3, and SIRT1. But treatment with berberine reduced the induction of these proteins [27].
ADT
One study showed that injection of ADT [5-(4-methoxyphenyl)-3H-1,2-dithiole-3-thione] at the beginning of reperfusion, which is a slow-releasing organic hydrogen sulfide (H2S) donor, caused a continuous increase in serum H2S levels and protected the heart against I/R damage. This protective effect of ADT was done by activating AMPK and thus restoring I/R-induced impair autophagy flux. As mentioned above, AMPK activates protective autophagy in myocardial ischemia. But in reperfusion phase, there is no increase in AMPK activity, but Beclin1 upregulation and decreased LAMP2 cause increase in autophagosomes, which intensifies infarct damage in this phase. It shows that myocardial I/R stimulates autophagosomes formation, but not lysosome-autophagosome formation. Therefore, it leads to the impaired clearance of autophagosomes, and thus aggravates myocardial I/R injury. It was found that slow release of H2S by administration of ADT after ischemia protected the heart against I/R damage by activating AMPK and thus restoring I/R-induced impair autophagy flux [82].
Basic fibroblast growth factor (bFGF).
Treatment with recombinant bFGF increased survival of cardiomyocytes and improved cardiac function via modulation of autophagy-mediated ubiquitination. bFGF exerted its protective effects by reducing excessive autophagy via activating PI3K/Akt/mTOR signaling pathway. bFGF also inhibited rapamycin-induced autophagy. It was found that bFGF caused increase in clearance of ubiquitinated proteins and p62 expression following cardiac I/R. In general, the beneficial effects of bFGF in reducing myocardial I/R injury were through inhibition of excessive autophagy and reversing the toxic accumulation of ubiquitinated proteins by p62 pathway, as well as reduction of apoptosis [78].
Sevoflurane.
Xie et al. showed that sevoflurane preconditioning led to the reduction of H/R-induced excessive autophagy partly by reducing Beclin1 expression and the number of autophagosomes and lysosomes, indicating the important role of Beclin1 in autophagocytosis in the heart. Sevoflurane preconditioning also increased Bcl-2 and decreased apoptosis in H9c2 cells. In general, sevoflurane preconditioning induced delayed protection in cardiomyocytes by inhibiting Beclin1-induced autophagic cell death [81].
Insulin, Tat-SabKIM1.
In I/R condition, increase in mitochondrial JNK (mito JNK) activation causes cell death through the releasing of cytochrome c from the mitochondria, and on the other hand causes myocardial injury via Bcl-2-regulated autophagy. However, treatment with insulin reduced cytochrome c release from the mitochondria via decreasing mitochondrial and cytosolic/nuclear JNK activation, thereby reduced myocardial injury following I/R. On the other hand, it decreased autophagy which was evidenced by decreased Beclin1 and LC3-II/LC3-I, and increased p62. Tat-SabKIM1 is a retro-inverse peptide which is capable to block JNK interaction with mitochondria. Treatment with Tat-SabKIM1 reduced mitochondria-mediated apoptosis by inhibiting mito JNK activation and reducing cytochrome c release from the mitochondria. It also reduced I/R-induced autophagy and myocardial I/R injury, and improved heart function. Thus, treatment with insulin or Tat-SabKIM1 was shown to have cardioprotective effect against I/R by inhibiting mito JNK and reducing apoptosis and Bcl-2-regulated autophagy [85].
Pyrrolidine dithiocarbamate (PDTC).
The results of a study by Zeng et al. showed that the expression of p65 protein and its phosphorylation in serine 536 were increased in myocardial area at risk (AAR), and caused upregulation of Beclin1 mRNA and protein levels. Thus, p65 is involved in I/R-induced autophagy by mediating Beclin1. It means that in response to ROS release, NF-κB promotes Beclin1-dependent autophagy and leads to heart damage in I/R condition. Inhibition of NF-κB activity may protect cardiomyocytes against damaging autophagy in myocardial I/R. Accordingly, administration of PDTC (as NF-κB inhibitor) inhibited ROS release and p65 activity in AAR. Therefore, PDTC inhibited I/R-induced Beclin1 activation and autophagosomes formation. In other words, PDTC treatment reduced heart injury via inhibiting p65-dependent autophagy [89].
Hydrogen sulfide (H2S).
A study showed that in porcine model of myocardial I/R, continuous infusion of H2S, which was started at the beginning of ischemia and continued during I/R, reduced apoptosis, necrosis and infarct size, as well as improved coronary microvascular reactivity and heart function. On the other hand, it caused an increase in p-eNOS. So it can be said that H2S infusion-induced improvement of coronary microvascular relaxation was associated with an increase in p-eNOS. H2S infusion also increased the phosphorylated form of GSK-3β and decreased apoptosis-inducing factor (AIF) expression, which were associated with cytoprotection and reduced infarct size. However, following bolus treatment within 10 s at the beginning of reperfusion, increase in extracellular signal-regulated kinase (ERK) 1/2 expression occured. H2S bolus also increased phosphorylated form of Bcl-2 associated death promoter (p-Bad) at serine 136 and decreased cleaved caspase 3. However, it was found that this reduction in apoptosis by H2S bolus did not improve infarct size, vascular reactivity or contractility. While H2S infusion improved these outcomes, although it had no effect on p-Bad and cleaved caspase 3. Therefore, it can be concluded that H2S infusion or bolus administration led to various intracellular events. Interestingly, it was shown that H2S treatment inhibited autophagy in myocardial I/R injury. Because H2S infusion increased p-mTOR and H2S bolus reduced Beclin1 expression. In general, the cardioprotective effect of H2S on I/R seems to be via improving coronary microvascular relaxation, increasing survival proteins expression, and decreasing autophagy and apoptosis. However, H2S infusion provided better cardioprotection than H2S bolus and was superior to bolus treatment [60].
Urocortin.
The results of in vitro experiment by Valentim et al. revealed that single cycle of cardiac I/R increased Beclin1 expression and autophagy in neonatal and adult myocytes. However, inhibition of autophagy by 3-methyl-adenine (3-MA) or Beclin1 knockdown inhibited cell death and increased cardiomyocytes survival after sI/R, suggesting that autophagy was probably an additional cell death mechanism following a single cycle of I/R, at least in this in vitro model. Pretreatment with urocortin, an endogenous cardiac peptide, reduced I/R-induced autophagy by reducing Beclin1 expression. The inhibitory effect of urocortin on Beclin1 expression was eliminated by PI3K inhibitors, which proved the inhibitory role of urocortin on Beclin1 via PI3K/Akt signaling pathway. Thus, the beneficial effects of urocortin against autophagocytosis were probably mediated through this mechanism. It was found that urocortin also provided cardioprotection against apoptosis through PI3K/Akt signaling pathway. However, by inhibiting mitogen-activated protein/extracellular signal-regulated kinase (MEK)1-ERK pathway, autophagic cell death was not modulated, while it had an effect on apoptotic cell death. Therefore, it can be concluded that PI3K played an important role in the inhibitory effects of urocortin on autophagy, but both PI3K and MEK kinase played important roles in cardioprotective effects of urocortin against cell death. It means that urocortin promoted survival of cardiac myocytes against sI/R injury through PI3K/Akt and MEK 1/2 p42/44 mitogen-activated protein kinase (MAPK) pathways. Urocortin, which was able to inhibit autophagy, apoptosis, and necrosis, could play a protective role against I/R injury. Thus, the clinical use of urocortin may provide more complete protection against myocardial I/R compared with agents that interfered with only one cell death pathway [75].
Conclusion and future directions
According to the findings presented in this review, it was revealed that higher or lower activity of autophagy is detrimental and is involved in myocardial I/R damage. Therefore, modulation of autophagy as a potential therapeutic target can significantly affect heart function. Further investigations and better clarifications of the complex function of autophagy in myocardial I/R may help to discover and develop novel therapeutic strategies in order to achieve cardioprotection [42]. It means that finding new therapeutic agents that stimulate or inhibit autophagy in the heart can lead to normal physiological range of autophagy in ischemia and reperfusion [25]. There are challenges in this regard which need to be considered and clarified carefully. If all these challenges are clarified and resolved successfully, the idea of “autophagy modulation as a treatment for myocardial I/R injury” will merit clinical researches. One of these challenges that can be mentioned is that the exact conditions and therapeutic window in which autophagy induction leads to cardioprotection have not been determined, and need further investigation. In other words, how to maintain appropriate autophagy flux and control excessive autophagy are important points for therapeutic interest. It should also be considered that pharmacological agents target autophagy selectively and specifically, and do not simply lead to the autophagosome augmentation and impair autophagy [14]. On the other hand, although many pharmacological agents showed promising results in animal studies, unfortunately most of them have failed in clinical setting. Because most of preclinical studies have been performed in young and healthy animals. But clinical studies include patients with co-morbidities such as metabolic syndrome, diabetes, hypertension, and aging. It has been observed that many of these co-morbidities may interfere with autophagy, which can lead to the failure of cardioprotection in patients [1, 8].
It can be concluded that therapeutic agents that directly target autophagy to induce controlled autophagy or inhibit inappropriate autophagy can be considered as potential therapeutic option to protect cardiomyocytes against I/R injury. As well as, clarification of specific cellular mechanisms which leads to the appropriate level of autophagy flux with appropriate timing will probably help to develop new cardioprotective strategies for ischemic heart diseases in the future [51].
References
Badalzadeh R, Azimi A, Alihemmati A, Yousefi B (2017) Chronic type-I diabetes could not impede the anti-inflammatory and anti-apoptotic effects of combined postconditioning with ischemia and cyclosporine A in myocardial reperfusion injury. J Physiol Biochem 73:111–120
Chen WR, Liu HB, Dai Chen Y, Sha Y, Ma Q, Zhu PJ, Mu Y (2018) Melatonin attenuates myocardial ischemia/reperfusion injury by inhibiting autophagy via an AMPK/mTOR signaling pathway. Cell Physiol Biochem 47:2067–2076
Chong ZZ, Shang YC, Maiese K (2011) Cardiovascular disease and mTOR signaling. Trends Cardiovasc Med 21:151–155
Davidson SM, Adameová A, Barile L, Cabrera-Fuentes HA, Lazou A, Pagliaro P, Stensløkken KO, Garcia-Dorado D, Action ECC (2020) Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J Cell Mol Med 24:3795–3806
Dong Y, Undyala VV, Gottlieb RA, Mentzer RM Jr, Przyklenk K (2010) Autophagy: definition, molecular machinery, and potential role in myocardial ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther 15:220–230
Duan Q, Yang W, Jiang D, Tao K, Dong A, Cheng H (2016) Spermine ameliorates ischemia/reperfusion injury in cardiomyocytes via regulation of autophagy. Am J Transl Res 8:3976–3985
Fan G, Yu J, Asare PF, Wang L, Zhang H, Zhang B, Zhu Y, Gao X (2016) Danshensu alleviates cardiac ischaemia/reperfusion injury by inhibiting autophagy and apoptosis via activation of mTOR signalling. J Cell Mol Med 20:1908–1919
Ferdinandy P, Schulz R, Baxter GF (2007) Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59:418–458
Fu H, Li X, Tan J (2018) NIPAAm-MMA nanoparticle-encapsulated visnagin ameliorates myocardial ischemia/reperfusion injury through the promotion of autophagy and the inhibition of apoptosis. Oncol Lett 15:4827–4836
Fujiwara Y, Furuta A, Kikuchi H, Aizawa S, Hatanaka Y, Konya C, Uchida K, Yoshimura A, Tamai Y, Wada K (2013) Discovery of a novel type of autophagy targeting RNA. Autophagy 9:403–409
Fujiwara Y, Kikuchi H, Aizawa S, Furuta A, Hatanaka Y, Konya C, Uchida K, Wada K, Kabuta T (2013) Direct uptake and degradation of DNA by lysosomes. Autophagy 9:1167–1171
Ghosh R, Pattison JS (2018) Macroautophagy and chaperone-mediated autophagy in heart failure: the known and the unknown. Oxid Med Cell Longev 2018:8602041
Gottlieb RA, Carreira RS (2010) Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol-Cell Physiol 299:C203–C210
Gottlieb RA, Mentzer RM Jr (2010) Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol 72:45–59
Guo X, Jiang H, Yang J, Chen J, Yang J, Ding J-W, Li S, Wu H, Ding H-S (2016) Radioprotective 105 kDa protein attenuates ischemia/reperfusion-induced myocardial apoptosis and autophagy by inhibiting the activation of the TLR4/NF-κB signaling pathway in rats. Int J Mol Med 38:885–893
Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK (2009) Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med 13:373–387
Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK (2010) Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res 86:103–112
Gustafsson ÅB, Gottlieb RA (2003) Mechanisms of apoptosis in the heart. J Clin Immunol 23:447–459
Hamacher-Brady A, Brady NR, Gottlieb RA (2006) The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther 20:445–462
Han Y, Wang H, Wang Y, Dong P, Jia J, Yang S (2021) Puerarin protects cardiomyocytes from ischemia–reperfusion injury by upregulating LncRNA ANRIL and inhibiting autophagy. Cell Tissue Res. https://doi.org/10.1007/s00441-021-03463-2
Hang P, Zhao J, Su Z, Sun H, Chen T, Zhao L, Du Z (2018) Choline Inhibits ischemia-reperfusion-induced cardiomyocyte autophagy in rat myocardium by activating Akt/mTOR signaling. Cell Physiol Biochem 45:2136–2144
Hariharan N, Zhai P, Sadoshima J (2011) Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 14:2179–2190
Hosseini L, Vafaee MS, Badalzadeh R (2020) Melatonin and nicotinamide mononucleotide attenuate myocardial ischemia/reperfusion injury via modulation of mitochondrial function and hemodynamic parameters in aged rats. J Cardiovasc Pharmacol Ther 25:240–250
Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM Jr, Gottlieb RA (2010) Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol-Heart Circ Physiol 298:H570–H579
Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Gottlieb RA (2010) Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 3:365–373
Huang K-y, Wang J-n, Zhou Y-y, Wu S-z, Tao L-y, Peng Y-p, Que J-q, Xue Y-j, Ji K-t (2019) Antithrombin III alleviates myocardial ischemia/reperfusion injury by inhibiting excessive autophagy in a Phosphoinositide 3-kinase/Akt-dependent manner. Front Pharmacol. https://doi.org/10.3389/fphar.2019.00516
Huang Z, Han Z, Ye B, Dai Z, Shan P, Lu Z, Dai K, Wang C, Huang W (2015) Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur J Pharmacol 762:1–10
Huang Z, Wu S, Kong F, Cai X, Ye B, Shan P, Huang W (2017) Micro RNA-21 protects against cardiac hypoxia/reoxygenation injury by inhibiting excessive autophagy in H9c2 cells via the Akt/mTOR pathway. J Cell Mol Med 21:467–474
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126:955–968
Jian J, Xuan F, Qin F, Huang R (2015) Bauhinia championii flavone inhibits apoptosis and autophagy via the PI3K/Akt pathway in myocardial ischemia/reperfusion injury in rats. Drug Des Devel Ther 9:5933
Jin P, Li L-H, Shi Y, Hu N-B (2021) Salidroside inhibits apoptosis and autophagy of cardiomyocyte by regulation of circular RNA hsa_circ_0000064 in cardiac ischemia-reperfusion injury. Gene 767:145075
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan K-L (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152:290–303
Kim J, Kundu M, Viollet B, Guan K-L (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141
Lee IH, Finkel T (2009) Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 284:6322–6328
Lekli I, Ray D, Mukherjee S, Gurusamy N, Ahsan MK, Juhasz B, Bak I, Tosaki A, Gherghiceanu M, Popescu LM (2010) Co-ordinated autophagy with resveratrol and γ-tocotrienol confers synergetic cardioprotection. J Cell Mol Med 14:2506–2518
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Li W-w, Li J, Bao J-k (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136
Li X, Hu X, Wang J, Xu W, Yi C, Ma R, Jiang H (2018) Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway contributes to the protection of hesperidin against myocardial ischemia/reperfusion injury. Int J Mol Med 42:1917–1924
Liang J, Shao SH, Xu Z-X, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL (2007) The energy sensing LKB1–AMPK pathway regulates p27 kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 9:218–224
Liang S, Ping Z, Ge J (2017) Coenzyme Q10 regulates antioxidative stress and autophagy in acute myocardial ischemia-reperfusion injury. Oxid Med Cell Longev 2017:9863181
Lin S-Y, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang S-M, Lian G, Liu Q, Ruan K (2012) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Sci 336:477–481
Lin XL, Xiao WJ, Xiao LL, Liu MH (2018) Molecular mechanisms of autophagy in cardiac ischemia/reperfusion injury. Mol Med Rep 18:675–683
Liu L, Jin X, Hu C-F, Li R, Shen C-X (2017) Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways. Cell Physiol Biochem 43:52–68
Lv XW, Wang MJ, Qin QY, Lu P, Qin GW (2021) 6-Gingerol relieves myocardial ischaemia/reperfusion injury by regulating lncRNA H19/miR-143/ATG7 signaling axis-mediated autophagy. Lab Invest. https://doi.org/10.1038/s41374-021-00575-9
Ma S, Wang Y, Chen Y, Cao F (2015) The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim Biophys Acta Mol Basis Dis 1852:271–276
Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A (2012) Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circ 125:3170–3181
Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP, Ho CY (2009) Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 301:1253–1259
Matsui Y, Kyoi S, Takagi H, Hsu C-P, Hariharan N, Ago T, Vatner SF, Sadoshima J (2008) Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy 4:409–415
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100:914–922
Mijaljica D, Prescott M, Devenish RJ (2011) Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7:673–682
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873
Mo Y, Tang L, Ma Y, Wu S (2016) Pramipexole pretreatment attenuates myocardial ischemia/reperfusion injury through upregulation of autophagy. Biochem Biophys Res Comm 473:1119–1124
Mokhtari B, Azizi Y, Abookheili AR, Aboutaleb N, Nazarinia D, Naderi N (2020) Human amniotic membrane mesenchymal stem cells-conditioned medium attenuates myocardial ischemia-reperfusion injury in rats by targeting oxidative stress. Iran J Basic Med Sci 23:1453–1461
Mokhtari B, Badalzadeh R, Aboutaleb N (2021) Modulation of autophagy as the target of mesenchymal stem cells-derived conditioned medium in rat model of myocardial ischemia/reperfusion injury. Mol Biol Rep. https://doi.org/10.1007/s11033-021-06359-0
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624
Naseroleslami M, Sharifi M, Rakhshan K, Mokhtari B, Aboutaleb N (2020) Nesfatin-1 attenuates injury in a rat model of myocardial infarction by targeting autophagy, inflammation, and apoptosis. Arch Physiol Biochem 7:1–9
Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K (2009) The role of autophagy in the heart. Cell Death Differ 16:31–38
Nishida K, Taneike M, Otsu K (2015) The role of autophagic degradation in the heart. J Mol Cell Cardiol 78:73–79
O’Neal W, Griffin W, Kent S, Virag J (2012) Cellular pathways of death and survival in acute myocardial infarction. J Clin Exp Cardiol. https://doi.org/10.4172/2155-9880.S6-003
Osipov RM, Robich MP, Feng J, Liu Y, Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C, Sellke FW (2009) Effect of hydrogen sulfide in a porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J Cardiovasc Pharmacol 54:287–297
Qiao S-g, Sun Y, Sun B, Wang A, Qiu J, Hong L, An J-z, Wang C, Zhang H-l (2019) Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism. Acta Pharmacol Sin 40:35–45
Sala-Mercado JA, Wider J, Reddy Undyala VV, Jahania S, Yoo W, Mentzer RM Jr, Gottlieb RA, Przyklenk K (2010) Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circ 122:S179–S184
Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596–603
Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760
Schiattarella GG, Hill JA (2016) Therapeutic targeting of autophagy in cardiovascular disease. J Mol Cell Cardiol 95:86–93
Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J (2011) Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol 32:275–281
Shang L, Chen S, Du F, Li S, Zhao L, Wang X (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci 108:4788–4793
Shaw RJ (2009) LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta physiol 196:65–80
Song H, Yan C, Tian X, Zhu N, Li Y, Liu D, Liu Y, Liu M, Peng C, Zhang Q (2017) CREG protects from myocardial ischemia/reperfusion injury by regulating myocardial autophagy and apoptosis. Biochim Biophys Acta Mol Basis Dis 1863:1893–1903
Sridhar S, Botbol Y, Macian F, Cuervo AM (2012) Autophagy and disease: always two sides to a problem. J Pathol 226:255–273
Takagi H, Matsui Y, Sadoshima J (2007) The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal 9:1373–1382
Tang Z, Yang L, Zhang X (2017) Vitexin mitigates myocardial ischemia reperfusion-induced damage by inhibiting excessive autophagy to suppress apoptosis via the PI3K/Akt/mTOR signaling cascade. RSC Adv 7:56406–56416
Thapalia BA, Zhou Z, Lin X (2014) Autophagy, a process within reperfusion injury: an update. Int J Clin Exp Pathol 7:8322–8341
Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A (2006) Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol 40:846–852
Wang J-L, Li Y-Z, Tao T-Q, Wang X-R, Wang Y, Song D-D, Liu X-H (2020) Postconditioning with calreticulin attenuates myocardial ischemia/reperfusion injury and improves autophagic flux. Shock 53:363–372
Wang L-q, Cheng X-s, Huang C-h, Huang B, Liang Q (2015) Rapamycin protects cardiomyocytes against anoxia/reoxygenation injury by inducing autophagy through the PI3k/Akt pathway. J Huazhong Univ Sci Technolog Med Sci 35:10–15
Wang ZG, Wang Y, Huang Y, Lu Q, Zheng L, Hu D, Feng WK, Liu YL, Ji KT, Zhang HY (2015) bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci Rep 5:1–12
Wei C, Li H, Han L, Zhang L, Yang X (2013) Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J Cardiovasc Pharmacol 61:416–422
Wu Y, Mao Q, Liang X (2020) Targeting the MicroRNA-490-3p-ATG4B-Autophagy Axis Relieves Myocardial Injury in Ischemia Reperfusion. J Cardiovasc Transl Res 14:173–183
Xie H, Liu Q, Qiao S, Jiang X, Wang C (2015) Delayed cardioprotection by sevoflurane preconditioning: a novel mechanism via inhibiting Beclin 1-mediated autophagic cell death in cardiac myocytes exposed to hypoxia/reoxygenation injury. Int J Clin Exp Pathol 8:217–226
Xie H, Xu Q, Jia J, Ao G, Sun Y, Hu L, Alkayed NJ, Wang C, Cheng J (2015) Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Comm 458:632–638
Xie M, Kong Y, Tan W, May H, Battiprolu P, Pedrozo Z, Wang Z, Jiang N, Warner J, Gillette TG (2013) HDAC inhibition blunts ischemia/reperfusion injury by normalizing cardiomyocyte autophagy. J Am Coll Cardiol 61:E2111
Xie M, Morales CR, Lavandero S, Hill JA (2011) Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol 26:216–222
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J, Zhang L, Tang Y, Liu J, Zhang X (2015) Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion. Biochim Biophys Acta Mol Basis Dis 1852:262–270
Xuan F, Jian J, Lin X, Huang J, Jiao Y, Huang W, Li J, Shi Z, Huang R (2017) 17-Methoxyl-7-Hydroxy-Benzene-Furanchalcone Ameliorates Myocardial Ischemia/Reperfusion Injury in Rat by Inhibiting Apoptosis and Autophagy Via the PI3K–Akt Signal Pathway. Cardiovasc Toxicol 17:79–87
Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W (2012) Function and molecular mechanism of acetylation in autophagy regulation. Sci 336:474–477
Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson ÅB, Mentzer RM, Gottlieb RA (2009) Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol 104:157–167
Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y, Chen J, Wang P, Fei Y (2013) NF-κB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem Biophys Res Comm 436:180–185
Zhang L, Wang H, Zhao Y, Wang J, Dubielecka PM, Zhuang S, Qin G, Chin YE, Kao RL, Zhao TC (2018) Myocyte-specific overexpressing HDAC4 promotes myocardial ischemia/reperfusion injury. Mol Med 24:37
Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang H, Ren J, Zang W-J (2013) Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol Biochem 32:601–613
Zhao R, Xie E, Yang X, Gong B (2019) Alliin alleviates myocardial ischemia-reperfusion injury by promoting autophagy. Biochem Biophys Res Comm 512:236–243
Zheng D, Liu Z, Zhou Y, Hou N, Yan W, Qin Y, Ye Q, Cheng X, Xiao Q, Bao Y (2020) Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol Res 153:104655
Zheng Y, Gu S, Li X, Tan J, Liu S, Jiang Y, Zhang C, Gao L, Yang H-T (2017) Berbamine postconditioning protects the heart from ischemia/reperfusion injury through modulation of autophagy. Cell Death Dis 8:e2577–e2577
Zhong Y, Zhong P, He S, Zhang Y, Tang L, Ling Y, Fu S, Tang Y, Yang P, Luo T (2017) Trimetazidine protects cardiomyocytes against hypoxia/reoxygenation injury by promoting amp-activated protein kinase–dependent autophagic flux. J Cardiovasc Pharmacol 69:389–397
Zuo Z, Zuo P-f, Sheng Z-l, Wang X, Ding J-d, Ma G-s (2019) Tetramethylprazine attenuates myocardial ischemia/reperfusion injury through modulation of autophagy. Life Sci 239:117016
Author information
Authors and Affiliations
Contributions
BM had the idea for the article. BM performed the literature search and data collecting, drafted the manuscript, and designed the figures and tables. RB edited the language and critically revised the manuscript. All gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
1. Higher or lower activity of autophagy may have harmful effects in the heart.
2. Excessive or insufficient autophagy flux is involved in myocardial I/R damage.
3. Modulation of autophagy is potential therapeutic target in myocardial I/R injury.
Rights and permissions
About this article

Cite this article
Mokhtari, B., Badalzadeh, R. The potentials of distinct functions of autophagy to be targeted for attenuation of myocardial ischemia/reperfusion injury in preclinical studies: an up-to-date review. J Physiol Biochem 77, 377–404 (2021). https://doi.org/10.1007/s13105-021-00824-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-021-00824-x