
Abstract
We investigated the potential role of miR-490-3p in ischemia reperfusion (IR) injury. We first determined the expression of miR-490-3p and autophagy-related 4B cysteine (ATG4B) in IR. Then, to explore whether miR-490-3p would affect autophagy, apoptosis, and IR injury, we evaluated apoptosis, autophagy, and infarct size via gain- and loss-of-function experiments. Furthermore, we used adenovirus to enhance or inhibit the expression of ATG4B, and then measured autophagy, apoptosis, and IR injury. miR-490-3p was downregulated in the hearts during the process of IR, while ATG4B was upregulated. The inhibition of miR-490-3p or overexpression of ATG4B could promote the expression of LC3II, increase the autolysosomes, inhibit the expression of p62, and reduce infarct size. On all accounts, the inhibition of miR-490-3p could promote autophagy to reduce myocardial IR injury by upregulating ATG4B, a finding that provides new insights for the protective mechanism of autophagy in IR.

Graphical Abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemia reperfusion (IR) is a pathological condition caused by the immediate recovery of perfusion of blood after the restricted blood supply to organs [1]. The nature of the injury caused by IR is very complex and influenced by various factors [2], and can lead to significant morbidity and mortality in myocardial infarction and ischemic stroke; in addition, IR injury represents a huge challenge for organ transplantation and cardiothoracic surgery [1]. As a deleterious aspect of myocardial reperfusion injury, reperfusion injury is a deadly myocardial injury aroused by coronary blood flow restoration after an ischemic episode [3]. Myocardial IR injury can cause myocardial cell death (necrosis, apoptosis, and autophagy), myocardial cell hypertrophy, cardiac fibrosis, and impaired angiogenesis [4]. For instance, as an atypical serine/threonine kinase, mammalian protein kinase or mechanism target rapamycin (mTOR) plays an important role in ischemic injury [5]. Before ischemia, endoplasmic reticulum stress–induced autophagy has been elucidated to diminish following deadly IR injury [6]. Additionally, B cell lymphoma-2/adenovirus E1B 19-kDa interacting protein 3 is implicated in cardiomyocyte death upon IR injury, which can be attenuated by stimulating autophagy [7]. Moreover, autophagy is regarded as a protective mechanism in IR injury, since it regulates the bulk degradation and recycling of cytoplasmic contents, which can help maintain cellular homeostasis [8]. A previous study also proved that some degree of IR injury is inevitable in the process of heart transplantation, which can result in graft failures and lower long-term survival rate of patients, while certain microRNAs (miRNAs) proved to be essential regulators of signaling pathways involved in IR injury [9].
miRNAs are small (22-nucleotide long) noncoding RNA molecules that negatively regulate the mRNA expression of target genes via binding to the 3′untranslated region (3′UTR) [10]. Moreover, previous work shows a pivotal role of miRNAs in acute and chronic cardiovascular disease processes [4]. A prior study has proven that the downregulation of miR-148b can reduce myocardial IR injury [11]. Additionally, the importance of numerous autophagy-related (ATG) genes including ATG4 is now recognized both in selective and in nonselective types of autophagy [12]. For instance, research findings now implicate ATG16L1 in the process of ischemic kidney injury [13]. Another study showed that inhibition of miR-665-3p reduced inflammation and cell death in intestinal IR injury by negatively regulating the expression of ATG4B [14]. In the current study, we aimed to investigate the function of miR-490-3p in myocardial cell autophagy and in the process of IR, as well as the regulatory relationship of miR-490-3p and ATG4B, hoping to introduce a novel approach for heart disease treatment.
Materials and Methods
Ethics Statement
The protocol of animal experiments was approved by the Institutional Animal Care and Use Committee of Nanjing Lishui People’s Hospital, Zhongda Hospital Lishui Branch, Southeast University.
Microarray-Based Analysis
Cardiac IR mice–related miRNA expression dataset GSE50885 profiles and annotation files were retrieved from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The Affy package of the R software (http://www.bioconductor.org/packages/release/bioc/html/affy.html) was applied for standardized pretreatment of the expression datasets, and the limma package (http://master.bioconductor.org/packages/release/bioc/html/limma.html) was used to screen the differentially expressed genes. Genes with |logFoldChange (FC)| > 1 and p value < 0.05 were identified as differentially expressed, and their heatmaps were plotted using the pheatmap package (https://cran.r-project.org/web/packages/pheatmap/index.html). Target genes of the miRNAs were predicted using the StarBase (http://starbase.sysu.edu.cn/) database after the IR differential genes of miRNA were screened.
Dual-Luciferase Reporter Gene Assay
First, a synthetic ATG4B 3′UTR gene fragment was introduced into the pGL3-reporter (Promega, Madison, WI, USA) using the endonuclease sites XhoI and BamH I. The complementary sequence mutation (Mut) site of the seed sequence was designed based on the ATG4B wild-type (WT) sequence using restriction endonucleases. After endonuclease digestion, the target fragments were inserted into the pGL3-reporter vector with the T4 DNA ligase. Subsequently, the constructed luciferase reporter plasmids pGL3-ATG4B-WT and pGL3-ATG4B-Mut were co-transfected with 490-3p mimic or mimic-NC respectively to HEK293 cells (ATCC, Manassas, VA, USA). After 48 h of transfection, the cells were harvested and lysed. Next, luciferase activities were measured respectively on a Luminometer TD-20/20 detector (E5311, Promega, Madison, WI, USA) using a Dual-Luciferase® Reporter Assay System kit (Promega, Madison, WI, USA).
Cell Culture and Plasmid Delivery
HEK293 cells (ATCC, Manassas, VA, USA) were inoculated in a 6-well plate (at a density of 1 × 105 cells/mL) and cultured in a humidified incubator with 5% CO2 in air at 37 °C. The Dulbecco’s modified Eagle’s medium (Sigma-Aldrich Chemical Company, St Louis, MO, USA) was replaced every 2 or 3 days, and cell growth was observed under an inverted microscope. When cell confluence reached 75%, the cells were treated with following plasmids according to the instructions of the Lipofectamine 2000 kit in 6-well plates: blank, mimic-NC, miR-490-3p mimic, inhibitor-NC, and miR-490-3p inhibitor. The plasmids we employed were purchased from GenePharma Co., Ltd. (Shanghai, China). The protein expression of ATG4B was determined by western blot analysis after 48-h transfection.
Construction of IR Mouse Model
A total of 255 C57BL/6 adult male mice (aged 6 weeks, weighing 19–25 g) were purchased from the Institute of Laboratory Animals Science, Chinese Academy of Medical Sciences & Peking Union Medical College (Beijing, China). The mice were anesthetized with 1% sodium pentobarbital (P3761, Sigma-Aldrich Chemical Company, St Louis, MO, USA) and ventilated with an HX-300S animal ventilator. A small incision was made on the left side of the chest, and the pericardial sac was removed from the chest. Next, the adenovirus (2 × 1010 m.o.i.) or miR-490-3p agomir/antagomir (80 mg/kg body weight) and corresponding control treatments were introduced via a catheter from the left ventricular apex into the aortic root, while the aorta and pulmonary artery were cross-clamped. The clamp was held for 20 s when the cardiac output was circulated in closed system. Afterwards, the chest was closed, and the mice were returned to their cages for recovery after removal of air and blood. The employed adenoviral vectors and miR-490-3p agomir/antagomir were purchased from GenePharma Co., Ltd. (Shanghai, China).
The mice were subjected to IR operation 5 days after adenovirus or miR-490-3p agomir/antagomir injection. Next, the mice were anesthetized, and their chests were opened to expose the heart for identifying the left anterior descending coronary artery (LAD). A suture line was passed under the left pinna of the LAD, and the artery was blocked by applying a vinyl tube with a ligature, with occlusion of the LAD by tightening the trap and fixing it using a hemostat. After a 45-min ischemia, the ligature was released, and the heart was reperfused. Sham operations were performed with the same procedures except that the trap remained untied. Subsequently, the mice were divided into the following 17 groups (n = 15 each): (1) normal (mice without any treatment), (2) sham (sham-operated mice), (3) oe-NC (mice injected with empty adenovirus vector), (4) oe-ATG4B (mice injected with adenovirus vector harboring overexpressed ATG4B), (5) sh-NC (mice injected with adenovirus vector harboring shRNA), (6) sh-ATG4B (mice injected with adenovirus vector harboring silenced ATG4B), (7) IR (mice induced with IR only), (8) IR + agomir-NC (mice injected with agomir-NC and induced with IR), (9) IR + miR-490-3p agomir (mice injected with miR-490-3p agomir and induced with IR), (10) IR + antagomir-NC (mice injected with antagomir-NC and induced with IR), (11) IR + miR-490-3p antagomir (mice injected with miR-490-3p antagomir and induced with IR), (12) IR + oe-NC (mice injected with empty adenovirus vector and induced with IR), (13) IR + oe-ATG4B (mice injected with adenovirus vector harboring overexpressed ATG4B and induced with IR), (14) IR + sh-NC (mice injected with adenovirus vector harboring shRNA and induced with IR), (15) IR + sh-ATG4B (mice injected with adenovirus vector harboring silenced ATG4B and induced with IR), (16) IR + miR-490-3p antagomir + sh-NC (mice injected with miR-490-3p antagomir + adenovirus vector harboring shRNA and induced with IR), and (17) IR + miR-490-3p antagomir + sh-ATG4B (mice injected with miR-490-3p antagomir + adenovirus vector harboring silenced ATG4B and induced with IR). After a 3-h reperfusion, 5 mice in each group were randomly selected, anesthetized with 1% sodium pentobarbital, and then euthanatized. The heart of the euthanized mice was quickly excised, fixed for 24 h by immersion in 4% paraformaldehyde, and then cut into paraffin sections.
Index of Myocardial Function
After the mouse models were established for 7 days, 5 mice in each group were randomly selected and anesthetized with 1% sodium pentobarbital for cardiac ultrasound using a small animal ultrasound system with a probe frequency of RMV 704 (40 MHz). The probe acquired a left ventricular M-mode ultrasound image through the parasternal long axis and the short axis. The following data were measured and recorded: left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic volume (LVEDV), and left ventricular end-systolic volume (LVESV). The left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated using following formula: LVEF = [(LVEDV-LVESV)/LVEDV] × 100, LVFS = [(LVEDD-LVESD)/LVEDD] × 100. The average of three consecutive cardiac cycles was used for these measurements.
Terminal-Deoxynucleoitidyl Transferase–Mediated Nick End Labeling Assay
The aforementioned paraffin sections were dewaxed, hydrated, and sliced. Each section was added with 1% proteinase K dilution (50 μL) and incubated for 30 min at 37 °C. The sections were added with 0.3% H2O2 methanol solution to eliminate endogenous peroxidase (POD) activity, and incubated at 37 °C for a further 30 min. Afterwards, the sections were added with TUNEL reaction solution, incubated in a humid chamber at 37 °C for 1 h, added with converter-POD (50 μL), and incubated in a wet box at 37 °C for 30 min. Next, the tissue pieces were added with 2% diaminobenzidine color solution, incubated at room temperature for 15 min, and then observed under a microscope. Hematoxylin was added for counterstaining, and distilled water was used to terminate the reaction. The sections were then dehydrated with gradient ethanol, cleared by xylene, and sealed with a neutral gum. These sections were observed under an optical microscope (× 200), with positive cells showing brownish yellow nuclei and healthy cells as blue. A total of 10 visual fields of each section were randomly selected, and the positive cells and myocardial cells were counted respectively. The averaged values were taken, and the ratio of brown-yellow cells and blue cells was regarded as the apoptotic index of myocardial cells.
Reverse Transcription Quantitative Polymerase Chain Reaction
Total RNA was extracted from tissues and cells using TRIzol (Invitrogen, Carlsbad, CA, USA). After measuring the absorbances at 260 nm and 280 nm for measuring RNA concentration in Nanodrop 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA), the total RNA was reverse transcribed into cDNA using a PrimeScript™ RT reagent with gDNA Eraser kit (RRO37A, Takara Bio Inc., Otsu, Shiga, Japan) following the manufacturer’s instructions. In the meantime, the miRNA was reverse transcribed using a miScript RT kit (Qiagen company, Hilden, Germany). The specific primers were miR-490-3p (5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAGCATGG-3′) and U6 (5′-AACGCTTCACGAATTTGCGT-3′). The expression of miR-490-3p and other genes was detected using the SYBR® Premix Ex Taq™ (Tli RNaseH Plus) kit (RR820A, Takara Bio Inc., Otsu, Shiga, Japan) following the manufacturer’s instructions. RT-qPCR was performed on an ABI7500 qPCR machine (Thermo Fisher Scientific Inc., Waltham, MA, USA). β-Actin was regarded as the internal reference for genes, and U6 was taken as the internal reference for miR-490-3p. The expression levels were calculated using the 2−ΔΔCt method [15]. The primers used in the reaction (Table 1) were supplied by GenePharma Co., Ltd. (Shanghai, China).
Western Blot Analysis
The heart tissues from each group were lysed with phenylmethylsulfonyl fluoride and phosphatase inhibitor to isolate the total protein content. Protein (50 μg) was dissolved in 2 × sodium dodecyl sulfate (SDS) loading buffer and boiled at 100 °C. After 5 min, each sample was subjected to 10% SDS-polyacrylamide gel electrophoresis, and the proteins were transferred to a polyvinylidene fluoride membrane using the wet transfer method. The samples were blocked with 5% skim milk powder at room temperature for 1 h, after which the PVDF membrane was incubated with the primary rabbit anti-ATG4B (ab225882, dilution ratio of 1:2000), light chain 3B (LC3B) (ab51520, dilution ratio of 1:3000), p62 (ab91526, dilution ratio of 1:1000), and β-actin (ab8226, dilution ratio of 1:1000) (all from Abcam, Cambridge, MA, USA). The membrane was then incubated with horseradish peroxidase–labeled secondary antibodies (mouse, HS201-01; rabbit, HS101-01, both from TransGen Biotech Co., Ltd., Beijing, China) for 1 h. The images were obtained by Bio-Rad image analysis system (Bio-Rad, Richmond, CA, USA) and analyzed with the Quantity One v4.6.2 software (Bio-Rad, Richmond, CA, USA). The ratio of the corresponding protein band gray value and the β-actin protein band gray value was regarded as the relative protein content.
Transmission Electron Microscope
TEM was performed as previously described [16]. The mouse heart tissues were immersed in 4% paraformaldehyde at 4 °C and then fixed with 0.1 M sodium cacodylate buffer solution containing 1.5% glutaraldehyde at 4 °C. The tissues were then fixed with 1% osmium tetroxide, dehydrated with ethanol, treated with propylene oxide, and embedded in EMbed 812/Araldite (Electron Microscopy Sciences, Fort Washington, PA, USA). The sections were mounted on a grid of copper troughs coated with parlodion, stained with uranyl acetate and lead citrate, and photographed using a Philips CM100 electron microscope (FEI, Hillsboro, OR, USA).
Hematoxylin-Eosin Staining
The mouse heart tissues were dewaxed, hydrated, sliced, and then stained with hematoxylin. Next, the tissues were differentiated with 70% ethanol containing 0.25% HCl for 1 min, returned to blue color with 2% sodium bicarbonate solution, stained with eosin and dehydrated with gradient ethanol, and cleared by xylene. Histomorphological changes in myocardial cells were observed under an optical microscope after sealing. A total of 5 view fields were randomly selected for photography, and the surface area of mouse myocardial cells in HE staining in each group was measured with a fully automatic morphometric apparatus (HPIAS 21000, China).
Evans Blue and 2,3,5-Triphenyl Tetrazolium Chloride Staining
Three hours after reperfusion, 5 mice were randomly selected from each group and subjected to Evans Blue (EB) and TTC staining following a previously reported method [17]. In brief, after reperfusion, the blood flow of the coronary artery was blocked, followed by myocardial staining. Then, 2 mL EB solution (2%) was injected into the left atrium to distinguish ischemic myocardial tissues from nonischemic myocardial tissues, and the hearts were immediately removed. The noncardiac tissues, left ventricle (LV), and right ventricle were removed, and the residual blood was washed off using cold saline. The heart was frozen at − 20 °C for 10 min after removal of excess moisture, and then sliced into 1.5-mm-thick sections oriented vertical to cardiac axis. The blue area indicated the IR zone, and the light red areas referred to the area at risk (AAR). After capturing micrographs, the slices were immersed in 1% TTC in phosphate buffer solution (pH = 74.4) and incubated at 37 °C for 20 min. After final fixation with 10% formalin for 24 h, samples were photographed, with areas of infarct size (IS) white-colored and non-IS areas brick red in color. The percentage of IS was quantified as (IS/AAR) × 100%; the percentage of AAR was expressed as (AAR/LV) × 100%; and the percentage of IS in LV was summarized as (IS/LV) × 100% using the Image-Pro Plus software.
Statistical Analysis
All data were analyzed using the SPSS 21.0 software (IBM Corp., Armonk, NY, USA). Measurement data were described as mean ± standard deviation. Comparisons between two groups with normal distribution and equal variance were carried out using the unpaired t test. One-way analysis of variance (ANOVA) was applied to compare the data between multiple groups, followed by Tukey’s post hoc test. A value of p < 0.05 indicated statistical significance.
Results
miR-490-3p Was Poorly Expressed in IR Hearts
Initially, we performed a differential gene expression analysis on the miRNA dataset GSE50885 of IR model mice to study the miRNAs related to myocardial cell function during IR. Subsequently, we plotted a heatmap of the top 10 differentially expressed miRNAs according to their p values in an ascending order (Fig. 1a), which illustrated that miR-490-3p presented with the largest FC in IR mice, and its expression in IR mice was lower than in normal mice (logFC = − 2.23). To verify the results of the biological analysis, we constructed a mouse model of myocardial IR using LAD ligation. By testing the cardiac function of the model mice, LVESD and LVEDD were significantly increased in the IR mice compared with that in the normal and sham-operated mice, while LVEF and LVFS were significantly decreased (Fig. 1b–e), indicating that the model was successfully constructed. RT-qPCR was applied to detect the expression of miR-490-3p in the mouse model, which showed that the expression of miR-490-3p was significantly decreased in the IR mice when compared with that in the normal and sham-operated mice (Fig. 1f). These results were consistent with the biological analysis, confirming that miR-490-3p expression was downregulated in the hearts during IR process.

miR-490-3p expression was downregulated in IR mouse heart. a A heatmap of the top 10 differentially expressed miRNAs in the GSE50885 dataset. The abscissa is the sample number, and the ordinate indicates the differential miRNA expression; the right upper histogram is the color scale. Each rectangle in the figure corresponds to a sample expression value. The number in parentheses after the gene is logFold Change. b–e LVESD, LVEDD, LVEF, and LVFS expression in each group (n = 5). f Expression of miR-490-3p in myocardial tissues of each group (n = 5). *p < 0.05 vs. the sham-operated mice. The data are measurement data and described as mean ± standard deviation. One-way ANOVA was used for analysis among multiple groups, with Tukey’s post hoc test
Inhibition of miR-490-3p Expression Promoted Autophagy, Inhibited Apoptosis, and Relieved Myocardial Injury in IR Mice
After confirming that miR-490-3p expression was downregulated during IR, we shifted our focus on investigating whether miR-490-3p affected autophagy, apoptosis, and myocardial injury induced by IR. Consequently, we overexpressed or silenced miR-490-3p in IR model mice with the heart pump, and later examined apoptosis, autophagy, and IS. Western blot analysis demonstrated that, in comparison with sham-operated mice, IR mice presented with an increased expression of autophagy-associated protein LC3II in the heart tissues of mice and a decreased p62 protein expression. A similar trend was observed in miR-490-3p antagomir-treated IR mice compared with that in antagomir-NC-treated IR mice (Fig. 2a).

miR-490-3p inhibition enhanced autophagy, suppressed apoptosis, and relieved myocardial injury in IR mice. a Expression of autophagy-related proteins (LC3II and p62) in myocardial tissues normalized to GAPDH determined by western blot analysis. b Apoptosis of myocardial tissues detected by TUNEL (× 200). c Autophagy in myocardial tissues detected by TEM (× 20,000). d The pathological condition of myocardial tissues detected by HE staining (× 200). e The myocardial IS of myocardial tissues detected by TTC: AAR is in light red, IS area is in white, non-IS area is brick red; the percentage of IS was quantified as (IS/AAR) × 100%, *p < 0.05 vs. the IR group (IR-modeled mice), #p < 0.05 vs. the IR + agomir-NC group (mice treated with agomir-NC and induced with IR), &p < 0.05 vs. the IR + antagomir-NC group (mice treated with antagomir-NC and induced with IR). The data are measurement data and described as mean ± standard deviation. One-way ANOVA was used for the data analysis between multiple groups. Tukey’s post hoc test was used. n = 5. The experiment was repeated three times independently
Additionally, the TUNEL assay showed that the apoptosis in the heart tissues increased in IR mice compared with that in sham-operated mice, and likewise in IR mice treated with miR-490-3p agomir compared with that in IR mice treated with agomir-NC, while apoptosis was lower in IR mice treated with miR-490-3p antagomir (Fig. 2b). Moreover, TEM observation revealed that the number of autolysosomes was increased in the heart tissues in IR mice when compared with that in normal or sham-operated mice, and were further increased in IR mice that were treated with miR-490-3p antagomir, but decreased in the mice treated with miR-490-3p agomir (Fig. 2c).
Furthermore, HE demonstrated significantly increased myocardial injury (evidenced by increased necrotic area and neutrophilic inflammation, worsened myocardium and interstitial edema, and broken and split myocardial fibers) in IR mice compared with that in sham-operated mice, and likewise in IR mice treated with miR-490-3p agomir compared with those treated with agomir-NC, while these effects were rescued by miR-490-3p antagomir (Fig. 2d). In the meantime, the detection of IS in the heart tissue by EB/TTC staining revealed no white infarcted areas in normal and sham-operated mice, but significantly increased IS/AAR in IR mice compared with that in sham-operated mice and likewise in IR mice treated with miR-490-3p agomir compared with that in IR mice treated with agomir-NC. However, we saw significantly decreased IS/AAR in IR mice treated with miR-490-3p antagomir (Fig. 2e). These results indicated that the inhibition of miR-490-3p expression could promote autophagy, inhibit apoptosis, and alleviate myocardial injury in IR mice.
miR-490-3p Targeted ATG4B and Regulated the Expression of ATG4B
As a posttranscriptional regulator, miRNA can inhibit mRNA expression of genes by targeting the 3′UTR. The StarBase database predicted the target genes of miR-490-3p, showing that the autophagy-related gene ATG4B could be a potential target gene of miR-490-3p (Fig. 3a). ATG4B contains a C-terminal immunoglobulin-like receptor sequence motif that is important for binding and cleavage of ATG8 family proteins [18]. HsAtg4B, as the only enzyme reported to cleave efficiently LC3 precursors and LC3-PE, is considered to have important functions in mammalian autophagy by processing and uncoupling of LC3 [19, 20]. We hypothesized that miR-490-3p might affect myocardial injury in IR by regulating the ATG4B expression.

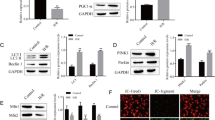
miR-490-3p targeted ATG4B and regulated the expression of ATG4B. a The targeted binding sequence of miR-490-3p to ATG4B predicted by StarBase. b The targeted binding relationship between miR-490-3p and ATG4B detected by dual-luciferase reporter gene assay. c The effects of miR-490-3p on the protein expression of ATG4B detected by western blot analysis. d Expression of ATG4B in myocardial tissues measured by RT-qPCR (n = 5). *p < 0.05 vs. the mimic-NC group (mice treated with mimic-NC), the blank group (mice treated with blank) or the normal group (normal mice), #p < 0.05 vs. the IR + agomir-NC group (mice treated with agomir-NC and induced with IR) or the inhibitor-NC group (mice treated with inhibitor-NC). The data are measurement data and described as mean ± standard deviation. The comparison between two groups was independent sample t test, and one-way ANOVA was used for analysis among multiple groups. Tukey’s post hoc test was used. The experiment was repeated three times independently
Results of the dual-luciferase reporter gene assay in HEK293 cells showed that luciferase activity was inhibited in cells co-transfected with miR-490-3p mimic and pGL3-ATG4B-WT when compared with the cells co-transfected with mimic-NC and pGL3-ATG4B-WT, while there were no significant differences in luciferase activity in cells co-transfected with miR-490-3p mimic and ATG4B-Mut (Fig. 3b). We then further performed a series of experiments to detect whether miR-490-3p could inhibit the protein expression of ATG4B in HEK293 cells. This showed decreased ATG4B expression in cells treated with miR-490-3p mimic compared with that in cells treated with mimic-NC, but increased expression in cells treated with miR-490-3p inhibitor compared with that in cells treated with inhibitor-NC (Fig. 3c). The expression of ATG4B was significantly increased in IR mice compared with that in the normal or sham-operated mice, while was decreased in IR mice treated with miR-490-3p agomir (Fig. 3d). These findings indicated that miR-490-3p could target ATG4B and regulate the expression of ATG4B.
miR-490-3p Inhibition Promoted Autophagy and Alleviated Myocardial Injury by Upregulating ATG4B in IR Mice
To verify whether miR-490-3p functioned via ATG4B in IR mice, we employed adenovirus to induce overexpression or inhibition of ATG4B in the mice. The results of RT-qPCR showed that ATG4B was upregulated in IR mice treated with oe-ATG4B, but downregulated in IR mice treated with sh-ATG4B compared with IR mice treated with NC (Fig. 4a). The results of western blot analysis and TUNEL assay revealed that the protein expression of p62 and apoptosis was significantly decreased in IR mice treated with oe-ATG4B in comparison with IR mice treated with oe-NC, while the protein expression of LC3II was elevated. However, IR mice treated with sh-ATG4B or miR-490-3p antagomir + sh-ATG4B exhibited opposite trends compared with the mice treated with NC (Fig. 4b, c).

miR-490-3p inhibition promoted autophagy and alleviated myocardial injury by upregulating ATG4B in IR mice. a Expression of ATG4B determined by RT-qPCR. b Expression of autophagy-related proteins (LC3II and p62) in myocardial tissues normalized to GAPDH detected by western blot analysis. c Apoptosis of the heart tissues detected by TUNEL (× 200). d Autophagy of the heart tissues detected by TEM (× 20,000). e IS in the heart tissues detected by TTC. f The pathological condition of the heart tissue detected by HE staining (× 200). *p < 0.05 vs. the oe-NC group (mice treated with oe-NC) or the IR + oe-NC group (mice treated with oe-NC and induced with IR), #p < 0.05 vs. the sh-NC group (mice treated with sh-NC) or the IR + sh-NC group (mice treated with sh-NC and induced with IR), &p < 0.05 vs. the IR + miR-490-3p antagomir + sh-NC group (mice treated with miR-490-3p antagomir + sh-NC and induced with IR). The data are measurement data presented as mean ± standard deviation. The comparison between two groups was independent sample t test, and Tukey’s post hoc test was used. The experiment was repeated three times independently
The results of TEM and EB/TTC assays further revealed that the number of autolysosome was increased, and IS/AAR was decreased in the heart tissues of IR mice treated with oe-ATG4B compared with that of IR mice treated with oe-NC, while opposite trends were observed when comparing IR mice treated with sh-ATG4B and IR mice treated with sh-NC as well as between IR mice treated with miR-490-3p antagomir + sh-ATG4B and IR mice treated with miR-490-3p antagomir + sh-NC (Fig. 4d, e).
Meanwhile, HE staining demonstrated that myocardial injury was alleviated in IR mice treated with oe-ATG4B. Myocardial injury in mice was worsened in IR mice treated with sh-ATG4B compared with that in IR mice treated with sh-NC, and similarly in IR mice treated with miR-490-3p antagomir + sh-ATG4B compared with IR mice treated with miR-490-3p antagomir + sh-NC (Fig. 4f). Above all, it was indicated that miR-490-3p inhibition promoted autophagy and alleviated myocardial injury by upregulating ATG4B in IR mice.
Discussion
IR injury remains a main challenge in organ transplantation and cardiothoracic surgery [1]. Autophagy is an essential process in the development and differentiation of cells, with effects in tumor suppression, and is probably associated with extended life span [12]. Moreover, autophagy is a protective mechanism in IR injury [8], and certain miRNAs also participate in IR injury [21]. Therefore, in the current study, we aimed to investigate the role of miR-490-3p in IR injury and myocardial cell autophagy. Our findings demonstrated that miR-490-3p inhibition promoted autophagy to reduce IR injury by upregulating ATG4B.
Preliminary findings in this project showed the expression of miR-490-3p was downregulated, while that of ATG4B was upregulated in the process of IR. As small endogenous noncoding RNAs, the miRNAs possess the ability to regulate gene expression by binding to complementary 3′UTR sequences [22]. A previous study further demonstrated that miR-125b acts as a biomarker implicated in renal IR injury [23]. ATG4B is an important enzyme for autophagy in mammals [24]. Furthermore, downregulated ATG4B inhibits autophagy induced by rapamycin [25]. Our results with the dual-luciferase reporter gene assay indicated that ATG4B was a potential target gene of miR-490-3p. A previous study has demonstrated that miR-665-3p knockdown can promote autophagic activity by upregulating ATG4B expression, thus alleviating intestinal IR injury [14]. Another research results show that doxorubicin-induced autophagy in pancreatic cancer is accompanied by diminished miR-137 expression, and overexpressed ATG5, a target gene of miR-137, can reverse the enhanced apoptosis and suppressed autophagy induced by overexpressed miR-137 [26].
Additionally, present research revealed that miR-490-3p inhibition promoted autophagy and inhibited cell apoptosis to reduce IR injury by upregulating ATG4B, as evidenced by increased LC3II and decreased p62 expression, in addition to reduced IS and apoptosis. LC3II is an essential protein in autophagy [27]. In addition, ATG4B is essential for the priming of a majority of LC3 subfamily isoforms (LC3B, LC3B2, and LC3C) [28]. In accordance with our results, a previous study also demonstrated that miR-204 expression decreases in cardiomyocyte autophagy induced by IR in parallel with upregulated LC3II [29]. Similar to LC3II protein, p62 is also regarded as a multifunctional protein associated with autophagy [30]. Moreover, a recent study proved that ATG4B expression correlates with that of p62, since ATG4B knockdown damages the formation of autophagosome and the delivery of p62 to lysosomes [28, 31, 32]. In summary, our findings demonstrated an important role of miR-490-3p in IR. More specifically, miR-490-3p inhibition can promote autophagy and inhibit apoptosis to alleviate myocardial IR injury by elevating the expression of ATG4B (Fig. 5). These promising results merit further investigation to identify the underlying mechanisms of the various specific miRNAs in myocardial IR injury.

miR-490-3p inhibition promoted autophagy to reduce myocardial IR injury by targeting ATG4B. In IR mice, low expression of miR-490-3p promoted LC3II expression and inhibited p62 expression by enhancing the expression of ATG4B, thereby promoting autophagy in myocardial cells. The autophagy reduced myocardial cell apoptosis to alleviate myocardial injury after reperfusion
References
Eltzschig, H. K., & Eckle, T. (2011). Ischemia and reperfusion—from mechanism to translation. Nature Medicine, 17(11), 1391–1401.
Rosero, O., Onody, P., Kovacs, T., Molnar, D., Fulop, A., Lotz, G., et al. (2017). Postconditioning: “toll-erating” mesenteric ischemia-reperfusion injury? Surgery, 161(4), 1004–1015.
Yellon, D. M., & Hausenloy, D. J. (2007). Myocardial reperfusion injury. The New England Journal of Medicine, 357(11), 1121–1135.
Fan, Z. X., & Yang, J. (2015). The role of microRNAs in regulating myocardial ischemia reperfusion injury. Saudi Medical Journal, 36(7), 787–793.
Sciarretta, S., Volpe, M., & Sadoshima, J. (2014). Mammalian target of rapamycin signaling in cardiac physiology and disease. Circulation Research, 114(3), 549–564.
Petrovski, G., Das, S., Juhasz, B., Kertesz, A., Tosaki, A., & Das, D. K. (2011). Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxidants & Redox Signaling, 14(11), 2191–2200.
Ma, X., Godar, R. J., Liu, H., & Diwan, A. (2012). Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy., 8(3), 297–309.
Jiang, M., Liu, K., Luo, J., & Dong, Z. (2010). Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. The American Journal of Pathology, 176(3), 1181–1192.
Zhou, L., Zang, G., Zhang, G., Wang, H., Zhang, X., Johnston, N., et al. (2013). MicroRNA and mRNA signatures in ischemia reperfusion injury in heart transplantation. PLoS One, 8(11), e79805.
Small, E. M., & Olson, E. N. (2011). Pervasive roles of microRNAs in cardiovascular biology. Nature, 469(7330), 336–342.
Yang, M., Kong, D. Y., & Chen, J. C. (2019). Inhibition of miR-148b ameliorates myocardial ischemia/reperfusion injury via regulation of Wnt/beta-catenin signaling pathway. Journal of Cellular Physiology, 234, 17757–17766.
Yang, Z., & Klionsky, D. J. (2009). An overview of the molecular mechanism of autophagy. Current Topics in Microbiology and Immunology, 335, 1–32.
Wang, I. K., Sun, K. T., Tsai, T. H., Chen, C. W., Chang, S. S., Yu, T. M., et al. (2015). MiR-20a-5p mediates hypoxia-induced autophagy by targeting ATG16L1 in ischemic kidney injury. Life Sciences, 136, 133–141.
Li, Z., Wang, G., Feng, D., Zu, G., Li, Y., Shi, X., et al. (2018). Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis in intestinal ischemia/reperfusion. Cell Death & Disease, 9(5), 483.
Livak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods, 25(4), 402–408.
Hamacher-Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., Gottlieb, R. A., & Gustafsson, A. B. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death and Differentiation, 14(1), 146–157.
Zhang, C. X., Cheng, Y., Liu, D. Z., Liu, M., Cui, H., Zhang, B. L., Mei, Q. B., & Zhou, S. Y. (2019). Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. Journal of Nanobiotechnology, 17(1), 18.
Skytte Rasmussen, M., Mouilleron, S., Kumar Shrestha, B., Wirth, M., Lee, R., Bowitz Larsen, K., et al. (2017). ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy, 13(5), 834–853.
Kabeya, Y., Mizushima, N., Yamamoto, A., Oshitani-Okamoto, S., Ohsumi, Y., & Yoshimori, T. (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. Journal of Cell Science, 117(Pt 13), 2805–2812.
Satoo, K., Noda, N. N., Kumeta, H., Fujioka, Y., Mizushima, N., Ohsumi, Y., et al. (2009). The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. The EMBO Journal, 28(9), 1341–1350.
Zheng, W., Men, H., Li, J., Xing, Y., Wu, B., Wang, Z., et al. (2016). Global microRNA expression profiling of mouse livers following ischemia-reperfusion injury at different stages. PLoS One, 11(2), e0148677.
Wojciechowska, A., Braniewska, A., & Kozar-Kaminska, K. (2017). MicroRNA in cardiovascular biology and disease. Advances in Clinical and Experimental Medicine, 26(5), 865–874.
Guclu, A., Kocak, C., Kocak, F. E., Akcilar, R., Dodurga, Y., Akcilar, A., et al. (2017). MicroRNA-125b as a new potential biomarker on diagnosis of renal ischemia-reperfusion injury. The Journal of Surgical Research, 207, 241–248.
Yang, Z., Wilkie-Grantham, R. P., Yanagi, T., Shu, C. W., Matsuzawa, S., & Reed, J. C. (2015). ATG4B (Autophagin-1) phosphorylation modulates autophagy. The Journal of Biological Chemistry, 290(44), 26549–26561.
Wu, Y., Dai, X., Ni, Z., Yan, X., He, F., & Lian, J. (2017). The downregulation of ATG4B mediated by microRNA-34a/34c-5p suppresses rapamycin-induced autophagy. Iranian Journal of Basic Medical Sciences, 20(10), 1125–1130.
Wang, Z. C., Huang, F. Z., Xu, H. B., Sun, J. C., & Wang, C. F. (2019). MicroRNA-137 inhibits autophagy and chemosensitizes pancreatic cancer cells by targeting ATG5. The International Journal of Biochemistry & Cell Biology, 111, 63–71.
Huang, J., Pan, W., Ou, D., Dai, W., Lin, Y., Chen, Y., et al. (2015). LC3B, a protein that serves as an autophagic marker, modulates angiotensin II-induced myocardial hypertrophy. Journal of Cardiovascular Pharmacology, 66(6), 576–583.
Agrotis, A., Pengo, N., Burden, J. J., & Ketteler, R. (2019). Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy, 15(6), 976–997.
Xiao, J., Zhu, X., He, B., Zhang, Y., Kang, B., Wang, Z., et al. (2011). MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. Journal of Biomedical Science, 18, 35.
Islam, M. A., Sooro, M. A. & Zhang, P. (2018). Autophagic regulation of p62 is critical for cancer therapy. International Journal of Molecular Sciences, 19(5),
McAlindon, E., Bucciarelli-Ducci, C., Suleiman, M. S., & Baumbach, A. (2015). Infarct size reduction in acute myocardial infarction. Heart, 101(2), 155–160.
Tang, Y., Zhong, Z., Wang, X., Wang, Y., Liu, Y., & Chang, Z. (2019). MicroRNA-497 inhibition mitigates myocardial infarction via enhancing wingless/integrated signal pathway in bone marrow mesenchymal stem cells. Journal of Cellular Biochemistry, 120(8), 13403–13412.
Acknowledgments
We would like to offer our sincere appreciation to the reviewers for their critical comments on this article.
Availability of Data and Materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
Yufu Wu and Qing Mao designed the study. Xiulin Liang collated the data. Yufu Wu, Qing Mao, and Xiulin Liang contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics Statement
The protocol of animal experiments was approved by the Institutional Animal Care and Use Committee of Nanjing Lishui People’s Hospital, Zhongda Hospital Lishui Branch, Southeast University.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Associate Editor Joost Sluijter oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, Y., Mao, Q. & Liang, X. Targeting the MicroRNA-490-3p-ATG4B-Autophagy Axis Relieves Myocardial Injury in Ischemia Reperfusion. J. of Cardiovasc. Trans. Res. 14, 173–183 (2021). https://doi.org/10.1007/s12265-020-09972-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-020-09972-9