
Abstract
It has been shown that diabetes modifies the myocardial responses to ischemia/reperfusion (I/R) and to cardioprotective agents. In this study, we aimed to investigate the effects of combined treatment with ischemic postconditioning (IPostC) and cyclosporine A (CsA) on inflammation and apoptosis of the diabetic myocardium injured by I/R. Eight weeks after induction of diabetes in Wistar rats, hearts were mounted on a Langendorff apparatus and were subsequently subjected to a 30-min regional ischemia followed by 45-min reperfusion. IPostC was induced at the onset of reperfusion, by 3 cycles of 30-s reperfusion/ischemia (R/I). The concentration of creatine kinase (CK), tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 were determined; the levels of total and phosphorylated glycogen synthase kinase 3 beta (p-GSK3β) and B-cell lymphoma 2 (Bcl-2) were quantified by western blotting, and the rate of apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining. Administration of either IPostC or CsA alone in nondiabetic animals significantly reduced CK, TNF-α, IL-1β, and IL-6 concentrations, increased the p-GSK3β and Bcl-2, and decreased the level of apoptosis (P < 0.05) but had no effect on diabetic hearts. However, in diabetic animals, after administration of CsA, the cardioprotective effects of IPostC in increasing the p-GSK3β and Bcl-2 and decreasing apoptosis and inflammation were restored in comparison with nonpostconditioned diabetic hearts. IPostC or CsA failed to affect apoptosis and inflammation and failed to protect the diabetic myocardium against I/R injury. However, combined administration of IPostC and CsA at reperfusion can protect the diabetic myocardium by decreasing the inflammatory response and apoptosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ischemic postconditioning (IPostC) is an effective cardioprotective strategy that works by rapid, short-lived, and intermittent interruptions of blood flow in the early phase of reperfusion [2]. IPostC stimulates the endogenous mechanisms that attenuate the multiple manifestations of reperfusion injury. This may activate pro-survival signaling pathways, including the extracellular signal-regulated kinase 1/2 (ERK1/2), phosphoinositide-3-kinase/protein kinase B (PI3K/Akt), and protein kinase C (PKC) pathways [20, 29]. In addition, IPostC has been shown to alter the activity of mitochondrial ATP-sensitive K+ channel and mitochondrial permeability transition pore (mPTP) [4, 27]. mPTP is highly integrated in apoptotic pathways; the opening of this pore during the onset of reperfusion or in other circumstances including diabetes results in cytochrome C release from the mitochondria into the cytosole, and this, in turn, induces the next phases of cell apoptosis [15, 19]. There is strong evidence that blocking the apoptotic process can reduce the incidence of heart failure by preventing the loss of contractile cells and minimizing cardiac ischemia/reperfusion (I/R) injury [1]. Moreover, IPostC-induced cardioprotection is linked to the inhibition of mPTP via upregulation of the PI3K/Akt survival signaling pathway and thereby inactivation of glycogen synthase kinase-3 beta (GSK3β ) in cardiac cells [6, 7]. Activation of PI3K/Akt/GSK3β pathway effectively reduces cardiomyocyte apoptosis and I/R injury [5, 28]. On the other hand, cyclosporine A (CsA) is an immunosuppressive agent that selectively inhibits mPTP opening [14]. As such, CsA exerts significant cardioprotective effects which have been proven in several previous studies on myocardial I/R injury in subjects and animal models yet without any comorbidities [17]. In addition, several studies have shown that IPostC-provided cardioprotection is offset by diabetes, although the effects of IPostC on diabetic heart have not been thoroughly investigated [21]. Diabetic patients face an increased risk of ischemic events and organ damage from myocardial infarction, and they may mount an inordinate response to I/R.
On the other hand, the inflammatory response has a crucial role in I/R injury [25]. The exaggerated inflammatory response to I/R which is induced by diabetes is concomitant with the accumulation of adherent leukocytes and high expression of adhesion molecules. Inflammatory signaling cascades triggered during reperfusion injury activate NF-κB and result in the overexpression of a range of important pro-inflammatory cytokine and chemokine genes, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-12, and IL-8, and this plays a pivotal role in myocardial inflammatory response [23, 31].
It has been argued that chronic diabetes sensitizes the heart to I/R injury [18]. In addition, the protective IPostC could not protect the diabetic myocardium from reperfusion injury. In our previous works, we showed that the IPostC or CsA could restore the hemodynamic function of I/R hearts in healthy but not diabetic rats [3]. Here, we have designed the same protocol with different animals to identify whether it is possible to protect the diabetic myocardium from I/R injury through combined postconditioning with ischemia and CsA and to explore the role of cardiomyocyte apoptosis and inflammation mediators in this context.
Materials and methods
Materials
Streptozotocin (STZ) was supplied by Tocris Company (London, UK). TNF-α, IL-6, and IL-1β detection kits were provided by eBioscience System (Austria) and Sigma-Aldrich (St. Louis, MO, USA). All constituents of Krebs–Henseleit solution were obtained from Merck company (Germany).
Animals
Male adult Wistar rats (250–350 g) were obtained from the animal house of Tabriz University of Medical Sciences (TUMS, Iran). All animals received humane treatment in accordance with the regulation of the Care and Use of Laboratory Animals (National Institute of Health, publication no. 85–23, revised 1996). The experimental procedures were approved by the animal ethics committee of the Tabriz University of Medical Sciences (ethics approval number: A473/1–11-2013). At the beginning of the experiment, the animals were randomly divided into two main categories of diabetic and nondiabetic (control) groups. The animals were housed under standard laboratory conditions at 24 ± 2 °C, relative humidity of 55 ± 5 %, and 12 h:12 h dark/light cycle. All animals were allowed free access to food and water.
Induction of diabetes
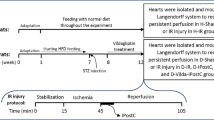
Diabetes was induced by a single 50 mg/kg body weight intraperitoneal injection of STZ. STZ was dissolved in a 0.1-M citrate buffer with pH 4.5. STZ disrupts the functioning of pancreatic islet cells and reduces the secretion of insulin, eventuating in the development of type-I diabetes. To confirm the development of diabetes, 72 h post injection of STZ, blood glucose levels were measured by a glucometer device by the blood samples acquired from scratching tails of rats. The animals with blood glucose levels higher than 300 mg/dl were considered diabetic [3]. Blood glucose measurements were performed again at fourth and eighth weeks after STZ injection. After 8 weeks post STZ administration (chronic diabetes), the diabetic as well as the control animals were sacrificed and all experiments were performed on isolated perfused beating hearts in a Langendorff setup.
Induction of regional I/R and IPostC
The isolated hearts were retrogradely perfused through the aorta with a Krebs–Henseleit solution as described previously [10]. The perfusion pressure of solution was adjusted to a constant 75 mmHg and the solution was gassed with a mixture of 95 % O2 and 5 % CO2 at 37 °C and pH 7.4. After a 15-min stabilization period, all isolated hearts were subjected to a 30-min regional ischemia followed by a 45-min reperfusion period. Regional I/R process was performed by blocking and re-opening of the left anterior descending (LAD) coronary artery, using a 5-0 silk ligature placed around the LAD close to its origin. An instantaneous drop (down to 30–40 % of its baseline value) in coronary flow at the onset of index ischemia and the subsequent recovery of the coronary flow in the reperfusion period indicated success in inducing the I/R cycle. IPostC was carried out by three intermittent cycles of 30-s reperfusion/ischemia (R/I) (3 cycles of 30-s R/I), respectively, applied immediately at the onset of reperfusion.
I/R experimental protocol
For I/R experiments, the control and diabetic animals were divided into eight subgroups (n = 8) as (1) control (C), (2) control with IPostC (C+IPostC), (3) control with CsA (C+CsA), (4) control with IPostC plus CsA (C+IPostC+CsA), (5) diabetic (D), (6) diabetic with IPostC (D+IPostC), (7) diabetic with CsA (D+CsA), and (8) diabetic with IPostC and CsA (D+IPostC+CsA). The isolated hearts in all groups were perfused with the Krebs–Henseleit solution and underwent 30 min of regional ischemia followed by 45 min of reperfusion. In IPostC-receiving groups, the hearts received 3 cycles of 30-s R/I at the onset of reperfusion according to the IPostC protocol. Furthermore, in CsA-receiving groups, 5 min before the onset of reperfusion up to 10 min after reperfusion, the hearts were perfused with a Krebs–Henseleit solution containing 0.01 mM CsA as an inhibitor of mPTP. The experimental groups which did not receive CsA were perfused with a pure Krebs–Henseleit solution for the designated period.
CK release measurement
Myocardial cellular damage was evaluated by measuring the creatine kinase (CK) release in coronary effluent collected 10 min after beginning of reperfusion. CK activity was measured spectrophotometrically with a commercially available kit (Roche Diagnostics, Germany). The absorbance CK solution was read at 340 nm. The results were reported in U/l.
Preparation of tissue homogenates
At the end of experiments, the heart samples (isolated from the ischemic zone of the left ventricle) were immediately frozen in liquid nitrogen and stored at −80 °C. Samples were prepared according to a modified protocol for tissue homogenization. Approximately 0.5 g of ventricular tissue was weighed and cut into pieces in about 5 ml of ice-cold lysis buffer containing 1 mM KH2PO4, 1 mM KCL, 50 mM Tris-HCl pH 7.4, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, triton X100, and protease inhibitor cocktail and was then homogenized with a Polytron PT-10/ST homogenizer. The homogenate was centrifuged at 10,000g for 10 min at 4 °C. The supernatants were removed from the homogenates and quickly frozen at −80 °C until determination of protein levels and cytokine activity. Bradford method was used to determine protein concentration in samples.
Determination of cardiac TNF-α, IL-1β, and IL-6 concentrations
Cardiac TNF-α, IL-1β, and IL-6 concentrations were measured using an ELISA and rat specific ELISA kits according to the kit instructions. A coating plate was then placed in the ELISA reader to read the absorbance of each sample at 450 nm. All optical density values were converted to the final concentration based on mg protein of each samples and expressed as picograms per milligram of total protein.
Determination of myocardial apoptosis
Myocardial apoptosis was monitored by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, as previously described in reference [20]. TUNEL staining was performed with fluorescein UTP according to manufacturer instructions (In Situ Cell Death Detection Kit, Roche Diagnostics) for apoptotic cell nuclei and 49 6-diamidino-2-henylindole (DAPI) (Sigma) stained all cell nuclei. Apoptotic index (AI) was calculated as the number of TUNEL-positive myocytes/total number of myocytes stained with DAPI from ten fields per heart. All of these assays were performed in a blinded manner.
Western blotting
Fifty microgram protein of each sample was separated on 12.5 % SDS-PAGE and electrotransferred to PVDF membranes (Millipore, Bedford, MA). The membranes were blocked with 5 % nonfat milk in PBS/T for 60 min at room temperature, then incubated overnight at 4 °C with monoclonal antibodies against GSK3β (1:2000, Cell Signaling Technology, USA), phospho(Ser9)GSK3β (1:2000, Cell Signaling Technology), B-cell lymphoma 2 (Bcl-2) (1:2000, Cell Signaling Technology), and β-actin (1:5000, Cell Signaling Technology) for overnight. After four 5-min washes with PBS/T, membranes were incubated with horseradish peroxidase-linked goat anti-mouse secondary antibody (1:2500, Cell Signaling) for 120 min at room temperature. After rinsing, the protein bands were visualized using the ECL chemiluminescence system (GE Healthcare). Protein band intensity were measured by ImageJ 1.6 software (National Institutes of Health, MD, USA) and normalized to its β-actin. To determine the amount of GSK3β phosphorylation in each sample, the ratio of the intensity of phospho(Ser9)GSK3β to total GSK3β was calculated.
Statistical analysis
All values were expressed as means ± SEM. Two-way analysis of variance (ANOVA) was used for comparison of parameters within groups. One-way ANOVA and post hoc Tukey’s test were used for multiple comparisons of parameters between different groups. Independent t test was used for comparison between two groups. Differences were considered statistically significant when p < 0.05.
Results
General characteristics of control and diabetic rats
Table 1 indicates the general characteristics and basic data of control and diabetic rats. Independent t test showed that 8 weeks of type-I diabetes led to significant hyperglycemia (F (1,58) = 43.88, P < 0.001) as well as reduced body weight (F (1,58) = 9.18, P < 0.001) and increased the ratio of heart weight to body weight (F (1,58) = 5.73, P < 0.001), in comparison with those of control animals.
Myocardial CK release
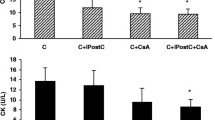
The alterations of CK levels can serve as an index for tissue injury. Myocardial CK release (in U/l) in experimental I/R hearts has been shown in Fig. 1. The two-way ANOVA indicated a significant effect of the treatment on myocardial CK levels (F (4,70) = 8.13, P < 0.001). The main effect of diabetes on CK levels was not significant (F (1,78) = 1.11, P = 0.29), but the interaction effect of treatment × diabetes was highly significant (F (4,70) = 3.75, P = 0.015). There was no significant difference in CK levels between control and diabetic sham groups, as indicated by one-way ANOVA and Tukey post hoc. Application of IPostC at the beginning of reperfusion in control hearts significantly reduced myocardial CK release (in U/l) as compared with those of the control I/R group (11.8 ± 2.1 vs. 17.5 ± 2.8, P = 0.04). Administration of CsA at the end of ischemia had an effect similar to IPostC effect (9.6 ± 1.2, P = 0.01). In addition, when both CsA and IPostC were administered simultaneously in control hearts, CK reduction was increased even to a greater extent (9.4 ± 0.9 vs. 17.5 ± 2.8, P = 0.007). In diabetic hearts, however, only the effect of combination therapy (IPostC+CsA) was statistically significant in comparison with the diabetic I/R group (8.6 ± 1.4 vs. 15.6 ± 2.7, P = 0.01).

Myocardial CK release (in U/l) in experimental I/R hearts. *P < 0.05 as compared with the control I/R group; #P = 0.01 as compared with the diabetic I/R group. Mean ± SEM. n = 8. I/R ischemic/reperfused, CsA cyclosporine A, IPostC ischemic postconditioning
Myocardial cytokines
The main effects of diabetes for IL-6 and IL-1β were significant (two-way ANOVA, F (1,62) = 29.07, P < 0.001; and F (1,62) = 19.38, P < 0.001, respectively). The significant main effects were found for treatments for IL-6 and IL-1β (two-way ANOVA, F (3,56) = 26.11, P < 0.001; and F (3,56) = 8.83, P = 0.021, respectively). In addition, results showed a significant interaction between treatment and diabetes for IL-6 (F (3,56) = 4.35, P = 0.04). One-way ANOVA showed that administration of IPostC or CsA in healthy (or control) I/R hearts significantly reduced the levels of IL-6, picograms per milligram of sample protein (42.802 ± 5.03 by IPostC and 42.625 ± 5.1 by CsA vs. 60.748 ± 6.3 in control I/R hearts, P < 0.001), and also significantly reduced the levels of IL-1β, picograms per milligram (30.49 ± 3.68, P = 0.032 by IPostC and 28.45 ± 6.49, P = 0.028 by CsA vs. 46.08 ± 2.76), in comparison with untreated control I/R hearts. However, in diabetic hearts, the positive effects of IPostC (P = 0.49) or CsA (P = 0.082) were not observed (Fig. 2a, b, respectively). Furthermore, the combination of IPostC and CsA, both in healthy and diabetic hearts, significantly decreased IL-6 and IL-1β levels in treated hearts compared to untreated I/R hearts and this effect was stronger than each single treatment along in both control and diabetic hearts. The mean ± SEM values of combination therapy vs. untreated I/R hearts and significance levels of multiple comparison by Tukey’s test for IL-6 levels were 33.72 ± 3.6 vs. 60.748 ± 6.3 pg/mg in control I/R hearts, P < 0.001 and 44.71 ± 3.18 vs. 73.78 ± 2.23 pg/mg in diabetic I/R hearts, P = 0.002 and for IL-1β levels were 25.03 ± 2.95 vs. 46.08 ± 2.76 pg/mg in control I/R hearts, P = 0.014 and 39.02 ± 3.56 vs. 58.43 ± 3.41 pg/mg in diabetic I/R hearts, P = 0.040.

Myocardial IL-6 (a), IL-1β (b), and TNF-α (c) levels (in pg/mg of sample protein) in experimental I/R hearts. *P < 0.05 as compared with the control I/R group; **P < 0.01 as compared with the control I/R group; #P < 0.05 as compared with the diabetic I/R group; ##P < 0.01 as compared with the diabetic I/R group. Mean ± SEM. n = 8. I/R ischemic/reperfused, CsA cyclosporine A, IPostC ischemic postconditioning
Moreover, the main effects of diabetes and treatments for TNF-α were significant (two-way ANOVA, F (1,62) = 12.67, P = 0.01 and F (3,56) = 3.18, P = 0.05, respectively). Also, two-way ANOVA results indicated a significant interaction between treatment and diabetes for TNF-α (F (3,56) = 5.03, P = 0.006). One-way ANOVA showed that in nondiabetic hearts, similar to the effects of combination treatment (P = 0.04), administration of single IPostC (P = 0.03) or CsA (P = 0.04) could significantly reduce the myocardial TNF-α levels compared with those of the control I/R group. In diabetic rats, administration of CsA alone (27.8 ± 2.54 pg/mg of sample protein, P = 0.04) or in combination with IPostC (25.1 ± 1.61 pg/mg of sample protein, P = 0.01) decreased TNF-α levels in comparison to untreated diabetic I/R hearts (40.5 ± 3.72 pg/mg) and the combination treatment had a potent effect on reduction of TNF-α levels (Fig. 2c).
Myocardial apoptosis
I/R hearts showed a higher rate of apoptosis than the other groups. There was a moderate number of apoptotic cells in I/R+IPostC and I/R+CsA groups of control hearts (Fig. 3). IPostC or CsA could not reduce the apoptotic cells in diabetic hearts (Fig. 4). On the other hand, a few apoptotic cells were seen in I/R+IPostC+CsA subgroups both in control and diabetic hearts (Figs. 3 and 4). In addition, AI was significantly higher in the diabetic sham group in comparison with the control sham (P = 0.04), according to the results of one-way ANOVA and Tukey post hoc analysis. Furthermore, IPostC or CsA alone (P = 0.01) or in combination (P < 0.001) significantly reduced the AI in control hearts compared with the corresponding I/R group (Fig. 5a). The effect of combination treatment was greater than those of single treatments, and only the combination treatment with CsA and IPostC reduced AI in diabetic hearts as compared with diabetic I/R hearts (P = 0.01).

Micrographs of myocytes indicating the process of apoptosis in experimental I/R hearts in control hearts. Myocardial apoptosis was detected by TUNEL staining. I/R ischemia/reperfusion, CsA cyclosporine A, IPostC ischemic postconditioning

Micrographs of myocytes indicating the process of apoptosis in experimental I/R diabetic hearts. Myocardial apoptosis was detected by TUNEL staining. I/R ischemia/reperfusion, CsA cyclosporine A, IPostC ischemic postconditioning

Apoptotic index, AI (a), phosphorylation of myocardial GSK3β (b), and activation of myocardial Bcl-2 (c) in experimental I/R hearts. ¥P < 0.05 as compared with the control sham group; *P < 0.05 as compared with the control I/R group; **P < 0.01 as compared with the control I/R group; #P < 0.05 as compared with the diabetic I/R group. Mean ± SEM. n = 8. I/R ischemic/reperfused, CsA cyclosporine A, IPostC ischemic postconditioning
Phosphorylation of myocardial GSK3β
Total and Ser9-phosphorylated forms of GSK3β, Bcl-2, and β-actin were measured in this study using an immunoblotting method (Fig. 5b, c). The total forms of GSK3β in diabetic or nondiabetic groups were similar and there was no significant differences between groups. Two-way ANOVA found significant effects of diabetes (F (1,62) = 9.48, P = 0.013), treatments (F (3,56) = 16.01, P < 0.001), and a diabetes × treatments interaction (F (3,56) = 3.63, P = 0.05) on the levels of phosphorylated to total form of GSK3β. In nondiabetic hearts, application of IPostC (1.05 ± 0.08, P = 0.03) and CsA (1.02 ± 0.09, P = 0.023) or their combination (1.2 ± 0.10, P = 0.002) significantly increased the levels of phosphorylated to total form of GSK3β (in arbitrary unit) as compared with the corresponding I/R group (0.60 ± 0.08). In diabetic hearts, however, the significant increase in the GSK3β phosphorylation was observed only under combination therapy (1.08 ± 0.09 vs. 0.67 ± 0.08, P = 0.018); the individual effects of IPostC or CsA in diabetic hearts were not significant as compared with those of diabetic I/R hearts (Fig. 5b).
Myocardial Bcl-2 protein expression levels
The levels of myocardial Bcl-2, normalized by β-actin bands, in diabetic and nondiabetic control hearts are shown in Fig. 5c. The results of two-way ANOVA analysis showed a significant main effect of treatment (F (3,56) = 9.88, P < 0.001) and a significant interaction effect between treatment and diabetes on Bcl-2 protein expression levels (F (3,56) = 4.69, P = 0.03). The main effect of diabetes on Bcl-2 levels was not significant (F (1,62) = 1.94). Moreover, in control healthy hearts, postconditioning protocols in both forms, alone (P < 0.04) or in combination (P = 0.003), significantly increased the relative expression of Bcl-2 protein as compared with those of the control I/R group. On the other hand, the only significant increase in the Bcl-2 protein level was achieved following combining both protocols IPostC and CsA as compared with the diabetic I/R group (P = 0.02). Treatment of diabetic hearts with IPostC (P = 0.99) or CsA (P = 0.90) could not significantly alter the expression levels of Bcl-2 (Fig. 5c).
Discussion
The results of the current study indicate that both IPostC and CsA could protect myocardium against reperfusion injury in normal nondiabetic animals by significantly increasing GSK3β phosphorylation and Bcl-2 activation and thereby decreasing apoptosis and inflammatory responses. However, IPostC or CsA fails to protect type-1 diabetic hearts. Nevertheless, the combination of IPostC and CsA in the diabetic animals reduced apoptosis and inflammatory response significantly and had more potent cardioprotective effects.
mPTP is a key event in cell death after I/R. Previous studies on rat hearts have demonstrated that the mPTP remains closed throughout ischemia but opens up during reperfusion [8, 16]. The opening of mPTP in the inner mitochondrial membrane disrupts the membrane potential and results in the uncoupling of the mitochondrial respiratory chain. Subsequently, the release of cytochrome C and other pro-apoptotic factors from the mitochondria can lead to apoptosis or necrosis [15]. In this regard, many studies have showed that inhibition of mPTP opening at reperfusion can provide significant protection against experimental I/R injury. Argaud et al. demonstrated that IPostC altered the threshold for mPTP opening, such that a greater Ca2+ load (as a stimulator of mPTP opening) will be required for opening mPTPs in rabbit hearts 1. Bopassa et al. have also demonstrated that keeping mPTP closed in isolated perfused rat hearts is somehow linked with PI3K activation, leading to the activation of survival kinase pathways [8].
There is some evidence suggesting that apoptosis is elicited by reperfusion after transient coronary artery occlusion. In a study, hypoxic postconditioning of isolated neonatal cardiomyocytes after a long hypoxic period has been shown to reduce cardiomyocyte apoptosis. Hypoxic postconditioning may possibly reduce cardiomyocyte apoptosis by exerting an overall anti-apoptotic effect through inhibition of caspases-3 and -9 and maintenance of the Bcl-2/Bax ratio [11, 12]. Zhao et al. have also suggested the inhibition of inflammatory response in reperfusion as yet another mechanism by which IPostC reduced reperfusion injury [30]. Preliminary studies have demonstrated that the level of pro-inflammatory cytokines such as TNF-α and IL-6 is reduced by IPostC. In the present study, we showed that the administration of single IPostC or CsA to nondiabetic hearts reduced the apoptosis and inflammatory cytokines in isolated I/R hearts; however, the combined IPostC+CsA treatment had a very potent favorable effect on those parameters in normal animals. On the other hand, in type-1 diabetic hearts, IPostC or CsA alone failed to provide any protection and, in other words, diabetes abolished the positive effects of IPostC or CsA on myocardium. However, combination of IPostC or CsA showed full cardioprotection and significant decrease in apoptosis and reduced the levels of inflammatory cytokines.
IPostC can activate many signaling pathways including PI3K/Akt, mitogen-activated protein kinase-ERK 1/2 (MEK-ERK1/2), PKC, and STAT3. The mentioned pathways are believed to function in concert to inhibit mPTP opening upon reperfusion. mPTP closure is achieved by phosphorylation and inhibition of GSK3β at Ser9. We showed that IPostC increased the phosphorylation of GSK3β and activation of Bcl-2 in normal hearts. On the other hand, in type-1 diabetic hearts, IPostC was not as effective. Interestingly, the diabetes-mediated pathogenic effects are associated with activation of GSK3β pathway [13, 24]. The lack of phosphorylation of GSK3β in diabetic hearts was associated with increased in cardiac injury and apoptosis in comparison with healthy hearts. The total amount of GSK-3β was the same in diabetic and nondiabetic groups of our study and this indicates that the chronic diabetes diminishes the power of IPostC to phosphorylate GSK-3β, keeping this detrimental protein kinase in its active form. Therefore, we hypothesized that type-1 diabetes mellitus might have a negative effect on these crucial pathways involved in IPostC. In this case, it has been reported that the activity of survival protein kinases, e.g., PI3K/Akt pathway, significantly decreases during diabetes and hyperglycemic conditions [9]. Thus, PI3K/Akt, ERK, and GSK3β, upstream of mPTP signal transduction, were not phosphorylated effectively by IPostC in diabetic animals in comparison with the healthy controls. These diabetes-induced intracellular changes along with greater oxidative stress, inflammation, and hyperglycemia may hinder the effect of IPostC on reduction of apoptosis and inflammatory responses during I/R insult. Consequently, future investigations can discover the exact contribution of each signaling mediator in interaction of diabetes with cardioprotection by IPostC.
Nevertheless, after administration of CsA, as an mPTP inhibitor, in combination with IPostC, the effect was stronger and their cardioprotection was enhanced and thus, this combination could overcome the diabetes-induced resistance to cardioprotection in the case of cardiac reperfusion injury. Although as a limitation, we could not evaluate the mitochondrial function in the present study; as a possible mechanism, the prolonged opening of mPTP during diabetes results in collapse of the mitochondrial membrane potential, matrix swelling, and uncoupling of the respiratory chain, leading to the disturbances in mitochondrial physiology and function [32]. These then facilitate the release of pro-apoptotic mediators and exacerbate the cellular integrity which may determine the consequence of myocardial reperfusion injury. Besides inhibiting the mPTP opening, CsA may also affect inflammatory responses and cellular signal transduction pathways terminating on mPTP [22, 26]. Therefore, when given together, CsA may enhance the potency of IPostC on activating the survival protein kinases and reduction of the mPTP opening, leading to the full cardioprotection.
In conclusion, the findings of the present study indicate that administration of IPostC or CsA in normal nondiabetic conditions, either as single or combination treatments, can protect the heart against I/R injury. However, they fail to decrease the apoptosis and inflammatory response in type-1 diabetic state and fail to protect the diabetic myocardium against I/R injury in comparison with normal animals. This loss of cardioprotection can be recovered by the concomitant administration of CsA and IPostC at reperfusion.
Abbreviations
- CsA:
-
Cyclosporine-A
- I/R:
-
Ischemia/reperfusion
- IPostC:
-
Ischemic postconditioning
- ERK:
-
Extracellular signal-regulated kinase
- MEK:
-
Mitogene-activated protein kinase/ERK kinase
- PI3K/Akt:
-
Phosphoinositide-3-kinase/protein kinase B
- PKC:
-
Protein kinase C
- mPTP:
-
Mitochondrial permeability transition pore
- GSK3β:
-
Glycogen synthase kinase-3 beta
- TNF-α:
-
Tumor necrosis factor-α
- IL-:
-
Interleukin-
- CK:
-
Creatine kinase
References
Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Béjui F, Robert D, Ovize M (2004) Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovas Res 61:115–122
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Postconditioning inhibits mitochondrial permeability transition. Circulation 111:194–197
Badalzadeh R, Mohammadi M, Najafi M, Ahmadiasl N, Farajnia S, Ebrahimi H (2012) The additive effects of ischemic postconditioning and cyclosporine-A on nitric oxide activity and functions of diabetic myocardium injured by ischemia/reperfusion. J Cardiovasc Pharmacol Ther 17:181–189
Badalzadeh R, Yousefi B, Majidinia M, Ebrahimi H (2014a) Anti-arrhythmic effect of diosgenin in reperfusion-induced myocardial injury in a rat model: activation of nitric oxide system and mitochondrial KATP channel. J Physiol Sci 64:393–400
Badalzadeh R, Yousefi B, Tajaddini A, Ahmadian N (2014b) Diosgenin-induced protection against myocardial ischaemia-reperfusion injury is mediated by mitochondrial KATP channels in a rat model. Perfusion 0267659114566064.
Badalzadeh R, Mokhtari B, Yavari R (2015a) Contribution of apoptosis in myocardial reperfusion injury and loss of cardioprotection in diabetes mellitus. J Physiol Sci 65:201–215
Badalzadeh R, Mohammadi M, Yousefi B, Faranjia S, Najafi M, Mohammadi S (2015b) Involvement of glycogen synthase kinase-3β and oxidation status in the loss of cardioprotection by postconditioning in chronic diabetic male rats. Adv Pharm Bull 5(3):321–327
Bopassa J-C, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M (2006) PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res 69:178–185
Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH (2011) Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3–and phosphatidylinositol 3-kinase–mediated inhibition. Anesthesiology 114:1364–1372
Ebrahimi H, Badalzadeh R, Mohammadi M, Yousefi B (2014) Diosgenin attenuates inflammatory response induced by myocardial reperfusion injury: role of mitochondrial ATP-sensitive potassium channels. J Physiol Biochem 70:425–432
Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J (2000) Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol 32:197–208
Gottlieb RA, Burleson K, Kloner RA, Babior B, Engler R (1994) Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94:1621
Hausenloy DJ (2009) Signalling pathways in ischaemic postconditioning. Thromb Haemost 101:626–634
Huhn R, Heinen A, Hollmann MW, Schlack W, Preckel B, Weber NC (2010) Cyclosporine A administered during reperfusion fails to restore cardioprotection in prediabetic Zucker obese rats in vivo. Nutr Metab Cardiovasc Dis 20:706–712
Karimian A, Ahmadi Y, Yousefi B (2016) Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA repair 42:63–71
Kin H, Zhao Z-Q, Sun H-Y, Wang N-P, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J (2004) Postconditioning attenuates myocardial ischemia–reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 62:74–85
Kupai K, Csonka C, Fekete V, Odendaal L, Van Rooyen J, Csont T, Ferdinandy P (2009) Cholesterol diet-induced hyperlipidemia impairs the cardioprotective effect of postconditioning: role of peroxynitrite. Am J Physiol Heart Circ Physiol 297:1729–1735
Miki T, Itoh T, Sunaga D, Miura T (2012) Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol 11:67
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol reviews 88:581–609
Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J, Yellon DM, Schulz R (2010) Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the working group of cellular biology of the Heart of the European Society of Cardiology. Cardiovasc Res 87:406–423
Przyklenk K, Maynard M, Greiner DL, Whittaker P (2011) Cardioprotection with postconditioning: loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid Redox Signal 14:781–790
Rafiee P, Heidemann J, Ogawa H, Johnson N, Fisher PJ, Li M et al (2004) Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun Signal 2:3
Ren J-Y, Song J-X, Lu M-Y, Chen H (2011) Cardioprotection by ischemic postconditioning is lost in isolated perfused heart from diabetic rats: involvement of transient receptor potential vanilloid 1, calcitonin gene-related peptide and substance P. Regul Pept 169:49–57
Schwartz LM, Lagranha CJ (2006) Ischemic postconditioning during reperfusion activates Akt and ERK without protecting against lethal myocardial ischemia-reperfusion injury in pigs. Am J Physiol Heart Circ Physiol 290:1011–1018
Song J-X, Wang L-H, Yao L, Xu C, Wei Z-H, Zheng L-R (2009) Impaired transient receptor potential vanilloid 1 in streptozotocin-induced diabetic hearts. Int J Cardiol 134:290–292
Wang SC, Tang CL, Piao HL, Zhu R, Sun C, Tao Y, Fu Q, Li DJ, Du MR (2013) Cyclosporine A promotes in vitro migration of human first-trimester trophoblasts via MAPK/ERK1/2-mediated NF-κB and Ca2+/calcineurin/NFAT signaling. Placenta 34(4):374–380
Yousefi B, Samadi N, Ahmadi Y (2014) Akt and p53R2, partners that dictate the progression and invasiveness of cancer. DNA repair 22:24–29
Zhao Z-Q, Vinten-Johansen J (2006) Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res 70:200–211
Zhao Z-Q, Corvera JS, Halkos ME, Kerendi F, Wang N-P, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 54:579–588
Zhao Z, Sun H, Wang N, Kin H, Guyton R, Vinten-Johansen J (2005) Hypoxic postconditioning attenuates cardiomyocyte apoptosis via inhibition of jnk and p38 kinases pathway. J Mol Cell Cardiol 38:870–870
Zheng L-R, Han J, Yao L, Sun Y-L, Jiang D-M, Hu S-J, Shao L, Sun Z-H, Wang L-H (2012) Up-regulation of calcitonin gene-related peptide protects streptozotocin-induced diabetic hearts from ischemia/reperfusion injury. Int J Cardiol 156:192–198
Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ (2009) Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res 83:213–225
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animals received humane treatment in accordance with the regulation of the Care and Use of Laboratory Animals (National Institute of Health, publication no. 85–23, revised 1996). The experimental procedures were approved by the animal ethics committee of the Tabriz University of Medical Sciences (ethics approval number: A473/1–11-2013).
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Badalzadeh, R., Azimi, A., Alihemmati, A. et al. Chronic type-I diabetes could not impede the anti-inflammatory and anti-apoptotic effects of combined postconditioning with ischemia and cyclosporine A in myocardial reperfusion injury. J Physiol Biochem 73, 111–120 (2017). https://doi.org/10.1007/s13105-016-0530-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-016-0530-4