
Abstract
This study investigated calciothermic cobalt boride (CoxB) powder production in molten salt, starting from cheap oxide compounds, without pre-treatment by in situ Ca production via electrolytic reduction of CaO. Firstly, calciothermic CoxB production in a molten salt environment was examined theoretically in detail, and the electrochemical behaviors of CaO and CoO in CaCl2 were examined via cyclic voltammetry (CV). Based on the CV results, the cathodic reduction of CoO occurred in one step at − 0.30 V, and CO2 formation occurred in two stages. After theoretical and CV analysis, the effects of temperature (1123–1323 K) and duration (30–60 min) on particle synthesis were investigated. While Co was found in the particles produced at low temperature and short durations, with increasing duration and temperature, B diffusion increased, and all Co was transformed into CoxB, and particles were found to be made up of Co2B and CoB phases.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cobalt borides (CoxB) are one of the significant metal borides (MexBy) due to their superior properties. They are used as catalysts mostly in fuel cell applications and hydrogen storage applications due to the electrochemical conversion properties of cobalt and its high charge–discharge capacity properties. In addition, its high oxidation resistance makes it possible to use in corrosion and wear-resistant surface coatings [1,2,3,4,5].
Processes including solid-state reactions, carbothermic reduction, hydrogen reduction, molten salt electrolysis, and metallothermic reduction are used to synthesize MexBy [6,7,8,9]. Among the mentioned processes, metallothermic method stand out with their low greenhouse gas emissions and scalable structure. Metallothermic MexBy production is carried out by reducing metal oxide (MeO) and boron oxide (B2O3) with a reductant (Al, Mg, Ca) in a ball mill or unique systems under a protective atmosphere [10,11,12]. In these processes, reduction occurs from the outside to the inside of particles, and a reductant oxide (RO) film is formed at the MeO/Me interface, which slows down the reactions. Removal of the RO is vital for the continuation of the reactions and obtaining high purity products with low oxide content. In conventional metallothermic systems, it is not possible to remove the RO from MeO/Me interface. However, it can be removed in molten salts with high RO solubility [10]. Al, Mg, and Ca are commonly utilized as reductants. In addition to the reductants' high reduction capabilities, the solubility of the oxidized components formed as a by-product in simple solvents is critical. The solubility of oxide compounds of Al, Mg in molten salts and simple solvents is known to be lower than that of oxide compounds of Ca. Although the oxide solubility of chloride salts is low, CaCl2 has a high CaO solubility of 20% by mole [10, 13]. CaCl2 is a vital salt widely used in electro-deoxidation processes due to its high decomposition voltage, high oxygen diffusion, and high Ca and CaO dissolving capabilities [14]. The high decomposition voltage of CaCl2 allows direct electrochemical or metallothermic reduction of compounds with high chemical stability, such as TiO2, SiO2, CaO, etc. Especially, electrolytic reduction of CaO in CaCl2 provides one step, controlled, safe calciothermic production. In recent years, studies on the production of high-temperature metals by electro-deoxidation and metallothermic reduction processes in the molten salt environments have become widespread [15]. In particular, Suzuki et al. studied the production of Ti from TiO2, TiS2 in detail by performing the electrochemical reduction of CaO, CaS in CaCl2 and showed that it is possible to produce Ti [16,17,18]. Similarly, Arriesgado et al. performed the calciothermic production of Ni from NiO by electrolysis of CaO in CaCl2 [19].
This study aims to examine the production of CoxB particles without pre-and post-treatment using cheap, easily accessible, technical purity oxidised raw materials. The theoretical calciothermic production of CoxB in molten salt was examined in detail, and the production of CoxB was carried out in CaCl2, with in situ Ca production from CaO by molten salt electrolysis.
2 Experimental
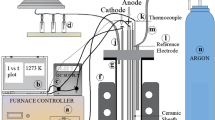
Experiments were carried out under a constant cell voltage of 3.0 V using technical purity materials (CoO, CaCl2, B2O3, CaO) supplied from Kent Kimya A.Ş, Turkey. Graphite crucible and graphite rods used as the anode, the counter electrode, reference electrode were supplied from Kares Seramik, Turkey. The electrolyte was formed by using constant proportions of calcium chloride (77 wt% CaCl2), boron oxide (13 wt% B2O3), calcium oxide (7 wt% CaO) and cobalt oxide (3 wt% CoO) in all experiments. Schematic depiction of the setup and the calciothermic production steps of CoxB particles in cell are shown in Fig. 1.

a Schematic depiction of the setup (a Data acquisition system, b DC supply, c induction furnace controller, d induction furnace, e alumina vessel, f gas outlet, g cathode connecting rod, h anode, k argon cylinder, l gas washing bottle) b Illustration of calciothermic reduction of CoO and formation of CoxB
Before constant voltage electrolysis, electrochemical behavior of CaCl2, CaO and CoO was investigated via CV. The CV measurements were conducted using a Gamry reference 3000 electrochemical workstation. In CV analyses, 3 mm diameter graphite rods were used as the working electrode (WE), counter electrode (CE) and the quasi-reference electrode (RE). The cyclic voltammograms of the system were obtained at 1223 K with a scanning rate of 200 mV/s. In constant voltage experiments, a graphite crucible (Ø 45 mm, h 100 mm) was polarized as the cathode, and a graphite rod (Ø 16 mm) was used as the anode, aiming to produce large amounts of Ca. CaCl2, CaO and B2O3 were fed into the graphite crucible and heated to the reaction temperature in a medium frequency induction furnace (50 kHz, 30 kW, 40 A) and kept at the reaction temperature for 30 min before the experiment to ensure homogeneity. CoO was added 5 min before the current was applied to the cell. The cell was swept with argon during the experiment at 50 mL/min. After the experiments, only hot water was used to remove the electrolyte surrounding the powders.
The composition, structure, morphology and magnetic properties of powders were characterized using atomic absorption spectrometry (Perkin Elmer Analyst 800), X-ray diffractometry (XRD-Phillips Model PW3710), scanning electron microscope (SEM-ZEISS EVO LS 10), and vibrating sample magnetometer (VSM-Lake Shore 7407).
3 Results and Discussion
3.1 Calciothermic Production Mechanism of CoxB
Ca formation in the cell occurs with the electrolytic reduction of CaO dissolved in CaCl2 on the large cathode surface, and 4% by mole of the produced Ca is dissolved in the electrolyte (Eq. 1). As soon as solid CoO (6.64 g/cm3) is added to the cell, CoO powders accumulate at the bottom of the graphite crucible polarized as cathode, due to its density. While the reduction of CoO occurs in a relatively large surface area with Ca dissolved in the electrolyte, it is also present in the electrolyte from a narrow surface area that is in contact with the cathode (Eq. 2). The Co (8.90 g/cm3) particles in contact with the graphite crucible polarized as the cathode form the new cathode surface. B2O3 is in contact with the entire cathode surface in its molten state under experimental conditions and elemental B is produced by calciothermic reduction of B2O3 at the Co electrolyte interface, which acts as the cathode (Eq. 6). B attaches to the Co surface acting as the cathode and first forms the low B containing Co2B (Eq. 7). Co2B compounds are transformed into CoB by increasing the B concentration gradient on the cathode surface and by thermochemical diffusion (Eq. 8a). CoB reacts with Co at the centre of the particle to form Co2B again (Eq. 8b). Since this transformation continues until the presence of Co in the particle is depleted, the dominant phase is usually Co2B. Due to the low Gibbs free energy of formation Co2B and CoB are formed as the main product in the cell, while a small amount of CaB6 is thought to occur (Eq. 9, Fig. 2b). Because using graphite anode, CO and CO2 gases are emitted on the anode (Eqs. 3, 4) [20].

a Change in the decomposition potentials (ΔE) for CaCl2, CaO, and CoO as a function of temperature, b Change in Gibbs free energy of boron, calcium and cobalt reactions as a function of temperature
The decomposition voltages of the components that make up the electrolyte, which change with temperature, are calculated from the Gibbs free energy of formation of the elements and given in Fig. 2a. The decomposition potentials (ΔE) of the materials used were calculated from Gibbs free energy of formation values using Eq. (10) [21]. In Eq. (10), n represents the number of electrons and F represents Faraday’s constant. It has been determined that the degradation potential of CaO occurs at potentials approximately 1.0 V lower than the theoretical degradation potential when using carbon anodes (Fig. 2a) [16].
In addition to the theoretical investigations, the electrochemical behavior of the electrolyte components was also investigated by CV analysis (Fig. 3). Voltammograms of pure CaCl2, CaCl2–CaO, and CaCl2–CaO–CoO are shown in Fig. 3. First, the behavior of pure CaCl2 was investigated, and no oxidation–reduction peaks were found except for Ca2+ reduction and Cl2− oxidation. Then CaO was added to pure CaCl2. On the voltammogram of CaCl2–CaO, no peak was observed on the reduction side except for Ca2+ reduction, while at + 0.87 V and + 1.25 V potentials, two peaks were detected, indicating the formation of CO2 on the oxidation region. The peak expressed with O1 denotes the adsorption of O2− to graphite, and the peak expressed by O2 denotes the desorption of CO2 consistent with the results of Mohamedi et al. Mohamedi et al. investigated the anodic behavior of the glassy carbon electrode in the CaCl2–CaO, and determined that the oxygen output occurs with electrochemical adsorption and electrochemical desorption steps that take place in two consecutive steps [22]. An examination of CaCl2–CaO–CoO voltammogram shows that CoO was reduced at − 0.30 V and oxidized at 0 V.

Cyclic voltammogram in pure CaCl2 and CaCl2–CaO and CaCl2–CaO–CoO mixture at 1223 K (WE: A = 0.6 cm2; scan rate = 200 mV/s)
3.2 Effect of Temperature on CoxB Synthesis
Firstly, the influence of temperature on the structure of CoxB particles was investigated by XRD analysis (Fig. 4). XRD analysis results of the particles produced at 1123 K shows that the particles were composed of Co, Co2B, and CoB phases. The presence of Co, Co2B, and CoB phases at low temperatures indicates the presence of sufficient B in the environment, and it is understood that the thermochemical diffusion of B in the particle is effective in the particle structure (Fig. 4). By increasing the temperature to 1223 K the thermochemical B diffusion in the particle increased, and accordingly, the Co ratio decreased, and the CoxB ratio increased. Co and CoxB phase composition amounts were analysed by AAS. The amount of Co decreased from 11 to 3.6 wt% by increasing the temperature from 1123 to 1323 K (Table 1). Furthermore, the decomposition voltages of CaO, CoO and B2O3 decreased with the increase in temperature (Fig. 2a). Temperature increase accelerated the reduction of CaO, B2O3, and CoO, increasing the time used for B diffusion. The graph of the variation of the current with time given in Fig. 5a also supports that the increase in temperature accelerates the degradation of CoB, B2O3, and CaO. Calık et al. investigated the boriding of pure Co with box boriding at 1173 and 1273 K, and determined that the Co and Co2B phases obtained at 1173 K on the surface were transformed into CoB by increasing the temperature to 1273 K [23].

XRD diffractograms of CoxB powders produced at different temperatures at constant 3.0 V cell voltage for 30 min

a Current variation versus time, SEM images of CoxB particles produced at constant 3.0 V cell voltage for 30 min at different temperatures: b 1223 K, c 1273 K, d 1323 K
As observed in the current variations with respect to electrolysis time given in Fig. 5a, the initial current values exhibited declining trends. The currents initiated with relatively high rates (I) and reached steady conditions (II) and remained almost unchanged for the rest of the experiment’s durations. In the first region, a rapid electrochemical CaO reduction and calciothermic CoO reduction occurred. In the 2nd stage, the B formed by the calciothermic reduction of B2O3 at the Co electrolyte interface diffused into the Co particles with temperature and formed CoxB. Cheng et al. carried out the electrochemical reduction of Co3O4 in the Na2CO3–K2CO3 electrolyte and observed a current regime consisting of two regions (a rapid current decrease and a flat plateau) [24]. While a current regime consisting of only two regions is observed in the electrochemical reductions of single oxides, the current regime may consist of three or more regions to reduce mixed oxides with different reduction potentials. Zheng et al. investigated the electro-deoxidation of the NiO–Co3O4 oxide mixture in the Na2CO3–K2CO3 electrolyte and found that the reduction occurs with a 3-domain current regime involving rapid reduction of oxides and alloy formation [25].
The impact of temperature on particle morphology was examined by SEM (Fig. 5). The morphologies of particles were highly variable, with particle sizes ranging from 1 to 50 µm. Large-sized, irregularly shaped particles were formed by the coalescence and sintering of small-sized spherical particles, as seen in SEM images of particles produced at 1123 K (Fig. 5b). By raising the temperature to 1223 K and 1323 K, small particles disappeared, and larger-sized denser particles were formed. The average particle sizes were determined to be 25 µm at 1123 K, 27 µm at 1223 K, and 33 µm at 1323 K. Similar particle morphologies have been discovered, particularly in particles formed by solid-state and metallothermic reactions [26, 27].
3.3 Effect of Process Duration on CoxB Synthesis
The effect of time on the current, phase and microstructure of the particles was investigated under constant 3.0 V cell voltage and 1323 K reaction temperature conditions in the range of 30–60 min. The influence of the experiment durations on the particle structure was examined by XRD analysis, and the results are given in Fig. 6. According to the XRD analysis results, B diffusion did not occur sufficiently in the particles in a short duration (30 min), and the particles were composed of Co, Co2B, and CoB. The experiment duration was raised to 45 and 60 min for the conversion of non-boride-forming Co particles to CoxB, and it was noted that after 45 min, the Co ratio in the particles acquired at the end of 30 min reduced all Co and was converted to CoxB in 60 min. Moreover, the boride and metallic phase compositions of the particles were also investigated by AAS. At the end of the 30 min experiment, the small amount of Co seen in the structure was completely transformed into CoxB by increasing the time to 60 min (Table 2). Calik et al. investigated the boronization of pure Co with box boriding for 1–6 h and determined that the Co, Co2B, and CoB phases in the structure were converted to Co2B with increasing time [23]. The transformation of the structure into a single-phase Co2B structure with increasing time and temperature is due to the reaction of CoB and Co (Eq. 8b) in the presence of Co in the structure.

XRD results of CoxB powders produced at constant 1323 K and 3.0 V for different durations
The current versus time variation curves obtained at different times at a constant 3.0 V cell voltage are given in Fig. 7a. The current regimes obtained at different durations were similarly composed of two regions containing only reduction and boronization regions. The influence of the duration on the particle morphology was examined by SEM (Fig. 7). The particles were determined to be made up of relatively large polygonal particles when the SEM images were examined. The particle morphology was found to be relatively unaffected by time.

a Current variation versus time graph, SEM images of CoxB particles produced at constant 3.0 V for 30 min. at different durations: b 30 min, c 45 min, d 60 min
The magnetic behaviour of CoxB powders, containing Co at the end of the 30 min. test period and without Co at the end of the 60 min test period, were investigated using VSM at room temperature conditions in the range of 0–1.5 T. From the magnetization curves given in Fig. 8a, it is understood that CoxB powders containing Co show soft ferromagnetic behaviour. It was determined that the saturation magnetization value of the powders was 76.011 emu/g, the coercivity value was 23.735 Oe, and the permanence value was 1.054 emu/g. The hysteresis curve of particles consisting entirely of Co2B and CoB without Co is given in Fig. 8b. The saturation magnetization value of the powders was 30.107 emu/g, the coercivity value was 40.210 Oe, and the permanence value was 0.764 emu/g, as determined by the hysteresis curve.

Magnetization curves of CoxB powders produced at 1323 K and 3.0 V for different durations: a 30 min, b 60 min
4 Conclusions
In this study, it has been shown that calciothermic CoxB can be produced in one step from oxide based materials without the need for pre-and post-treatment by in situ Ca production via electrolytic reduction of CaO. According to the results reported, the following conclusions can be drawn:
-
(a)
According to the CV results, the cathodic reduction of CoO occurred in one step at − 0.30 V, and CO2 formation occurred in two steps at + 0.87 V and + 1.25 V potentials.
-
(b)
Temperature was determined to accelerate the conversion of Co to CoxB. At 1123 K, the powder structure contained 11 wt% Co, but this ratio dropped to 3.6 percent at 1323 K.
-
(c)
The duration of the electrolysis had no effect on particle size or morphology. However, the conversion of Co to CoxB increased with electrolysis time. The Co ratio in the particles decreased from 3.6 percent to 0 percent by increasing the experiment time from 30 to 60 min.
-
(d)
The magnetic properties of the Co2B and CoB particles obtained at the end of 30 min and 60 min were investigated by VSM. The saturation magnetization, permanent magnetization, and coercivity values of the particles obtained at the end of the 30 min experiment period were determined as 76,011 emu/g, 23,735 Oe, 1.054 emu/g, respectively. These values were 76,011 emu/g, 23,735 Oe, and 1.054 emu/g, respectively, in the particles obtained at the end of 60 min and consisting entirely of Co2B and CoB.
References
Ali A Z O, Masoudi A, and Soliyman R, J Nanomed Nanotechnol (2017). https://doi.org/10.4172/2157-7439.1000477.
Zieschang A M, Bocarsly J D, Schuch J, Reichel C V, Kaiser B, Jaegermann W, et al., Inorg Chem 58 (2019) 16609. https://doi.org/10.1021/acs.inorgchem.9b02617.
Baris M, Simsek T, and Akkurt A, Bull Mater Sci 39 (2016) 1119. https://doi.org/10.1007/s12034-016-1231-x.
Wu C, Wu F, Bai Y, Yi B, and Zhang H, Mater Lett 59 (2005) 1748. https://doi.org/10.1016/j.matlet.2005.01.058.
Campos-Silva I, Franco-Raudales O, Meda-Campaña J A, Espino-Cortés F P, and Acosta-Pavón J C. High Temp Mater Process 38 (2019) 158. https://doi.org/10.1515/htmp-2018-0013.
Kaptay G and Kuznetsov S A, Plasmas Ions 2 (2000) 45. https://doi.org/10.1016/S1288-3255(00)87686-8.
Aĝaoĝullari D, Gökçe H, Duman I, and Öveçoĝlu M L. J Eur Ceram Soc 32 (2012) 1457. https://doi.org/10.1016/j.jeurceramsoc.2011.03.037.
Khanra A K, Pathak L C, Mishra S K, and Godkhindi M M, Mater Lett 58 (2004) 733. https://doi.org/10.1016/j.matlet.2003.06.003.
Robert F, The Physics and Chemistry of Carbides, Nitrides and Borides, Kluwer Academic Publishers (1989). https://doi.org/10.1007/978-94-009-2101-6.
Okabe T H, Extr. Metall. Titan. Conv. Recent Adv. Extr. Prod. Titan. Met. (2019). https://doi.org/10.1016/B978-0-12-817200-1.00008-9.
Okabe T H and Takeda O. Extr. Metall. Titan. Conv. Recent Adv. Extr. Prod. Titan. Met. (2019). https://doi.org/10.1016/B978-0-12-817200-1.00005-3.
Bugdayci M, Deniz G, Ziyreker C, Turan A, and Oncel L, Eng Sci Technol Int J 23 (2020) 1259. https://doi.org/10.1016/j.jestch.2020.03.003.
Govinda Rajulu G, Girish Kumar M, Hari Babu B, Srinivasa Rao K, and Nagesh C R V S, Trans Indian Inst Met 69 (2016) 999. https://doi.org/10.1007/s12666-015-0600-2.
Mohanty J and Behera P K, Trans Indian Inst Met 72 (2019) 859. https://doi.org/10.1007/s12666-018-1544-0.
Pradip S, Gautham B P, Reddy S, and Runkana V, Trans Indian Inst Met 72 (2019) 2159. https://doi.org/10.1007/s12666-019-01790-1.
Suzuki R O. JOM 59 (2007) 68. https://doi.org/10.1007/s11837-007-0014-7.
Ono K, Okabe T H, and Suzuki R O. Mater Trans 58 (2017) 313. https://doi.org/10.2320/matertrans.MK201604.
Suzuki N, Tanaka M, Noguchi H, Natsui S, and Kikuchi T, ECS Trans 75 (2013) 507.
Descallar-Arriesgado R F, Kobayashi N, Kikuchi T, and Suzuki R O, Electrochim Acta 56 (2011) 8422. https://doi.org/10.1016/j.electacta.2011.07.027.
Suzuki R O, Ono K, and Teranuma K. Metall Mater Trans B 34 (2003) 287. https://doi.org/10.1007/s11663-003-0074-1.
Vignes A, Electrometallurgical Extraction Processes, in Extractive Metallurgy, (ed) Vignes A, LONDONISTE Ltd and John Wiley & Sons, Inc (2011), vol 2, p 87–116.
Mohamedi M, Børresen B, Haarberg G M, and Tunold R. J Electrochem Soc 146 (1999) 1472. https://doi.org/10.1149/1.1391789.
Çalik A, Karakas M S, Ucar N, and Ünüvar F, Kov Mater 52 (2014) 107. https://doi.org/10.4149/km20142107.
Cheng X, Tang D, Tang D, Zhu H, and Wang D, J Electrochem Soc 162 (2015) E68. https://doi.org/10.1149/2.0991506jes.
Zheng K, Cheng X, Dou Y, Zhu H, and Wang D. J Electrochem Soc 164 (2017) E422. https://doi.org/10.1149/2.1141713jes.
Kovalev D Y, Potanin A Y, Levashov E A, and Shkodich N F. Ceram Int 42 (2016) 2951. https://doi.org/10.1016/j.ceramint.2015.10.078.
Korchagin M A, Dudina D V., Bokhonov B B, Bulina N V, Ukhina A V, and Batraev I S, J Mater Sci 53 (2018) 13592. https://doi.org/10.1007/s10853-018-2290-8.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kartal, L. Synthesis of Cobalt Boride Particles by Molten Salt Assisted Calciothermic Reduction. Trans Indian Inst Met 76, 757–764 (2023). https://doi.org/10.1007/s12666-022-02762-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12666-022-02762-8