
Abstract
Methylphenidate (MPH) abuse can cause serious neurological damages. The neuroprotective effects of topiramate (TPM) have been reported already, but its mechanism of action still remains unclear. The current study evaluates in vivo role of CREB/BDNF in TPM protection of the rat hippocampal cells from methylphenidate-induced apoptosis, oxidative stress, and inflammation. A total of 60 adult male rats were divided into six groups. Groups 1 and 2 received normal saline (0.7 ml/rat) and MPH (10 mg/kg) respectively for 14 days. Groups 3 and 4 were concurrently treated with MPH (10 mg/kg) and TPM 50 and 100 mg/kg respectively for 14 days. Groups 5 and 6 were treated with 50 and 100 mg/kg TPM only respectively. After drug administration, open field test (OFT) was used to investigate motor activity. The hippocampus was then isolated and the apoptotic, antiapoptotic, oxidative, antioxidant, and inflammatory factors were measured. Expression of the total and phosphorylated CREB and BDNF in gene and protein levels, and gene expression of Ak1, CaMK4, MAPK3, PKA, and c-Fos levels were also measured. MPH significantly decreased motor activity in OFT. TPM (50 and 100 mg/kg) decreased MPH-induced motor activity disturbance. Additionally, MPH significantly increased Bax protein level, CaMK4 gene expression, lipid peroxidation, catalase activity, mitochondrial GSH, IL-1β, and TNF-α levels in isolated hippocampal cells. Also CREB, in total and phosphorylated forms, BDNF and Bcl-2 protein levels, Ak1, MAPK3, PKA and c-Fos gene expression, superoxide dismutase, glutathione peroxidase, and glutathione reductase activities decreased significantly by MPH. TPM (50 and 100 mg/kg), both in the presence and absence of MPH, attenuated the effects of MPH. Immunohistochemistry data showed that TPM increased localization of the total and phosphorylated forms of CREB in dentate gyrus (DG) and CA1 areas of the hippocampus. It seems that TPM can be used as a neuroprotective agent against apoptosis, oxidative stress, and neuroinflammation induced by frequent use of MPH. This might be probably mediated by the CREB/BDNF and their upstream signaling pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methylphenidate (MPH) is a neural stimulant widely used for the management of hyperactive children (Challman and Lipsky 2000; Motaghinejad et al. 2016c). MPH inhibits reuptake of dopamine and norepinephrine into presynaptic terminals (Motaghinejad et al. 2015c; Patrick and Markowitz 1997) and has properties similar to those of cocaine and methamphetamine, which render it a high risk substance in terms of abuse and dependency (Huss and Lehmkuhl 2001; Tagaya 2010). Previous studies have suggested that oxidative stress, inflammation, and apoptosis could be induced by chronic MPH abuse in brain cells, especially in the hippocampus, but its mechanism of action and signaling pathway are not clear (Motaghinejad et al. 2016c; Motaghinejad et al. 2016d; Réus et al. 2013; Réus et al. 2014). Topiramate (TPM), an anticonvulsant successfully used in the treatment of alcoholism, cocaine, and methamphetamine addiction (Arnone 2005; Garnett 2000; Johnson et al. 2013; Motaghinejad et al. 2016a; Oncken et al. 2014), has antioxidant, anti-inflammatory, antiapoptotic, immunomodulatory, and neuroprotective properties (Armaǧan et al. 2008; Dudley et al. 2011; Mao et al. 2015). These studies have shown that TPM could act as a protective compound in therapy of few neurodegenerative disorders. However, the exact mechanism of action of TPM neuroprotective effect and signaling pathways still remain unclear (Motaghinejad and Motevalian 2016; Shank et al. 2000). On the other hand, previous studies have shown that cyclic AMP response element binding protein (CREB) acts as a major transcription factor in brain development and neurogenesis (Blendy 2006; Gao et al. 2011; Lee et al. 2005). CREB is activated in a phosphorylated form and some protein kinases such as Ak1, CaMK4, MAPK3, PKA enzymes phosphorylate this transcription factor and convert CREB to its active form (Aguiar et al. 2011; Carlezon et al. 2005; Kitagawa 2007; Réus et al. 2011; Terranova et al. 2016). CREB act on DNA and prompts the production of BDNF and c-Fos proteins, which are important in neurogenesis and the development of neurons (Aguiar et al. 2011; Almeida et al. 2010; Réus et al. 2011; Zuo et al. 2016). Considering the role of hippocampus in depression, cognition, and anxiety-like behaviors, the aim of this research is to study the effects of TPM against MPH-induced apoptosis, oxidative stress, and inflammation and clarify the role of the phosphorylated-CREB signaling pathway in this type of neuroprotection in both dentate gyrus (DG) and CA1 areas of hippocampus. We also attempted to indicate the possible roles of Ak1, CaMK4, MAPK3, and PKA, as the upstream proteins of CREB signaling pathway, on gene expression and the phosphorylation and activation of CREB protein and c-Fos as well as BDNF as downstream products of CREB.
Materials and Methods
Animals
Sixty adult male wistar rats (mean weight 210 ± 10 g, 10 weeks old) were obtained from animal house of Iran University of Medical Sciences (IUMS, Tehran, Iran) and transferred to the laboratory. The animals were kept for 2 weeks before the start of the experiment at room temperature with free access to standard food and tap water in a standard dark/light cycle. Our experimental protocol was approved by the ethical committee in research deputy of Iran University of Medical Sciences.
Drug
MPH and TPM were obtained from Sigma-Aldrich (USA) and dissolved in normal saline for injection, freshly prepared just before use, and the volume of injection was 0.7 ml/rat.
Experimental Design
Sixty adult male rats were divided randomly into six groups. Group 1 (as negative control) was treated with normal saline (0.7 ml/rat, ip) for 14 days. Group 2 (as positive control) received MPH (10 mg/kg) for 14 days. Groups 3 and 4 were treated concurrently for 14 days with MPH (10 mg/kg, ip) and TPM at doses of 50 and 100 mg/kg (ip), respectively. Groups 5 and 6 received only TPM at doses of 50 and 100 mg/kg respectively for 14 days. On day 15 after drug administration, open field test (OFT), a standard behavioral method used for the study of hippocampal degeneration, was performed to evaluate motor activity in animals and then hippocampus was isolated. In addition, using the mentioned behavioral protocol, some markers of oxidative stress, inflammation, and also some proteins which are involved in apoptosis were also measured in the hippocampus. Considering the importance of CREB and BDNF signaling in neuroprotection, we evaluated the effects of TPM on MPH-induced disturbances in the CREB/BDNF signaling pathway by using real-time reverse transcriptase-PCR (RT-PCR) and western Blot methods. Also, the localization of CREB proteins, both CREB and phosphorylated CREB (P-CREB), in DG and CA1 regions of hippocampus were evaluated by immunohistochemistry. In addition, the alterations in expression of some genes coding for protein kinases such as Ak1, CaMK4, MAPK3, PKA, which are involved in CREB phosphorylation, were measured in hippocampus. Furthermore, cell densities of the mentioned areas were evaluated by hematoxylin and eosin (H&E) staining.
Open Field Test
OFT was used as a standard test for the assessment of anxiety and locomotor activity in rodents. For performing the test, an apparatus was used with the bottom divided into 16 equally spaced squares bordered by opaque walls, 65.90 cm in length. The bottom was painted black, excluding the 6-mm broad white lines, which divided the ground into 16 squares. During the experiment, all parts of the room except for the open field were kept dark, and the apparatus was illuminated by a 100-W bulb that focused on the field from a height of about 110 cm from the ground. The open field maze was cleaned between experiments for each rat using 70% ethyl alcohol. Behavior was scored with Hindsight for MS-dos (version 1.5, California, USA), and each trial was recorded for latter analysis, using a video camcorder (Hitachi, VM-7500LA Tokyo, Japan) positioned above the apparatus. Measures of ambulation distance and line crosses and other behaviors were obtained with an automated camera-based computer tracking system (Limelight, Actimetrics, Wilmette, Illinois, USA) on an IBM PC computer with the camera fixed to the ceiling, 2.1 m above the apparatus. To assess the process of habituation to the novelty of the environment, rats were exposed to the apparatus for 5 min 1 day before the experiment (day 14). On the day of experiment (day 15), each animal was centrally positioned in the field for a maximum of 5 min and the following behaviors were monitored:
-
1.
Ambulation distance: the distance with which the rat crossed one of the grid lines.
-
2.
Center square entries: frequency with which the rat crossed one of the red lines with all four paws into the central square.
-
3.
Center square duration: duration of the time rat spent in the central square.
-
4.
Rearing: frequency with which the rat stood on their hind legs in the maze. This assay was used to evaluate locomotor activity and anxiety in rodents according to the references (Motaghinejad et al. 2016b; Motaghinejad et al. 2016c; Schwarting et al. 1999)
Mitochondrial Preparation
By administration of 50 mg/kg of thiopental, all animals were anesthetized and euthanized. Then, the brain tissues were detached after the skull was dissected out for isolation of hippocampus. The isolated hippocampal tissues were homogenized separately in cold homogenization buffer (20 mM 4-morpholinepropanesulfonic acid, pH 7.2, 10 mM MgCl2, 250 mM sucrose, 0.05 mM EGTA) using a homogenizer. Homogenated tissues were centrifuged at 400×g for 10 min, and the supernatant obtained was re-centrifuged at 12000×g for 15 min. The sediments were suspended again two more times in homogenization buffer and centrifuged at 12000×g for 10 min. Finally, the remaining sediments were mixed with homogenization buffer and stored in the refrigerator (3 °C). The total concentration of mitochondrial protein was determined by the Bradford method. The homogenized cell solutions were analyzed for the measurement of oxidative stress and apoptotic factors (Aliev et al. 2003; Motaghinejad et al. 2016d; Motaghinejad et al. 2015a; Tagaya 2010).
Determination of Lipid Peroxidation
The amount of the total lipid peroxidation was measured by evaluating the amount of thiobarbituric acid reactive compounds (TBARS). The quantity of this compound was calculated using an extension coefficient of 165 mM−1 cm−1 at 530 nm. Results were expressed as nmol/mg protein (Amador et al. 2001; Fortunato et al. 2006; Motaghinejad et al. 2016e).
Determination of Manganese Superoxide Dismutase Activity
Activity of manganese superoxide dismutase (MnSOD) was evaluated using a special kit (Cayman Chemical Company, USA). The absorbance was obtained at 450 nm, and the findings were expressed as U/ml/mg protein (Bongiovanni et al. 2007; Motaghinejad et al. 2016a; Pomierny-Chamioło et al. 2013; Motaghinejad et al. 2015b)
Study of Catalase (CAT) Activity
The CAT activity was measured by a method that employs hydrogen peroxide (H2O2) to generate H2O and O2 (Aebi 1984). Brain tissue was sonicated in 50 mmol/l phosphate buffer (pH 7.0), and the resulting suspension was centrifuged at 3000×g for 10 min. The sample aliquot (20 μL) was added to 980 μL of the substrate mixture. The substrate mixture contained 0.3 mL of hydrogen peroxide in 50 ml of 0.05 M phosphate buffer (pH 7.0). Initial and final absorbance was recorded at 240 nm after 1 and 6 min, respectively. A standard curve was established using purified catalase (Sigma, MO, USA) under similar conditions (Bongiovanni et al. 2007; Johansson and Borg 1988).
IL-1β and TNF-α Measurements
Concentrations of TNF-α and IL-1β in supernatant of hippocampal cells were measured using a commercially available ELISA kit (Genzyme Diagnostics, Cambridge, UK). Briefly, 96 well microtitre plates (Nunc) were coated with sheep anti-rat TNF-α and IL-1β polyclonal antibodies (2 mg/ml in bicarbonate coating buffer; 0.1 M NaHCO3, 0.1 M NaCl, pH 8.2, for 20 h at 48 °C), then washed three times with washing buffer (0.5 M NaCl, 2.5 mM NaH2PO4, 7.5 mM Na2HPO4, 0.1% Tween 20, pH 7.2). 100 μl of a 1% (w/v) ovalbumin (Sigma Chemical Co., Poole, Dorset, UK) solution in bicarbonate as coating buffer was added to each well and incubated at 37 °C for 1 h. After three washes, 100 μl of samples and standards were added and plates were incubated at 48 °C for 20 h. After three washes, 100 μl of the biotinylated sheep anti-rat IL-1β or TNF-α antibody (1:1000 dilutions in washing buffer containing 1% sheep serum, Sigma Chemical Co., Poole and Dorset, UK) was added to each well. The further incubation was carried out for 1 h at room temperature. After three washes, 100 μl avidin-HRP (Dako Ltd., UK) (1:5000 dilution in wash buffer) was added to each well, and the plates were incubated at room temperature for 15 min. After three washes, 100 μl of 3, 3′, 5, 5′-tetramethylbenzidine (TMB) substrate solution (Dako Ltd., UK) was added to each well, and the plates were incubated for 10 min at room temperature. At the end of the incubation period, 100 μl of 1 M H2SO4 was added to each well to stop the reaction and to facilitate the color development. Absorbance was read at 450 nm on a microtitre plate reader. The detection limit of the assay was determined to be 4.3 pg/ml. Results were expressed as ng IL-1β/ml or TNF-α/ml (Erickson and Banks 2011; Marquette et al. 2003).
Real-Time Reverse Transcriptase-PCR Studies
The total RNA was extracted from hippocampus (~200 μg) using ONE STEP-RNA Reagent (Bio Basic, Canada Inc.) according to the manufacturer’s instructions, and the quantity and quality of RNA were analyzed using a nanodrop (ND-1000, Thermo Scientific Fisher, US) and gel electrophoresis. RNA was treated with DNase I (Qiagen, Hilden, Germany) to eliminate any genomic contamination, as described by the manufacturer. The total RNA (1 μg) was used to synthesize cDNA. GAPDH primers as housekeeping were used to examine the integrity and quality of cDNA. RT-PCR was carried out to evaluate the differences in expression patterns of BDNF gene among samples of each group. The specific primers corresponding to the coding sequence including BDNF Forward: 5′-GGAGGCTAAGTGGAGCTGAC-3′; BDNF Reverse: 5′-GCTTCCGAGCCTTCCTTTAG-3′; Akt1 Forward: 5′-AAGGAGATCATGCAGCACCG-3′; Akt1 Reverse: 5′-GGTGGGCTCACCTTCTTCTC-3′; CaMK4 Forward: 5′-AGCAGCAGTCACACCAACAT-3′; Reverse: 5′-TCTGTCTTGTCCTTGCCGTC-3′; MAPK3 Forward: 5′-TATCAACACCACCTGCGACC-3′; Reverse: 5′-ATGATCTCTGGGGCTCGGTA-3′; c-Fos Forward: 5′-GGGAGCTGACAGATACGCTC-3′; Reverse: 5′-TTGGCAATCTCGGTCTGCAA-3′; CREB1 Forward: 5′-CAGACAACCAGCAGAGTGGA-3′; Reverse: 5′-CTGGACTGTCTGCCCATTG-3′; GAPDH Forward: 5′-AGACAGCCGCATCTTCTTGT; Reverse: 5′-CCGTTCACACCGACCTTCA-3′; PKA Forward: 5′-GCAGGAGAGCGTGAAAGAGT-3′; Reverse:5′-CTGAGAAGGGGTCTCCCATTT-3′, were designed by Primer 3 software version 0.4 (frodo.wi.mit.edu). Real-time RT-PCR was performed in 20-μl reactions containing 1 μl cDNA target, 100 nM forward and reverse primers, and 1× SYBR® Premix Ex Taq™ II (Takara, Tokyo, Japan). Experiments were carried out in triplicate using a CFX96™ Real-Time System (C1000TM Thermal Cycler) (Bio-Rad, Hercules, CA, USA). Amplification conditions were as follow: initial denaturation at 95 °C for 10 min, followed by 40 cycles (denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 60 s). The relative value of the mRNA expression level of CREB, Ak1, CaMK4, MAPK3, PKA, BDNF, and c-Fos gene was calculated by comparing the cycle thresholds (CTs) of the target gene with that of housekeeping gene (GAPDH) using the 2−ΔΔct method and REST 2009 software. Serial dilutions of cDNAs were used to calculate the efficiencies of the primer sets on real-time PCR. In this regard, it was found that the efficiencies of the various primer sets were similar (Motaghinejad et al. 2016a; Peng et al. 2012).
Western Blot
We studied the immunoreactivity, CREB, total and phosphorylated, BDNF, Bax, Bcl-2, and caspase3 contents of isolated hippocampus from treated rats by western blotting. Electrotransfer of the resolved bands from gel to polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, USA) was fulfilled in 90 min at 0.7 mA/cm2 using a semi-dry transfer apparatus (PeQlab). After transferring step, the membrane was weakly stained for about 3 min with Coomassie blue G-250 (Sigma-Aldrich, UK) 1 μg/100 ml distilled water without methanol. Then the membrane was dried and cut into 2-mm wide stripes. After destaining with methanol, the strips washed and blocked with 2% BSA overnight at 4 °C and then added with 1:100 diluted human or bovine sera at room temperature (RT) for 2 h on a shaker. The membranes then were washed with PBS-T (three washing steps in a total of 10 min) and incubated with the following conjugated polyclonal anti-rabbit antibody: BDNF, CREB, total and phosphorylated, (1:500 dilutions in BSA, 360 min, RT; Sigma-Aldrich, Germany) Bax, Bcl-2 (1:1000 dilutions in BSA, 240 min, RT; Sigma-Aldrich, Germany) then all strips was exposed by secondary HRP conjugated polyclonal Rabbit anti-sheep antibody (1:5000 dilution in BSA, 120 min, RT; Sina Biotech, Iran). The strips were washed and incubated with chemiluminescent substrate (Luminol and H2O2) for 2 min in RT. Finally, the reactive bands were detected on X-ray film within 10–20 s under safelight condition (Motaghinejad et al. 2016a; Woo et al. 2005).
Immunohistochemistry
Immunohistochemical detection of CREB, total and phosphorylated, was performed on tissue slides. Tissue slides were prepared based on previous studies (Madjd et al. 2013; Motaghinejad et al. 2016a). After deparaffinization of slides in xylene and rehydration through graded alcohol, slides were immersed in methanol/hydrogen peroxide for 10 min to block endogenous peroxidase activity. Antigen was retrieved by autoclaving tissue sections for 10 min in sodium citrate buffer (pH 6.0). The sections were then incubated with primary antibody with an optimal dilution of 1:300 for the total CREB and 1:50 for phosphorylated CREB for overnight at +4 °C. After washing, the sections were incubated with anti-rabbit/anti-mouse Envision (Dako, Denmark) secondary antibody for 1 h. Color was developed with the addition of 3, 3′-diaminobenzidine (DAB, Dako) to achieve visualization of the antigen. In the final step, sections were lightly counterstained with hematoxylin (Dako, Denmark), dehydrated in alcohol, cleared in xylene, and mounted for examination. The omission of primary antibody and its replacement with TBS (Tris-buffered saline) was used as negative reagent control. After processing, sections were mounted on glass slides, dehydrated, and cover slipped. Immunoreactivity was visualized using a Nikon Eclipse E600 microscope, and images were captured with a QImaging Retiga 1300 camera using Image-Pro Plus software, and cell counting was performed using a 400× oil-immersion lens on a Nikon Eclipse E600 microscope (Media Cybernetics) (Madjd et al. 2013). A computer image analysis system morphometric software (Optikavision pro, Italy) was used to determine the average CREB and/or P-CREB positive cells in the hippocampal DG and CA1 regions (Motaghinejad et al. 2016c).
Histological Studies (H&E Staining)
For histological studies, isolated hippocampus was fixed in formalin, washed in physiologic serum and dehydrated by ethanol, embedded in paraffin wax, sectioned at 5 μm thicknesses, and stained with Mayer’s hematoxylin and eosin (H&E). The morphological changes and the cell density were observed in the DG and CA1 respectively with the use of cell counting performed by morphometric software (Optikavision pro, Italy) (Motaghinejad et al. 2016c).
Statistical Analysis
The data obtained was statistically analyzed by Graph Pad PRISM Software (version 6). In every experimental group, the data were averaged and expressed as means ± standard errors of the mean (SEM). Then, the differences between controls and treatment groups were evaluated by one-way ANOVA. Differences between severities of behaviors in groups were evaluated by Tukey’s post-hoc test. P < 0.05 was taken as statistically significant.
Results
Results of Open Field Test
As shown in Table 1, the MPH (10 mg/kg) treated group had a lower frequency of central square entries, with F(5,42) = 5.908, and also spent a shorter period of time in the central region of the OFT, with F(5,42) = 3.350, compared to the negative control group (P < 0.05) (Table 1).
On the other hand, TPM (50 and 100 mg/kg) can diminish this effect of MPH and significantly increased the frequency of central square entries, with F(5,42) = 5.908, as well as the time spent in the central region of the OFT, with F(5,42) = 3.350, in comparison with the MPH only treated group (P < 0.05) (Table 1). The MPH treated group had less ambulation distance, with F(5,42) = 2.412, and rearing, with F(5,42) = 1.960, in OFT in comparison to the negative control group. But in the MPH treated groups concomitantly treated with 50 and 100 mg/kg of TPM, a greater ambulation distance, with F(5,42) = 2.412, and rearing number in OFT, with F(5,42) = 1.960, were observed in comparison with MPH (10 mg/kg) only treated group (P < 0.05) (Table 1). On the other hand, the groups treated with TPM (50 and 100 mg/kg), in the absence of MPH, had more ambulation distance, rearing number, frequency of central square entries, and also spent more time in OFT with F(5,42) = 2.412, F(5,42) = 1.960, F(5,42) = 5.908, and F(5,42) = 3.350 respectively in comparison with groups treated with MPH in combination with TPM (P < 0.05) (Table 1). Also, our data showed that there is no significant difference in OFT behavior in groups treated with TPM (50 and 100 mg/kg) alone in comparison with negative control group (Table 1).
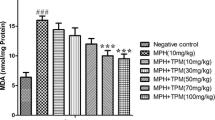
Effects of TPM on MPH-Induced Alterations in Oxidative Stress Biomarkers in Mitochondria
As shown in Table 2, the MPH treated group (10 mg/kg) had an increased MDA level, with F(5,42) = 76.89, and catalase activity, with F(5,42) = 7.170, and decreased SOD activities, with F(5,42) = 6.253, in comparison to the negative control group (P < 0.05) (Table 2).
On the other hand, TPM (50 and 100 mg/kg) can diminish this effect of MPH and decrease MDA level, with F(5,42) = 76.89, and catalase activity, with F(5,42) = 7.170, and increase SOD activity, with F(5,42) = 6.253, (P < 0.05) (Table 2). In addition, the group treated with TPM (50 and 100 mg/kg), in the absence of MPH, significantly decreased MDA level, with F(5,42) = 76.89, and catalase activity, with F(5,42) = 7.170, and increased SOD activity, with F(5,42) = 6.253, in comparison to the groups treated with MPH in combination with TPM (P < 0.05) (Table 2). Our data also showed that there are no significant differences in MDA level, SOD, and CAT activities in groups treated with TPM (50 and 100 mg/kg) alone in comparison with negative control group (Table 2).
Effects of TPM on MPH-Induced Alterations of Inflammation Biomarkers
As shown in Table 2, the MPH (10 mg/kg) treated group had an increased IL-1β, with F(5,42) = 11.34, and TNF-α, with F(5,42) = 19.48, levels in comparison to the negative control group (P < 0.05) (Table 2). On the other hand, TPM (50 and 100 mg/kg) can attenuate this effect of MPH and decrease IL-1β, with F(5,42) = 11.34, and TNF-α, with F(5,42) = 19.48, levels (P < 0.05) (Table 2). Also, groups treated with TPM (50 and 100 mg/kg) in the absence of MPH, showed a significantly decreased level of IL-1β, with F(5,42) = 11.34, and TNF-α, with F(5,42) = 19.48, levels in comparison to groups treated with MPH in combination with TPM (P < 0.05) (Table 2). According to our data, there is a significant difference with F(5,42) = 19.48 in TNF-α in groups treated with TPM (50 and 100 mg/kg) alone in comparison with the negative control group (P < 0.05) (Table 2). While about the IL-1β, this significant difference was observed in group treated with TPM 100 mg/kg alone with F(5,42) = 11.34 when compared with the control group(P < 0.05) (Table 2).
Results of RT-PCR
CREB Expression
MPH decreased CREB gene expression, with F(5,12) = 7.815, in comparison with the control group (P < 0.05). In the MPH treated group, TPM 50 and 100 mg/kg increased CREB gene expression in comparison with the MPH only treated group, with F(5,12) = 7.815 and (P < 0.05), while the group treated with TPM (50 and 100 mg/kg), without MPH, increased CREB gene expression, with F(5,12) = 7.815, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 1). According to our data, there is also a significant difference in CREB gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 7.815, in comparison to the negative control group (P < 0.05) (Fig. 1).

Alterations in expression (RT-PCR) of CREB in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
Ak1 Expression
MPH-decreased Ak1 gene expression, with F(5,12) = 18.02, in comparison with the control group (P < 0.05). In the MPH treated group, TPM 50 and 100 mg/kg increased Ak1 gene expression, with F(5,12) = 18.02, in comparison with the MPH only treated group (P < 0.05), while the group treated only with TPM (50 and 100 mg/kg), without MPH, increased Ak1 gene expression, with F(5,12) = 18.02, in comparison with the group treated with TPM in combination with MPH (P < 0.05) (Fig. 2). According to our data, there is a significant difference in Ak1 gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 18.02, in comparison with the negative control group (P < 0.05) (Fig. 2).

Alterations in expression (RT-PCR) of Ak1 in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
CaMK4 Expression
MPH increased CaMK4 gene expression, with F(5,17) = 29.94, in comparison with the control group (P < 0.05). In the MPH treated group, TPM 50 and 100 mg/kg decreased CaMK4 gene expression, with F(5,17) = 29.94, in comparison with the MPH only treated group (P < 0.05), while groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, caused a decrease of CaMK4 gene expression, with F(5,17) = 29.94, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 3). Our data also indicated that there is a significant difference in CaMK4 gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,17) = 29.94, in comparison with the negative control group (P < 0.05) (Fig. 3).

Alterations in expression (RT-PCR) of CaMK4 in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
MAPK3 Expression
MPH decreased MAPK3 gene expression, with F(5,12) = 16.63, in comparison with the control group (P < 0.05). In the MPH treated group, TPM 50 and 100 mg/kg increased MAPK3 gene expression, with F(5,12) = 16.63, in comparison with the MPH only treated group (P < 0.05), while the group only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased MAPK3 gene expression, with F(5,12) = 16.63, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 4). Our data indicated that there is a significant difference in MAPK3 gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 16.63, in comparison with the negative control group (P < 0.05) (Fig. 4).

Alterations in expression (RT-PCR) of MAPK3 in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
PKA Expression
MPH-decreased PKA gene expression, with F(5,12) = 16.85, in comparison with the control group (P < 0.05) (Fig. 5). In the MPH treated group, TPM at doses of 50 and 100 mg/kg increased PKA gene expression, with F(5,12) = 16.85, in comparison with the MPH only treated group (P < 0.05), while groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased PKA gene expression, with F(5,12) = 16.85, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 5). Our data indicated that there is a significant difference in PKA gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 16.85, in comparison with the negative control group (P < 0.05) (Fig. 5).

Alterations in expression (RT-PCR) of PKA in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
C-Fos Expression
MPH decreased c-Fos gene expression, with F(5,12) = 7.30, in comparison with the control group (P < 0.05) (Fig. 6). In the MPH treated group, TPM 50 and 100 mg/kg increased c-Fos gene expression, with F(5,12) = 7.30, which was statistically significant in 50 mg/kg of TPM in comparison with the MPH only treated group (P < 0.05), while the group treated with only TPM (50 and 100 mg/kg), in the absence of MPH, increased c-Fos gene expression, with F(5,12) = 7.30, in comparison with groups treated with TPM in combination with MPH (10 mg/kg) (P < 0.05) (Fig. 6). Our data also indicated that there is a significant difference in c-Fos gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 7.30, in comparison with the negative control group (P < 0.05) (Fig. 6).

Alterations in expression (RT-PCR) of c-FOS in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). †Shows significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
BDNF Expression
MPH decreased BDNF gene expression, with F(5,12) = 10.07, in comparison with the control group (P < 0.05) (Fig. 7). In the MPH treated group, TPM 50 and 100 mg/kg increased BDNF gene expression, with F(5,12) = 10.07, in comparison with the MPH only treated group (P < 0.05), while groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased BDNF gene expression, with F(5,12) = 10.07, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 7). Our data also indicated that there is a significant difference in BDNF gene expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 10.07, in comparison with the negative control group (P < 0.05) (Fig. 7).

Alterations in expression (RT-PCR) of BDNF in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows significant difference from negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
Results of Western Blot
Total and Phosphorylated CREB (Ser133), Protein Expression
MPH (10 mg/kg) decreased the total, with F(5,12) = 32.56, and phosphorylated CREB (Ser133), with F(5,12) = 31.71, protein expressions in comparison with the control group (P < 0.05) (Figs 8 and 9). In the MPH treated group, TPM 50 and 100 mg/kg increased total, with F(5,12) = 32.56, and phosphorylated CREB (Ser133), with F(5,12) = 31.71, protein expressions in comparison with the MPH only treated group (P < 0.05), while groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased total, with F(5,12) = 32.56, and phosphorylated CREB (Ser133), with F(5,12) = 31.71, protein expressions in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Figs. 8 and 9). According to our data, there are also significant differences in total, with F(5,12) = 32.56, and phosphorylated CREB (Ser133), with F(5,12) = 31.71, protein expressions in groups treated with TPM (50 and 100 mg/kg) alone in comparison with the negative control group (P < 0.05) (Figs. 8 and 9).

Alterations in expression (WB) of the total CREB in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate

Alterations in expression (WB) of phosphorylated CREB in hippocampus in control and groups under treatment with 10 mg/kg of MPH in absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
BDNF Protein Expression
MPH (10 mg/kg) decreased BDNF protein expression, with F(5,12) = 6.904, in comparison with the control group (P < 0.05) (Fig. 10). TPM (50 and 100 mg/kg) in the MPH-treated group increased BDNF protein expression, with F(5,12) = 6.904, in comparison with the MPH only treated group (P < 0.05) (Fig. 10). Groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased BDNF protein expression, with F(5,12) = 6.904, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 10). According to our data, there is no significant difference in BDNF protein expression in groups treated with TPM (50 and 100 mg/kg) alone in comparison to the negative control group (Fig. 10).

Alterations in expression (WB) of BDNF in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
Bax Protein Expression
MPH (10 mg/kg) increased Bax protein expression, with F(5,12) = 46.52, in comparison with the control group (P < 0.05) (Fig. 11). TPM (50 and 100 mg/kg) in the MPH-treated group decreased Bax protein expression, with F(5,12) = 46.52, in comparison with the MPH only treated group (P < 0.05) (Fig. 11). Groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, decreased Bax protein expression, with F(5,12) = 46.52, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 11). According to our data, there is no significant difference in Bax protein expression in groups treated with TPM (50 and 100 mg/kg) alone in comparison to the negative control group (Fig. 11).

Alterations in expression (WB) of Bax in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
Bcl-2 Protein Expression
MPH (10 mg/kg) decreased Bcl-2 protein expression, with F(5,12) = 32.56, in comparison with the control group (P < 0.05) (Fig. 12). TPM (50 and 100 mg/kg) in the MPH-treated group increased Bcl-2 protein expression, with F(5,12) = 32.56, in comparison with the MPH only treated group (P < 0.05) (Fig. 12). Groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased Bcl-2 protein expression, with F(5,12) = 32.56, in comparison with groups treated with TPM in combination with MPH (P < 0.05) (Fig. 12). According to our data, there is also a significant difference in Bcl-2 protein expression in groups treated with TPM (50 and 100 mg/kg) alone, with F(5,12) = 32.56, in comparison to the negative control group (P < 0.05) (Fig. 12).

Alterations in expression (WB) of Bcl-2 in hippocampus in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
Results of IHC
Total and Phosphorylated CREB Protein (Ser-133), Expression, and Localization
MPH (10 mg/kg) caused a dramatic loss of positive CREB total, with F(5,42) = 141.3, and phosphorylated (Ser-133) of granular cells, with F(5,42) = 133.8, in DG [Figs. 13, 16a, 17, and 20a]. TPM at doses of 50 and 100 mg/kg in the MPH-treated groups increased a positive CREB total, with F(5,42) = 141.3, and phosphorylated (Ser-133), with F(5,42) = 133.8, of granular cells in DG in comparison with the MPH only treated group (P < 0.05). Groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased a positive CREB total and phosphorylated (Ser-133), with F(5,42) = 141.3 and F(5,42) = 133.8 respectively, of granular cells in DG in comparison with groups treated with TPM (50 or 100 mg/kg) in combination with MPH (10 mg/kg) (P < 0.05) [Figs. 13, 16a, 17, and 20a].

Expression and localization of the positive total CREB in dentate gyrus (DG) cell layer of the hippocampus in A1 (positive control = ×400), B1 (positive control = ×200), C1 (negative control), D1 (group received 10 mg/kg of methylphenidate), E1 and F1 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G1 and H1 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive CREB total. (Magnification ×400; scale bar 100 μm)
On the other hand, MPH in the mentioned doses significantly decreased a positive CREB total, with F(5,42) = 101.5 in small pyramidal and F(5,42) = 147.5 in large pyramidal and phosphorylated (Ser-133), with F(5,42) = 184.7 in small pyramidal and F(5,42) = 266.9 in large pyramidal of small and large pyramidal cells in CA1 [Figs. 14, 15, 16b, 16c, 18, 19, 20b, and 20c]. TPM at doses of 50 and 100 mg/kg in the MPH-treated group increased a positive CREB total, with F(5,42) = 101.5 in small pyramidal and F(5,42) = 147.5 in large pyramidal, and phosphorylated (Ser-133), with F(5,42) = 184.7 in small pyramidal and F(5,42) = 266.9 in large pyramidal small and large pyramidal cells in CA1 in comparison with the MPH only treated group (P < 0.05) [Figs. 14, 15, 16b, 16c, 18, 19, 20b, and 20c]. Groups only treated with TPM (50 and 100 mg/kg), in the absence of MPH, increased a positive CREB total, with F(5,42) = 101.5 in small pyramidal and F(5,42) = 147.5 in large pyramidal, and phosphorylated (Ser-133), with F(5,42) = 184.7 in small pyramidal and F(5,42) = 266.9 in large pyramidal, small and large pyramidal cells in CA1 in comparison with groups treated with TPM (50 or 100 mg/kg) in combination with MPH (10 mg/kg) (P < 0.05) [Figs. 14, 15, 16b, 16c, 18, 19, 20b, and 20c]. According to our data, there are no significant differences in the number of CREB and P-CREB positive cells in both DG and CA1 parts of hippocampus in groups treated with TPM (50 and 100 mg/kg) alone in comparison to the negative control group [Figs. 13, 14, 15, 16a, b, c, 17, 18, 19, 20a, b, c].

Expression and localization of the positive total CREB in small pyramidal cell layer of the CA1 hippocampus in A2 (positive control = ×400), B2 (positive control = ×200), C2 (negative control), D2 (group received 10 mg/kg of methylphenidate), E2 and F2 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G2 and H2 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive CREB total. (Magnification ×400; scale bar 100 μm)

Expression and localization of the positive total CREB in large pyramidal cell layer of the CA1 hippocampus in A3 (positive control = ×400), B3 (positive control = ×200), C3(negative control), D3 (group received 10 mg/kg of methylphenidate), E3 and F3 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G3 and H3 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive CREB total. (Magnification ×400; scale bar 100 μm)

Alterations in expression (IHC) of the total CREB in granular cells of DG (a), small pyramidal cells of CA1 (b) and large pyramidal cells of CA1 (c) in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate

Expression and localization of positive phosphorylated CREB in dentate gyrus (DG) cell hippocampus in A4 (positive control = ×400), B4 (positive control = ×200), C4 (negative control), D4 (group received 10 mg/kg of methylphenidate), E4 and F4 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G4 and H4 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive phosphorylated CREB. (Magnification ×400; scale bar 100 μm)

Expression and localization of positive phosphorylated CREB in small pyramidal cell layer of the CA1 hippocampus in A5 (positive control = ×400), B5 (positive control = ×200), C5 (negative control), D5 (group received 10 mg/kg of methylphenidate), E5 and F5 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G5 and H5 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive phosphorylated CREB. (Magnification ×400; scale bar 100 μm)

Expression and localization of positive phosphorylated CREB in large pyramidal cell layer of the CA1 hippocampus in A6 (positive control = ×400), B6 (positive control = ×200), C6 (negative control), D6 (group received 10 mg/kg of methylphenidate), E6 and F6 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and G6 and H6 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. The arrow pointing up shows a positive phosphorylated CREB. (Magnification ×400; scale bar 100 μm)

Alterations in expression (IHC) of phosphorylated CREB in granular cells of DG (a), small pyramidal cells of CA1 (b) and large pyramidal cells of CA1 (c) in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (50 and 100 mg/kg). All data are expressed as mean ± SEM (n = 10). The asterisk shows a significant difference from the negative control group (P < 0.05). The number sign shows a significant difference from the methylphenidate only treated group (P < 0.05). The dagger shows a significant difference from groups under concomitant treatment with MPH (10 mg/kg) and TPM (50 and 100 mg/kg) (P < 0.05). MPH methylphenidate, TPM topiramate
H&E Staining Results
MPH (10 mg/kg), remarkably decreased the cell density of DG granular cells, with F(5,42) = 28.66, and small and large pyramidal cells in CA1 region, with F(5,42) = 29.79, in comparison with the control group (P < 0.05) (Table 3), while TPM (50 and 100 mg/kg) inhibited this reduction caused by MPH and increased number of mentioned cells in DG granular cells, with F(5,42) = 28.66, and small and large pyramidal cells in CA1 region, with F(5,42) = 29.79, in comparison with the MPH only treated group (P < 0.05) (Table 3). TPM (50 and 100 mg/kg), in the absence of MPH, increased the number DG granular cells, with F(5,42) = 28.66, and small and large pyramidal cells in CA1 region with F(5,42) = 29.79, which were statistically significant in comparison with the group treated with TPM (50 or 100 mg/kg) in combination with MPH (10 mg/kg) (P < 0.05) (Table 3). Degenerated shrunken dark cells with condensed nuclei were observed in granular cells and pyramidal cells in groups treated with 10 mg/kg of MPH. TPM (50 and 100 mg/kg), in the presence of MPH, inhibited such MPH-induced histomorphological changes (Figs. 21, 22, and 23). TPM (50 and 100 mg/kg), in the absence of MPH, did not show any of the mentioned histomorphological changes in the mentioned areas of hippocampus (Figs. 21, 22, and 23). According to our data, there are significant differences in cell density and shapes in both DG, with F(5,42) = 28.66, and CA1, with F(5,42) = 29.79, parts of hippocampus in groups treated with TPM (50 and 100 mg/kg) alone in comparison to the negative control group (P < 0.05) (Table 3) (Figs. 21, 22, and 23).

Hematoxylin and eosin staining shows dentate gyrus (DG) cell layer of the hippocampus in A1, B1, C1, D1, E1, and F1 which respectively shows a group of control A1 (negative control), B1 (group received 10 mg/kg of methylphenidate), C1 and D1 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and E1 and F1 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. Cell layers are different in all groups and show marked vacuolations (arrow pointing up) in DG in B1 (magnification ×400; scale bar 100 μm)

Hematoxylin and eosin staining shows small pyramidal layer of CA1 of the hippocampus in A2, B2, C2, D2, E2, and F2 which respectively shows a group of control A2 (negative control), B2 (group received 10 mg/kg of methylphenidate), C2 and D2 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg respectively), and E2 and F2 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. Cell layers are different in all groups and show marked necrosis, vacuolization, and shrunken (arrow pointing up) in CA1 in B2. (Magnification ×400; scale bar 100 μm)

Hematoxylin and eosin staining shows large pyramidal layer of CA1 of the hippocampus in A3, B3, C3, D3, E3, and F3 which respectively shows a group of control A3 (negative control), B3 (group received 10 mg/kg of methylphenidate), C3 and D3 groups received 10 mg/kg of methylphenidate in the presence of TPM (50 and 100 mg/kg, respectively), and E3 and F3 groups received 50 and 100 mg/kg of TPM in the absence of methylphenidate. Cell layers are different in all groups and show marked necrosis, vacuolization, and shrunken (arrow pointing up) in CA1 in B3. (Magnification ×400; scale bar 100 μm)
Discussion
The result of this study indicated that TPM administration can diminish apoptosis, oxidative stress, and inflammation induced by chronic MPH in the isolated hippocampus of adult rats. In addition, our study suggested that the neuroprotection of TPM might be mediated by the `signaling pathway. The present study demonstrated that MPH administered chronically (10 mg/kg) might alter some behavioral parameters, like motor activity in OFT. Also, according to our results, MPH can increase lipid peroxidation, catalase activity, and decrease SOD activity. Furthermore, it can increase IL-1β and TNF-α levels in the isolated rat hippocampus. On the other hand, MPH can activate apoptosis, increase Bax, and decrease Bcl-2. MPH in the mentioned dose can inhibit CREB, total and in active phosphorylated form, in gene and protein levels, and can also change Ak1, CaMK4, MAPK3, PKA proteins which are involved in CREB signaling pathway and its phosphorylation. This agent also causes inhibition of BDNF and c-Fos as a protein product of the CREB’s effect on DNA. Our data also showed that MPH can change the localization of CREB, total and active phosphorylated form, in DG and CA1 areas of the hippocampus. This data suggests that MPH can induce neurodegeneration by inhibiting the CREB/BDNF signaling pathway. Our study suggests that TPM can decrease lipid peroxidation and catalase activities and increase SOD activity; furthermore, it can decrease the levels of IL-1β and TNF-α in the isolated hippocampus of rats. This neuroprotective agent can ameliorate apoptosis, decrease Bax, and increase Bcl-2 in the isolated hippocampus of MPH treated rats.
TPM can increase CREB, total and active phosphorylated form, in gene and protein levels, and also it can change Ak1, CaMK4, MAPK3, and PKA protein expression in MPH-treated rats. TPM also increased BDNF and c-Fos proteins. TPM can inhibit MPH-induced changes in the localization of CREB, total and active phosphorylated form, in DG and CA1 areas of the hippocampus. This data suggests that by inhibiting the CREB/BDNF signaling pathway, MPH can induce its neurodegenerative effects. In addition, our data showed that TPM, in the absence of MPH, can inhibit apoptosis, inflammation, and oxidative stress, activates the CREB/BDNF signaling pathway, and increases CREB, total and active phosphorylated form, localization in DG and CA1 areas of the hippocampus. Other studies have indicated the use of MPH in the management of hyperactivity in attention—deficit disorders in children and demonstrated that MPH is an amphetamine like compound with neural stimulant activity (Huss and Lehmkuhl 2001; Motaghinejad et al. 2016c). MPH has structural similarities to cocaine and amphetamines (Klein-Schwartz 2002; Motaghinejad et al. 2016c; Motaghinejad et al. 2016d). MPH binds the norepinephrine transporter and to a higher extent to the dopamine transporter, and inhibits the reuptake of norepinephrine and dopamine into presynaptic terminals (Klein-Schwartz 2002; Motaghinejad et al. 2015c). Its high potential for addiction and abuse is for the similarity to amphetamines, methylenedioxymethamphetamine (MDMA), and cocaine (Huss and Lehmkuhl 2001; Motaghinejad et al. 2015d). The data obtained in this study have shown decreases in ambulation number and ambulation distance in OFT caused by MPH (10 mg/kg), in the brain of adult rats. Furthermore, the concomitant use of TPM (50 and 100 mg/kg) with MPH increased ambulation number and ambulation distance in comparison to MPH only treated rats. According to the other findings, MPH can alter motor activity in juvenile rats (Davids et al. 2002; Juárez and Guerrero-Álvarez 2015), and the result of the present study also showed that administration of MPH (10 mg/kg) could decrease time spent in central square and the number of central square entries in OFT, which suggests that chronic MPH administrations might induce anxiety like behavior in adult rats. The anxiety induced by MPH and amphetamine have already been reported in different studies (Motaghinejad et al. 2016c; Vendruscolo et al. 2008). Also, concomitant use of TPM (50 and 100 mg/kg( by MPH increased time spent in central square and central square entries in OFT compared to rats treated only by MPH. Also, other studies have shown that a few anticonvulsant agents have anxiolytic effects and can reduce anxiety and depressive-like behavior in rodents (Khan and Liberzon 2004; Molina-Hernández et al. 2010; Motaghinejad et al. 2016a). Furthermore, anxiety and motor activity disturbance induced by MPH have been also documented already (Davids et al. 2002; Motaghinejad et al. 2015c; Vendruscolo et al. 2008). These results suggest that non-pharmacologic and toxic doses of MPH might induce neurobehavioral changes and anxiety-like behavior in rats (Motaghinejad et al. 2016c; Vendruscolo et al. 2008). On the other hand, antidepressant and anti-anxiety activities of topiramate have been approved in many previous works (Cagetti et al. 2004; Motaghinejad et al. 2016a; Mula et al. 2007). These studies suggested that TPM can diminish the anxiety and depression induced by amphetamine and alcohol and can modulate the disturbances in motor activity induced by these drugs (Cagetti et al. 2004; Siniscalchi et al. 2015). Based on our present findings, TPM at doses of 50 and 100 mg/kg, in the absence of MPH, can also increase the number of rearing, ambulation distance, central square entry, and the time spent in central square in OFT which confirms the findings of the previous studies. MPH (10 mg/kg) can alter oxidative stress situation, and our data showed that MPH can decrease SOD activity while increasing MDA level, as a marker of lipid peroxidation, and catalase activity in the hippocampus. Previous studies showed that chronic administration of MPH caused mitochondrial dysfunction and alteration in respiratory enzymes in brain cells of adult and juvenile rats; these studies suggested that MPH can induce oxidative stress in the brain of rats (Lau et al. 2000; Martins et al. 2006; Motaghinejad et al. 2016c). TPM at doses of 50 and 100 mg/kg can inhibit the changes induced by MPH in SOD and catalase activities and MDA level. Previous studies confirmed that TPM has neuroprotective effects in rat brain (Gibbs et al. 2006; Motaghinejad et al. 2016a; Noh et al. 2006); these results showed a decrease in lipid peroxidation by TPM in the brain of rats treated with pentylenetetrazol (PTZ), and it was highlighted that TPM has a neuroprotective effect mediated by the inhibition of free radicals (Agarwal et al. 2011; François et al. 2006; Mao et al. 2015). Our data showed that TPM at doses of 50 and 100 mg/kg, in the absence of MPH, can activate antioxidants and inhibit oxidative damages. The results of the present study about the the increase of CAT activity by MPH and the decrease of its activity by TPM is debatable, as noted in previous studies, chronic abuse of MPH can cause production of free radicals such as H2O2 and body cells in order to compensate and metabolize the H2O2, increased CAT production. While by TPM administration, the production of free radicals induced by MPH would be inhibited, and the compensatory increases of CAT were not necessary (Armaǧan et al. 2008; Nazirolu et al. 2008). While for SOD activity, this phenomenon does not occur, and according to previous studies, MPH directly inhibits SOD activity, and TPM probably inhibits this reduction which showed in present and previous studies (Kutluhan et al. 2009; Nazıroğlu et al. 2009). The inflammatory markers in the hippocampus were increased by MPH administration at a dose of 10 mg/kg. Other studies indicated that chronic administration of MPH (10 mg/kg) increases pro-inflammatory markers such as TNF-α and IL-1β which are in consistency with our findings (Kuczenski and Segal 2001; Motaghinejad et al. 2016c). This study suggested that the increase in pro-inflammatory markers is responsible for neurodegenerative effects of MPH (Kuczenski and Segal 2001; Motaghinejad et al. 2016c). Many previous studies have shown that methamphetamine and similar compounds can activate neuroinflammatory pathways and increase TNF-α and IL-1β (Wells et al. 2016; Yamamoto and Raudensky 2008).Furthermore, our study revealed that TPM (50 and 70 mg/kg) could significantly decrease the inflammatory biomarkers in MPH-treated rats. Some previous studies showed that TPM has a protective effect against inflammation; these studies suggested that TPM can attenuate TNF-α and TGF-β1 and also reduce the inflammation and injury of the liver and kidneys (Koçer et al. 2009; Motaghinejad et al. 2016c). In consistency with oxidative stress biomarkers, our data showed that TPM (50 and 100 mg/kg), in the absence of MPH, can inhibit inflammation biomarkers which confirms the results of recent studies, conferring the anti-inflammatory properties of TPM (Motaghinejad and Motevalian 2016; Motaghinejad et al. 2016a). Based on our data, MPH can increase Bax and decrease Bcl-2 expression in hippocampus. A recent study suggests that MPH at doses of 2 and 10 mg/kg in young and adult rats can activate apoptosis, increase Bax and some genes involved in apoptosis, and reduce Bcl-2 in some brain areas (Réus et al. 2014). Our data showed that TPM (50 and 100 mg/kg) can inhibit MPH-induced apoptosis, which confirms the findings of recent studies, confirming the antiapoptotic properties of TPM. TPM at doses of 50 and 100 mg/kg, in the absence of MPH, decreases Bax and increases Bcl-2 expressions, which confirms the previous findings, suggesting that the neuroprotective properties of TPM might be mediated by the inhibition of pro-apoptotic pathways and activation of antiapoptotic proteins (Cheng and Li 2014; Schubert et al. 2005).
As we have noted, TPM can inhibit MPH-induced oxidative stress, inflammation, and apoptosis in hippocampal cells, but to clearly define its mechanism of action, we have tried to evaluate its molecular basis and signaling pathways, thus, evaluating the CREB/BDNF pathway. Our study has shown that MPH-inhibited CREB gene expression and protein production in the hippocampus, while TPM at doses of 50 and 100 mg/kg can activate CREB expression in gene and protein levels in hippocampus. In addition to the evaluation of CREB, it has been shown that MPH decreases BDNF expression in gene and protein levels. On the other hand, TPM (50 and 100 mg/kg) increases BDNF in gene and protein levels and can probably cause neuroprotection by activating neurotrophic factors. TPM, 50 and 100 mg/kg, in the absence of MPH, can also activate CREB and BDNF expression in both gene and protein levels.
The protective role of CREB, as a transcription factor, has been established by many previous studies, and this transcription factor can affect DNA and cause production of BDNF and c-Fos and probably by this mechanism, can initiate its protective effect. CREB/BDNF protects brain cells against neurodegeneration (Almeida et al. 2005; Almeida et al. 2010; Dworkin and Mantamadiotis 2010; Huang et al. 2015; Wang et al. 2014). It has been shown by many previous studies that amphetamine abuse decreases CREB and BDNF expression and through this mechanism, disrupts cell survival and triggers neurodegeneration (Guo et al. 2011; Meredith et al. 2002). Furthermore, the neuroprotective effect of TPM has been established in many previous studies, and based on our results, we believe that the protective properties of TPM could be mediated by activating the CREB/ BDNF pathway (Pandey et al. 2005).
CREB, as a main transcription factor, was activated in phosphorylated form and its phosphorylation takes place by some upstream kinases in signaling pathway including Ak1, CaMK4, MAPK3, and PKA (Mayr and Montminy 2001; Saeed et al. 2009; Silva et al. 1998; Wei et al. 2009). Our data indicates that MPH can decrease Ak1, MAPK3, and PKA while increasing CaMK4 at the gene level. On the other hand, our data showed that TPM, 50 and 100 mg/kg, can increase Ak1, MAPK3, and PKA while decreasing CaMK4 at the gene level. Based on these findings, we can conclude that the inhibition of CREB phosphorylation could be done by the inhibition of either Ak1 or MAPK3, and PKA kinases and TPM can prevent this inhibition. However, we have tried to show the involvement of kinases in CREB phosphorylation, and our aim was to evaluate the role of CREB and its product, BDNF, in TPM neuroprotection against MPH-induced neurodegeneration. TPM, 50 and 100 mg/kg, in the absence of MPH, can also increase Ak1, MAPK3, and PKA while decreasing CaMK4 at the gene level and probably by this mechanism can increase CREB/BDNF at gene and protein levels and activate CREB/BDNF signaling pathway. We can discuss this data by basic concept that TPM can inhibit glutamate receptor and activate GABA receptor in brain cells. By the inhibition of glutamate receptor, TPM can inhibit influx of exorbitance Ca2+ in neural cells, which is responsible for some neurodegenerative effects of methamphetamine type stimulants. In other neurodegenerative disorders, in fact this inhibition of MPH-induced Ca2+ influx, by TPM attenuates CaMK4 formation (Arundine and Tymianski 2003; Mattson 2007). On the other hand, probably TPM by inhibition of Ca2+ influx can decrease oxidative stress, inflammation, and cell death induced by influx (Poulsen et al. 2004). Also TPM (50 and 100 mg/kg) could decrease CaMK4 expression and possibly inhibit the degeneration caused by exorbitance Ca2+ influx (Poulsen et al. 2004). Also, some similar data showed that activation of GABA or inhibition of glutamate by TPM and other similar agents can affect on some intracellular kinases such as Ak1 or MAPK3 and PKA and potentiate their protective role in phosphorylation and formation of some key proteins such as CREB (Lonze and Ginty 2002; Raskin et al. 2004), and by this effects changed the cells and tissues behavior and chemical in response to the TPM and other agents (Lonze and Ginty 2002). In the present study, we have tried to show the involvement of the kinase enzymes in CREB phosphorylation in order to evaluate the role of CREB and its product, BDNF and c-Fos, in TPM neuroprotection against MPH-induced neurodegeneration. In consistency with the current data, the role of BDNF and c-Fos are indicated in many previous studies (Carlezon et al. 2005; Lonze and Ginty 2002).
Based on our data, we can suggest that CREB/BDNF could be a signaling candidate for TPM neuroprotection. For the detection of CREB expression and its localization in hippocampus, the immunohistochemistry (IHC) data, in qualitative and quantitative manner, showed that TPM can be responsible for the localization and expression of CREB in granular cells of DG and also small and large pyramidal cells of CA1; this data showed that TPM can cause an increase in the number of total and phosphorylated CREB positive cells in both mentioned areas of hippocampus. On the other hand, TPM at doses of 50 and 100 mg/kg can increase its localization in granular cells of DG and also small and large pyramidal cells of CA1. This data showed that TPM in the mentioned doses caused an increase in the number of total and phosphorylated CREB positive cells in both mentioned areas of hippocampus in MPH-treated rat. Although TPM can increase CREB expression, its intensity in localization in granular cells of DG is more significant than the small and large pyramidal cells of CA1. TPM at doses of 50 and 100 mg/kg, in the absence of MPH, intensifies CREB (in both forms) localization and increases total and phosphorylated CREB positive cells in granular cells of DG and also in small and large pyramidal cells of CA1, and this intensity is more significant in granular cells of DG. Moreover, our study showed that despite all these changes, MPH can cause cell loss and degeneration and death in the hippocampus in DG and CA1 areas, and this confirms the results of our behavioral studies. It also suggests that the mentioned apoptosis, oxidative stress, and inflammation by mediation of CREB/BDNF pathway could be the cause of these types of damages, while TPM (50 and 100 mg/kg) can inhibit MPH-induced cell death and degeneration of hippocampal cells in DG and CA1 areas. In addition, our histopathological evaluation revealed that, following the consumption of MPH, cell density and the number of cell layers in these two areas of the hippocampus dropped which suggests a decrease in hippocampal performance and can be responsible for the drop in animal performance evaluated by the behavioral assays. However, TPM (50 and 100 mg/kg) can increase cell density and the number of cell layers in the MPH-treated groups and this confirms the findings of previous studies (Motaghinejad and Motevalian 2016; Motaghinejad et al. 2016a; Qian and Noebels 2003). This also confers the results of previous studies, demonstrating that the amphetamine abuse can cause cell damage and death in rat hippocampus (Brown and Yamamoto 2003; Cippitelli et al. 2010). On the other hand, our data showed that there is no significant difference in OFT behavior and oxidative stress biomarkers (MDA level, SOD, and CAT activity) in group under treatment with TPM (50 and 100 mg/kg) alone when compared to the negative control group, while in inflammatory biomarkers (IL-1β and TNF-α) and in expression of CREB and its upstream kinases such as, MAPK3, CaMK4, Akt-1, and PKA and also downstream products such as BDNF and c-Fos, these changes were significantly different compared to control group. Also at protein level in total and phosphorylated CREB and also Bcl-2 level, these differences were significant in TPM (50 and 100 mg/kg) alone in comparison to the control group. Also, about the localization of CREB in DG and CA1 areas, the changes were not significant compared to the negative control group and in the case of the cell density and shapes, a significant difference was observed in TPM (50 and 100 mg/kg) treated alone in comparison to control group. This concept that TPM alone has not affect the OFT behavior, motor activity and oxidative stress balances, shows that TPM has useful properties because it does not have a disruptive effect on behavior and oxidative stress on its own (Armaǧan et al. 2008; Arnone 2005), but as soon as oxidative and behavioral changes occurred by MPH, TPM can inhibit this MPH-induced malicious effect (Motaghinejad et al. 2016a). About the anti-inflammatory effects of TPM, previous data showed that TPM by itself has anti-inflammatory properties, and these effects are explainable by its role in activation of GABAA, which has an anti-inflammatory role itself (Raskin et al. 2004). About the role of TPM alone on CREB and up- and downstream genes, similar previous studies showed the role of TPM on gene expression and cell signaling. Some of these studies claimed that TPM neuroprotective effect, when used alone, was mediated by CREB/BDNF pathway. In the presence of MPH, this neuroprotective role is more visible, and the role of TPM administered alone in activation of Bcl-2 and inhibition of cell death in DG and CA1 can be explained by this neuroprotective role (Demirci et al. 2013; Park et al. 2008).
Conclusion
The present study supports the beneficial effects of TPM against oxidative stress, inflammation, and apoptosis induced by MPH and probably could be applied for therapy of patients abusing MPH and prevent its neurodegenerative consequences. Also, our results showed that this neuroprotection is possibly mediated by the activation of phosphorylated-CREB/BDNF in both DG and CA1 areas of the hippocampus. Based on our results, we suggested that phosphorylation and activation of CREB by TPM might be mediated by Ak1, MAPK3, and PKA genes, and neuroprotection of TPM may have been mediated by c-Fos as well as BDNF, downstream products of the CREB expression, although further studies are required with human subjects.
Change history
25 July 2024
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1007/s12640-024-00714-2
References
Agarwal NB, Agarwal NK, Mediratta PK, Sharma KK (2011) Effect of lamotrigine, oxcarbazepine and topiramate on cognitive functions and oxidative stress in PTZ-kindled mice. Seizure 20(3):257–262
Aguiar AS, Castro AA, Moreira EL, Glaser V, Santos AR, Tasca CI et al (2011) Short bouts of mild-intensity physical exercise improve spatial learning and memory in aging rats: involvement of hippocampal plasticity via AKT, CREB and BDNF signaling. Mech Ageing Dev 132(11):560–567
Aliev G, Smith MA, Obrenovich ME, Jack C, Perry G (2003) Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Alzheimer disease. Neurotox Res 5(7):491–504
Almeida R, Manadas B, Melo C, Gomes J, Mendes C, Graos M et al (2005) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 12(10):1329–1343
Almeida S, Cunha-Oliveira T, Laço M, Oliveira CR, Rego AC (2010) Dysregulation of CREB activation and histone acetylation in 3-nitropropionic acid-treated cortical neurons: prevention by BDNF and NGF. Neurotox Res 17(4):399–405
Amador FC, Santos MS, Oliveira CR (2001) Lipid peroxidation and aluminium effects on the cholinergic system in nerve terminals. Neurotox Res 3(3):223–233
Armaǧan A, Kutluhan S, Yılmaz M, Yılmaz N, Bülbül M, Vural H et al (2008) Topiramate and vitamin E modulate antioxidant enzyme activities, nitric oxide and lipid peroxidation levels in pentylenetetrazol-induced nephrotoxicity in rats. Basic Clin Pharmacol Toxicol 103(2):166–170
Arnone D (2005) Review of the use of topiramate for treatment of psychiatric disorders. Ann General Psychiatry 16(4):1–8
Arundine M, & Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Arundine M et al Cell Calcium 34(5): 325–337.
Blendy JA (2006) The role of CREB in depression and antidepressant treatment. Biol Psychiatry 59(12):1144–1150
Bongiovanni B, De Lorenzi P, Ferri A, Konjuh C, Rassetto M, De Duffard AE et al (2007) Melatonin decreases the oxidative stress produced by 2, 4-dichlorophenoxyacetic acid in rat cerebellar granule cells. Neurotox Res 11(2):93–99
Brown JM, Yamamoto BK (2003) Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther 99(1):45–53
Cagetti E, Baicy KJ, Olsen RW (2004) Topiramate attenuates withdrawal signs after chronic intermittent ethanol in rats. Neuroreport 15(1):207–210
Carlezon WA, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28(8):436–445
Challman TD, Lipsky JJ (2000) Methylphenidate: its pharmacology and uses. Mayo Clin Proc 75:711–721
Cheng X, Li M (2014) Effect of topiramate on apoptosis-related protein expression of hippocampus in model rats with Alzheimers disease. Eur Rev Med Pharmacol Sci 18(16):761–768
Cippitelli A, Damadzic R, Frankola K, Goldstein A, Thorsell A, Singley E et al (2010) Alcohol-induced neurodegeneration, suppression of transforming growth factor-β, and cognitive impairment in rats: prevention by group II metabotropic glutamate receptor activation. Biol Psychiatry 67(9):823–830
Davids E, Zhang K, Tarazi FI, Baldessarini RJ (2002) Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacology 160(1):92–98
Demirci S, Kutluhan S, Nazıroğlu M, Uğuz AC, Yürekli VA, Demirci K (2013) Effects of selenium and topiramate on cytosolic Ca2+ influx and oxidative stress in neuronal PC12 cells. Neurochem Res 38(1):90–97
Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP et al (2011) Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med 3:96ra76–96ra76
Dworkin S, Mantamadiotis T (2010) Targeting CREB signalling in neurogenesis. Expert Opin Ther Targets 14(8):869–879
Erickson MA, Banks WA (2011) Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: multiplex quantification with path analysis. Brain Behav Immun 25(8):1637–1648
Fortunato JJ, Agostinho FR, RÉus GZ, Petronilho FC, Dal-Pizzol F, Quevedo J (2006) Lipid peroxidative damage on malathion exposure in rats. Neurotox Res 9(1):23–28
François J, Koning E, Ferrandon A, Nehlig A (2006) The combination of topiramate and diazepam is partially neuroprotective in the hippocampus but not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy. Epilepsy Res (3):147–163
Gao H-L, Xu H, Xin N, Zheng W, Chi Z-H, Wang Z-Y (2011) Disruption of the CaMKII/CREB signaling is associated with zinc deficiency-induced learning and memory impairments. Neurotox Res 19(4):584–591
Garnett WR (2000) Clinical pharmacology of topiramate: a review. Epilepsia 41(5):61–65
Gibbs JE, Walker MC, Cock HR (2006) Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 47(3):469–478
Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF et al (2011) Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One 6(5):e19351–e19351
Huang W, Cao J, Liu X, Meng F, Li M, Chen B et al (2015) AMPK plays a dual role in regulation of CREB/BDNF pathway in mouse primary hippocampal cells. J Mol Neurosci 56(4):782–788
Huss M, Lehmkuhl U (2001) Methylphenidate and substance abuse: a review of pharmacology, animal, and clinical studies. J Atten Disord 6:S65–S71
Johansson LH, Borg LH (1988) A spectrophotometric method for determination of catalase activity in small tissue samples. Anal Biochem 174(1):331–336
Johnson BA, Ait-Daoud N, Wang X-Q, Penberthy JK, Javors MA, Seneviratne C et al (2013) Topiramate for the treatment of cocaine addiction: a randomized clinical trial. JAMA psychiatry 70(12):1338–1346
Juárez J, Guerrero-Álvarez Á (2015) Effects of methylphenidate and atomoxetine on impulsivity and motor activity in preadolescent rats prenatally-treated with alcohol. Behav Neurosci 129(6):756
Khan S, Liberzon I (2004) Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology 172(2):225–229
Kitagawa K (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J 274(13):3210–3217
Klein-Schwartz W (2002) Abuse and toxicity of methylphenidate. Curr Opin Pediatr 14(2):219–223
Koçer A, Memişoğullari R, Domaç FM, Ilhan A, Koçer E, Okuyucu Ş et al (2009) IL-6 levels in migraine patients receiving topiramate. Pain Practice 9(3):375–379
Kuczenski R, Segal DS (2001) Locomotor effects of acute and repeated threshold doses of amphetamine and methylphenidate: relative roles of dopamine and norepinephrine. J Pharmacol Exp Ther 296(3):876–883
Kutluhan S, Nazıroğlu M, Çelik Ö, Yılmaz M (2009) Effects of selenium and topiramate on lipid peroxidation and antioxidant vitamin levels in blood of pentylentetrazol-induced epileptic rats. Biol Trace Elem Res 129(1–3):181–189
Lau JW, Senok S, Stadlin A (2000) Methamphetamine-induced oxidative stress in cultured mouse astrocytes. Ann N Y Acad Sci 914:146–156
Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K (2005) Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci 25(5):1137–1148
Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35(4):605–623
Madjd Z, Akbari ME, Zarnani AH, Khayamzadeh M, Kalantari E, Mojtabavi N (2013) Expression of EMSY, a novel BRCA2-link protein, is associated with lymph node metastasis and increased tumor size in breast carcinomas. Asian Pac J Cancer Prev 15(4):1783–1789
Mao X-Y, Cao Y-G, Ji Z, Zhou H-H, Liu Z-Q, Sun H-L (2015) Topiramate protects against glutamate excitotoxicity via activating BDNF/TrkB-dependent ERK pathway in rodent hippocampal neurons. Prog Neuro-Psychopharmacol Biol Psychiatry 60:11–17
Marquette C, Linard C, Galonnier M, Van Uye A, Mathieu J, Gourmelon P et al (2003) IL-1 β, TNF α and IL-6 induction in the rat brain after partial-body irradiation: role of vagal afferents. Int J Radiat Biol 79(10):777–785
Martins MR, Reinke A, Petronilho FC, Gomes KM, Dal-Pizzol F, Quevedo J (2006) Methylphenidate treatment induces oxidative stress in young rat brain. Brain Res 1078(1):189–197
Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6(1):337–350
Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature reviews Nat Rev Mol Cell Biol 2(8):599–609
Meredith GE, Callen S, Scheuer DA (2002) Brain-derived neurotrophic factor expression is increased in the rat amygdala, piriform cortex and hypothalamus following repeated amphetamine administration. Brain Res 949(1–2):218–227
Molina-Hernández M, Téllez-Alcántara NP, Olivera-Lopez JI, Jaramillo MT (2010) Antidepressant-like or anxiolytic-like actions of topiramate alone or co-administered with intra-lateral septal infusions of neuropeptide Y in male Wistar rats. Peptides 31(6):1184–1189
Motaghinejad M, Karimian M, Motaghinejad O, Shabab B, Yazdani I, Fatima S (2015a) Protective effects of various dosage of Curcumin against morphine induced apoptosis and oxidative stress in rat isolated hippocampus. Pharmacol Rep 67(2):230–235
Motaghinejad M, Karimian SM, Motaghinejad O, Shabab B, Asadighaleni M, Fatima S (2015b) The effect of various morphine weaning regimens on the sequelae of opioid tolerance involving physical dependency, anxiety and hippocampus cell neurodegeneration in rats. Fundam Clin Pharmacol 29(3):299–309
Motaghinejad M, Motevalian M (2016) Involvement of AMPA/kainate and GABA A receptors in topiramate neuroprotective effects against methylphenidate abuse sequels involving oxidative stress and inflammation in rat isolated hippocampus. Eur J Pharmacol 784:181–191
Motaghinejad M, Motevalian M, Ebrahimzadeh A (2015c) Reduction of methylphenidate induced anxiety, depression and cognition impairment by various doses of venlafaxine in rat. Int J Prev Med 784:181–191
Motaghinejad M, Motevalian M, Larijani SF, Khajehamedi Z (2015d) Protective effects of forced exercise against methylphenidate-induced anxiety, depression and cognition impairment in rat. Adv Biomed Res 27(4):134. doi:10.4103/2277-9175
Motaghinejad M, Motevalian M, Shabab B (2016c) Effects of chronic treatment with methylphenidate on oxidative stress and inflammation in hippocampus of adult rats. Neurosci Lett 21(619):106–113
Motaghinejad M, Motevalian M, Shabab B (2016a) Neuroprotective effects of various doses of topiramate against methylphenidate induced oxidative stress and inflammation in rat isolated hippocampus. Clin Exp Pharmacol Physiol 43(3):360–371
Motaghinejad M, Motevalian M, Shabab B (2016b) Possible involvements of glutamate and adrenergic receptors on acute toxicity of methylphenidate in isolated hippocampus and cerebral cortex of adult rats. Fundam Clin Pharmacol 14. doi:10.1111/fcp.12250
Motaghinejad M, Motevalian M, Shabab B, & Fatima S (2016e) Effects of acute doses of methylphenidate on inflammation and oxidative stress in isolated hippocampus and cerebral cortex of adult rats. J Neural Transm (Vienna) Sep 28.
Motaghinejad M, Seyedjavadein Z, Motevalian M, Asadi M (2016d) The neuroprotective effect of lithium against high dose methylphenidate: possible role of BDNF. Neurotoxicology 56:40–54
Mula M, Pini S, Cassano GB (2007) The role of anticonvulsant drugs in anxiety disorders: a critical review of the evidence. J Clin Psychopharmacol 27(3):263–272
Nazıroğlu M, Kutluhan S, Uğuz AC, Çelik Ö, Bal R, Butterworth PJ (2009) Topiramate and vitamin E modulate the electroencephalographic records, brain microsomal and blood antioxidant redox system in pentylentetrazol-induced seizure of rats. J Membr Biol 229(3):131–140
Nazirolu M, Kutluhan S, Yilmaz M (2008) Selenium and topiramate modulates brain microsomal oxidative stress values, Ca2+ − ATPase activity, and EEG records in pentylentetrazol-induced seizures in rats. J Membr Biol 225(1–3):39–49
Noh M-R, Kim SK, Sun W, Park SK, Choi HC, Lim JH et al (2006) Neuroprotective effect of topiramate on hypoxic ischemic brain injury in neonatal rats. Exp Neurol 201(2):470–478
Oncken C, Arias AJ, Feinn R, Litt M, Covault J, Sofuoglu M et al (2014) Topiramate for smoking cessation: a randomized, placebo-controlled pilot study. Nicotine Tob Res 16(3):288–296
Pandey SC, Chartoff EH, Carlezon WA, Zou J, Zhang H, Kreibich AS et al (2005) CREB gene transcription factors: role in molecular mechanisms of alcohol and drug addiction. Alcohol Clin Exp Res 29(2):176–184
Park HJ, Kim HJ, Park HJ, Ra J, Zheng LT, Yim SV et al (2008) Protective effect of topiramate on kainic acid–induced cell death in mice hippocampus. Epilepsia 49(1):163–167
Patrick KS, Markowitz JS (1997) Pharmacology of methylphenidate, amphetamine enantiomers and pemoline in attention-deficit hyperactivity disorder. Hum Psychopharmacol 12(2):527–546
Peng J, Sarkar S, Chang SL (2012) Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend 124(3):223–228
Pomierny-Chamioło L, Moniczewski A, Wydra K, Suder A, Filip M (2013) Oxidative stress biomarkers in some rat brain structures and peripheral organs underwent cocaine. Neurotox Res 23(1):92–102
Poulsen CF, Simeone TA, Maar TE, Smith-Swintosky V, White HS, Schousboe A (2004) Modulation by topiramate of AMPA and kainate mediated calcium influx in cultured cerebral cortical, hippocampal and cerebellar neurons. Neurochem Res 29(1):275–282
Qian J, Noebels JL (2003) Topiramate alters excitatory synaptic transmission in mouse hippocampus. Epilepsy Res 55(3):225–233
Raskin P, Donofrio P, Rosenthal N, Hewitt D, Jordan D, Xiang J et al (2004) Topiramate vs placebo in painful diabetic neuropathy analgesic and metabolic effects. Neurology 63(5):865–873
Réus GZ, Scaini G, Furlanetto CB, Morais MO, Jeremias IC, Mello-Santos LM et al (2013) Methylphenidate treatment leads to abnormalities on Krebs cycle enzymes in the brain of young and adult rats. Neurotox Res 24(2):251–257
Réus GZ, Scaini G, Jeremias GC, Furlanetto CB, Morais MO, Mello-Santos LM et al (2014) Brain apoptosis signaling pathways are regulated by methylphenidate treatment in young and adult rats. Brain Res 1583:269–276
Réus GZ, Stringari RB, Ribeiro KF, Ferraro AK, Vitto MF, Cesconetto P et al (2011) Ketamine plus imipramine treatment induces antidepressant-like behavior and increases CREB and BDNF protein levels and PKA and PKC phosphorylation in rat brain. Behav Brain Res 221(1):166–171
Saeed U, Karunakaran S, Meka DP, Koumar RC, Ramakrishnan S, Joshi SD et al (2009) Redox activated MAP kinase death signaling cascade initiated by ASK1 is not activated in female mice following MPTP: novel mechanism of neuroprotection. Neurotox Res 16(2):116–126
Schubert S, Brandl U, Brodhun M, Ulrich C, Spaltmann J, Fiedler N et al (2005) Neuroprotective effects of topiramate after hypoxia–ischemia in newborn piglets. Brain Res 1058(1–2):129–136
Schwarting RK, Sedelis M, Hofele K, Auburger GW, Huston JP (1999) Strain-dependent recovery of open-field behavior and striatal dopamine deficiency in the mouse MPTP model of Parkinson’s disease. Neurotox Res 1(1):41–56
Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE (2000) An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 41:3–9
Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21:127–148
Siniscalchi A, Bonci A, Biagio Mercuri N, Pirritano D, Squillace A, De Sarro G et al (2015) The role of topiramate in the management of cocaine addiction: a possible therapeutic option. Curr Neuropharmacol 13(6):815–818
Tagaya H (2010) Methylphenidate: pharmacology, indication and potential of abuse. Nihon Rinsho 68(8):1550–1555
Terranova C, Rizzo V, Rajan T, Naro A, Ahmad A, Ghilardi M et al (2016) 58. Repetitive transcranial magnetic stimulation induces neuroprotection via activating CaMKIIα-CREB-Bcl-2 pathway in rat brain. Clin Neurophysiol 127:e146
Vendruscolo LF, Izídio GS, Takahashi RN, Ramos A (2008) Chronic methylphenidate treatment during adolescence increases anxiety-related behaviors and ethanol drinking in adult spontaneously hypertensive rats. Behav Pharmacol 19(1):21
Wang B, Zhao J, Yu M, Meng X, Cui X, Zhao Y et al (2014) Disturbance of intracellular calcium homeostasis and CaMKII/CREB signaling is associated with learning and memory impairments induced by chronic aluminum exposure. Neurotox Res 26(3):52–63
Wei Z, Chigurupati S, Bagsiyao P, Henriquez A, Chan SL (2009) The brain uncoupling protein UCP4 attenuates mitochondrial toxin-induced cell death: role of extracellular signal-regulated kinases in bioenergetics adaptation and cell survival. Neurotox Res 16(1):14–29
Wells PG, Bhatia S, Drake DM, Miller-Pinsler L (2016) Fetal oxidative stress mechanisms of neurodevelopmental deficits and exacerbation by ethanol and methamphetamine. Birth Defects Res C Embryo Today: Reviews 10(2):108–130
Woo NH, Teng HK, Siao C-J, Chiaruttini C, Pang PT, Milner TA, et al. (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression.Nat Neurosci 8(8): 1069–1077.
Yamamoto BK, Raudensky J (2008) The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J NeuroImmune Pharmacol 3(4):203–217
Zuo D, Lin L, Liu Y, Wang C, Xu J, Sun F et al (2016) Baicalin attenuates ketamine-induced neurotoxicity in the developing rats: involvement of PI3K/Akt and CREB/BDNF/Bcl-2 pathways. Neurotox Res 30(2):1–14
Acknowledgements
This research was financially supported by the Research Deputy of Iran University of Medical Sciences (code 26217).
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Motaghinejad, M., Motevalian, M., Abdollahi, M. et al. RETRACTED ARTICLE: Topiramate Confers Neuroprotection Against Methylphenidate-Induced Neurodegeneration in Dentate Gyrus and CA1 Regions of Hippocampus via CREB/BDNF Pathway in Rats. Neurotox Res 31, 373–399 (2017). https://doi.org/10.1007/s12640-016-9695-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-016-9695-4