
Abstract
The hippocampus is vulnerable to damage under conditions of ischemia and hypoxia, causing multiple mental illnesses. cAMP response element-binding protein (CREB) plays a pivotal role in preventing the apoptosis of neurons and many other cells. Here, we found that AMP-activated protein kinase (AMPK) and CREB are oppositely regulated in mouse primary hippocampal neurons impaired by hypoxia-hypoglycemia. AMPK overexpression reduced the CREB level by upregulating SIRT1 and was negatively posttranscriptionally regulated by miR-134, suggesting a negative regulatory role of AMPK in the expression of CREB. Interestingly, the downstream genes of CREB, brain-derived neurotrophic factor (BDNF), and Bcl-2 remained unchanged when CREB was downregulated by AMPK expression. In addition, in AMPK−/− primary hippocampal neurons, comparisons between the effect of upregulation and silencing of miR-134 on the expression of CREB, BDNF, and Bcl-2 were made. The results reveal that AMPK is crucial for the activation of CREB via phosphorylation. Therefore, AMPK plays a dual role in the regulation of CREB in mouse primary hippocampal cells: a negative effect on total CREB expression by elevating SIRT1/miR-134 and a positive effect on activity via phosphorylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The hippocampus is a major component of the brains of higher animals and plays important roles in recognition, memory, and spatial memory. Owing to its active metabolism, high oxygen consumption, and large blood flow volume, the hippocampus is vulnerable to damage under conditions of ischemia and hypoxia, causing mental illness like Alzheimer’s disease, epilepsy, etc. Our understanding of the cell and molecular mechanisms of hippocampal neuron damage in the hypoxic-ischemic state remains incomplete.
The cAMP response element-binding protein (CREB) is an important cellular transcription factor which binds to certain DNA sequences called cAMP response elements. Evidence is emerging that CREB plays a pivotal role in preventing apoptosis in neurons and other cells (Aggarwal et al. 2008; Kim et al. 2009; Özgen et al. 2009). CREB is regarded as a key intracellular target of biological or physicochemical factors in studies on neuron apoptosis and repair (Chun et al. 2009; Lebesgue et al. 2009; Li and Liu 2010). Currently, several CREB-mediated signaling pathways have been reported to be activated or dysregulated in impaired neurons: aluminum chloride impaired mouse long-term memory through downregulating the cAMP-PKA-CREB signaling pathway (Zhang et al. 2014); serotonin transporter interactions with the CREB/brain-derived neurotrophic factor (BDNF) signaling pathway changes the risk of developing a major depressive disorder (Ignácio et al. 2014); benzyl butyl phthalate exposure attenuates neurotransmission through the cAMP-PKA-CREB signaling pathway and impairs learning and memory (Min et al. 2014); estradiol treatment stimulates the ERK/MAPK signaling pathway, which in turn maintains CREB activity in the ischemic CA1, rescuing hippocampal neurons from ischemia-induced cell death (Lebesgue et al. 2009). In these signaling pathways, cell proliferation and apoptosis were modulated by the CREB upregulation or downregulation, or CREB phosphorylation promotion or suppression. Recently, a novel pathway regulating memory and plasticity was confirmed. In this pathway, activation of SIRT1 enhanced, whereas its loss-of-function impaired, synaptic plasticity mediated via negative posttranscriptional regulation of CREB expression by brain-specific miR-134 (Gao et al. 2010). This SIRT1/miR-134/CREB pathway was the source of inspiration for our study.
AMP-activated protein kinase (AMPK) is a typical metabolic fuel gauge in eukaryotes which senses changes in the intracellular AMP/ATP ratio (Hardie 2007). In general, activation of AMPK acts to maintain cellular energy stores and enhances oxidative metabolism and mitochondrial biogenesis in adipose tissue, muscle, or liver (Fryer et al. 2002; Zhou et al. 2001; Zong et al. 2002). Evidence also indicates that AMPK can activate the NAD1-dependent type III deacetylase SIRT1 to control the expression of genes involved in energy metabolism in mouse skeletal muscle and adipocytes (Cantó et al. 2009; Chau et al. 2010). Additionally, AMPK is important for phosphorylation-mediated activation of a few genes including CREB, but in non-nervous tissues or cells (Mair et al. 2011; Salminen and Kaarniranta 2012).
In this study, we found that AMPK was upregulated, whereas CREB was downregulated, in mouse primary hippocampal neurons in a hypoxic-hypoglycemic (HO-HG) state. AMPK overexpression reduced the CREB level via upregulating SIRT1 and negative posttranscriptional regulation by miR-134, suggesting a negative regulatory role of AMPK in the expression of CREB. However, it was interesting that the downstream genes, BDNF and Bcl-2, remained unchanged. In AMPK−/− hippocampal neurons, a comparison between the effect of upregulation and silencing of miR-134 on the expression of CREB, BDNF, and Bcl-2 revealed that AMPK is crucial for the phosphorylation and activation of CREB. Therefore, AMPK plays a dual role in the regulation of CREB in mouse primary hippocampal cells: negative in terms of total CREB expression through elevating SIRT1/miR-134, while positively influencing CREB activity via phosphorylation.
Materials and Methods
Ethics Statement
All animal procedures described herein were approved by the Shaanxi Laboratory Animal Surveillance and Detection Center. Due to the nature of the experimental design, the animals were subjected to a restraint stress procedure. At other times, the mice were monitored in their home cage in a stress-free environment where they were given food and water ad libitum in a humidity- and temperature-controlled room under a 12-h light-dark cycle. After experimentation, to minimize pain without drugs, the mice were rapidly euthanized by cervical dislocation and decapitation by an experienced animal handler.
Hippocampal Neuron Culture
Six C57BL/6J wild-type suckling mice and six systemic AMPKα2−/− suckling mice (purchased from the Institute of Zoology, Chinese Academy of Medical Sciences) born within a 24-h period were sacrificed by cervical dislocation, and their hippocampi were collected in a sterile cabinet. The hippocampi without meninges were rinsed twice and dissected in anatomy liquid at 2 °C. Then, 0.1 % collagenase I (Invitrogen) and 0.1 % trypsin (Invitrogen) were added to digest the hippocampi at 37 °C for 15 min in 15-mL Falcon tubes. The hippocampi were washed twice with DF12 containing 10 % FBS to stop the digestion. Next, 2 mL of DF12 containing 10 % FBS was added, and the digest was triturated using a 0.1-1000-μL Finnpipette. After 2 min, the upper layer was transferred to a new tube. Another 1 mL of DF12 was added, and the sublayer was triturated again to get more cells. The cells were transferred into the same tube. After the cell number was counted, the cells were seeded on poly-D-lysine-treated 6-well plates at the density of 105/mL. The cells were cultured in DF12 with 10 % FBS at 37 °C, with 5 % carbon dioxide.
Constructs and Transfection
The full length cds sequence of SIRT1 mRNA was amplified by PCR using primers containing Kpn I and Xho I sites. SIRT1 F: 5′-CGG GGT ACC TAT GCT ATG AAC AAT GGA AG-3′, R: 5′-CCG CTC GAG TTG CCT GTT GAG GAT TTG GT-3′. The product was cleaved and ligated onto corresponding sites of the pcDNA3.1 plasmid, confirmed by sequencing.
pcDNA-AMPK was kindly gifted by Prof. Antero Salminena from the Department of Neurology, Institute of Clinical Medicine, University of Eastern Finland.
To perform the transfection, 2 μg of pcDNA-SIRT1, 2 μg of pcDNA-AMPK, or 6 pmol of miR-134 inhibitor (in a 6-well plate) was, respectively, transfected or cotransfected into the cells with Lipofectamine3000 (Invitrogen) according to the manufacturer’s instructions. The miR-134 inhibitor was a specific single-strand oligonucleotide inhibitor for miR-134, which was chemically modified, and was of high structural stability.
Hypoxia-Hypoglycemia Treatment
After culture for 24 h, the medium was replaced with low glucose DF12 (1 mg/mL) supplemented with 300 μM CoCl2 (to simulate a hypoxic environment in vitro). Hippocampal neurons cultured in DF12 with 4.5 mg/mL glucose and 300 μM DMSO were regarded as the control.
Cell Viability Assay
The cell viability assay was performed using a Cell Titer-Blue H Cell Viability Assay Kit (Promega, Madison, Wisconsin) according to the manufacturer’s instructions.
Real-Time Quantitative PCR
Extracted RNA was treated with DNaseI (Fermentas) to exclude the possibility of DNA contamination. The RNA concentration was quantified using a spectrophotometer measuring the OD260/280 ratio (1.80–1.95). The integrity of RNA was checked by electrophoresis on a 1.0 % agarose gel with ethidium bromide staining. Real-time qPCR reactions were carried out in a final volume of 25 μL, using SYBR Premix Ex Taq (TaKaRa), with 0.4 mM of each primer and 200 ng of the cDNA template. Primers applied in the reactions are as follows: AMPK (5′-GCC GAG AAG CAG AAA C-3′, 5′-TGT CAC CCA GAA TGT AGT G-3′); SIRT1 (5′-ATT CTG CTAT TAC AAG TT-3′, R: 5′-AAG TAC ATG TCT CCC AT-3′); CREB (5′-TCA GCC GGG TAC TAC CAT TC-3′, 5′-TTC AGC AGG CTG TGT AGG AA-3′); BDNF (5′-ATG GGA CTC TGG AGA GCC TGA A-3′, 5′-CGC CAG CCA ATT CTC TTT TTG C-3′); Bcl-2 (5′-ATT GTG GCC TTC TTT GAG TTC G-3′, 5′-CAT CCC AGC CTC CGT TAT CC-3′); β-actin (5′-TTG TTG CCA TCA ACG ACC CC-3′, 5′-ATG AGC CCT TCC ACA ATG CC-3′). The miR-134 stem-loop primer and quantitative primers were designed and produced by Invitrogen. Each individual sample was run in triplicate wells. PCR amplification cycles were performed using the iQ™5 Multicolor Real-Time PCR Detection System (Bio-Rad) and SYBR Premix Ex Taq II kit (Invitrogen). The reactions were initially denatured at 95 °C for 3 min followed by 40 cycles of 95 °C for 10 s, 55 °C for 30 s, and 72 °C for 20 s. The change in transcript abundance of all tested genes was calculated using the 2−ΔΔCt method. All mRNA levels were normalized to GAPDH.
Western Blotting
Cells were lysed in lysis buffer (Beyotime) supplemented with 1 mM PMSF. The protein concentration was determined using the BCA protein assay (Tiangen). Twenty micrograms of protein in each sample were separated by 12 % SDS-PAGE and electro-transferred to PVDF membranes (Millipore) for immunoblotting analysis. The following primary antibodies were used: anti-AMPK α2 (1:300, ab3760, Abcam), anti-CREB (1:300, ab32515, Abcam), anti-CREB phosphor S133 (1:300, ab32096, Abcam), anti-SIRT1 (1:300, ab32441, Abcam), anti-BDNF (1:200, ab6201, Abcam), anti-Bcl-2 (1:400, ab117115, Abcam), and anti-β-actin (1:800, Santa Cruz), which was used as the internal reference. After incubation with the appropriate HRP-conjugate secondary antibody, proteins were detected using a ChemiDoc XRS imaging system and analysis software Quantity One (Bio-Rad).
Statistical Analysis
All data were obtained from at least three independent experiments. Values are expressed as mean ± SEM. Statistics were calculated using SPSS 19.0. Multiple comparisons were assessed by one-way ANOVA followed by Dunnett’s tests. Differences between groups were considered statistically significant if P < 0.05.
Results
AMPK was upregulated, and CREB was downregulated in hypoxia-hypoglycemia in primary hippocampal neurons.
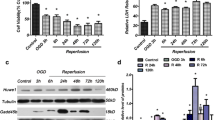
Primary hippocampal neurons obtained from C57BL/6J wide-type suckling mice were seeded onto poly-D-lysine-treated 6-well plates at the density of 105/mL and cultured in DF12 with 10 % FBS at 37 °C, with 5 % carbon dioxide. After culture for 24 h, the medium was replaced with low glucose DF12 (1 mg/mL) supplemented with 300 μM CoCl2 to simulate a hypoxic environment in vitro. Hippocampal neurons cultured in DF12 with 4.5 mg/mL glucose and 300 μM DMSO were regarded as the control. After another 4 h, the viability of the cells and the expression of several genes were assessed. The hippocampal neurons under hypoxia-hypoglycemia (HO-HG) displayed significant lower viability compared to the control (Fig. 1a). Real-time qPCR and Western blotting analyses showed that the expression of AMPK, SIRT1, and miR-134 were upregulated, while the level of CREB, a target gene of miR-134 that has a protective effect on hippocampal neurons, was dramatically decreased (Fig. 1b, c). Expression of BDNF and the anti-apoptosis gene Bcl-2 was resultantly decreased, suggesting that the cells were induced to apoptosis under HO-HG condition (Fig. 1b, c).

AMPK was upregulated, and CREB was downregulated in mouse primary hippocampal neurons impaired by hypoxia-hypoglycemia (HO-HG). a Cell viability of mouse primary hippocampal neurons after culture in HO-HG conditions for 4 h. Gene expression on the mRNA level (b) and protein level (c) was detected after culture in HO-HG conditions for 4 h. Asterisk indicates significantly different (P < 0.05)
AMPK overexpression reduced total CREB levels but did not alter the levels of BDNF and Bcl-2. Next, pcDNA-AMPK was transfected into primary hippocampal neurons under normal conditions to explore its effect on the expression of CREB. After 48 h, in response to AMPK overexpression, SIRT1 and miR-134 levels were upregulated, and as a result, total CREB was reduced (Fig. 2a, b). Surprisingly, the levels of Bcl-2 and the CREB downstream gene BDNF were not changed (Fig. 2a, b). As a result, cell viability was not changed by AMPK overexpression (Fig. 2c). Phosphorylated CREB (pCREB) elevation might be a legitimate interpretation (Fig. 2b). These results suggested that AMPK negatively regulated CREB expression through SIRT1/miR-134, but it had a positive effect on phosphorylation of CREB.

AMPK overexpression reduced total CREB but did not alter cell viability in mouse primary hippocampal neurons. After pcDNA-AMPK was transfected into primary hippocampal neurons derived from WT mice in a normal state for 48 h, then expression of AMPK, SIRT1, miR-134, CREB, BDNF, and Bcl-2 and cell viability were detected. a Detection of gene expression on the mRNA level by real-time qPCR. b Detection of gene expression on the protein level by Western blotting. c Detection of cell viability using the Cell Titer-Blue H Cell Viability Assay Kit. Asterisk indicates significantly different (P < 0.05)
SIRT1 Elevation Reduced CREB and BDNF in the Absence of AMPK
To investigate the impact of the absence of AMPK, primary hippocampal neurons were isolated from systemic AMPKα2−/− mice. Then, pcDNA-SIRT1 was transfected into AMPK−/− primary hippocampal neurons. As expected, the total CREB level was reduced via miR-134-mediated posttranscriptional regulation (Fig. 3a, b). But, unlike in the WT neurons (Fig. 2a, b), in the absence of AMPK, CREB abatement caused a reduction in BDNF and Bcl-2 expression (Fig. 3b) and cell viability (Fig. 4b). Additionally, pCREB levels were very low in both pcDNA-SIRT1 transfected and control cells (Fig. 3b). AMPK is crucial for the phosphorylation of CREB and hippocampal neuron growth.

SIRT1 overexpression reduced CREB and BDNF in AMPK−/− primary hippocampal neurons. pcDNA-SIRT1 was transfected into the AMPK−/− primary hippocampal neurons isolated from systemic AMPKα2−/− mice. After transfection for 48 h, expression of genes was detected. a Levels of SIRT1, miR-134, CREB, BDNF, and Bcl-2 were detected by real-time qPCR. b Protein levels of SIRT1, CREB, BDNF, Bcl-2, and pCREB were detected by Western blotting. Asterisk indicates significantly different (P < 0.05)

Silence of miR-134 induced expression of CREB but had no effect on BDNF in AMPK−/− neurons. A single-strand oligonucleotide miR-134 inhibitor was transfected into AMPK−/− primary hippocampal neurons. a After transfection for 48 h, protein levels of CREB, BDNF, Bcl-2, and pCREB were detected by Western blotting. b Cell viability after transfected with pcDNAs or anti-miR-134 for 48 h under normal condition. WT primary hippocampal neurons and AMPK−/− primary hippocampal neurons were transfected with pcDNA-AMPK, or pcDNA-SIRT1, or anti-miR-134. After incubated under normal condition (5 % CO2, 37 °C) for 48 h, cell viability of each group was detected with the Cell Titer-Blue H Cell Viability Assay Kit. c Cell viability after transfected with pcDNAs or anti-miR-134 for 48 h under HO-HG condition. WT primary hippocampal neurons and AMPK−/− primary hippocampal neurons were transfected with pcDNA-AMPK, or pcDNA-SIRT1, or anti-miR-134. After incubated under normal condition (5 % CO2, 37 °C) for 48 h, cell viability of each group was detected with the Cell Titer-Blue H Cell Viability Assay Kit. Different letters suggest that there are statistically significant differences between the groups (P < 0.05). d AMPK plays a dual role in the regulation of CREB in mouse primary hippocampal cells. On one hand, AMPK negatively regulates total CREB expression through elevating SIRT1/miR-134; on the other hand, it promotes the phosphorylation of CREB
Silencing miR-134 induced the expression of CREB but had no effect on BDNF in AMPK−/− neurons under normal condition and HO-HG condition.
Finally, an miR-134 inhibitor was applied to treat AMPK−/− primary hippocampal neurons. Under normal condition, the total CREB level was significantly increased by the miR-134 inhibitor (Fig. 4a), while the expression of BDNF was not changed (Fig. 4a). Cell viability was also unchanged after transfection with the miR-134 inhibitor (Fig. 4b). Interestingly, AMPK overexpression caused a distinct increase in the viability of AMPK−/− neurons, although we knew that the total CREB level was decreased (Fig. 4b). Under HO-HG condition, the changes of cell viability were similar to those of normal condition, whereas the overall cell viability was lower than that of normal condition (Fig. 4c).
These all data revealed a dual role of AMPK in the regulation of CREB: negative in terms of total levels through elevating SIRT1/miR-134, while positive in terms of activity via phosphorylation (Fig. 4d).
Discussion
To simulate an environment of ischemia and hypoxia in vitro, we treated primary hippocampal neurons with low glucose (1 mg/mL) and 300 μM CoCl2. The neurons were seriously impaired after treatment, with a marked decrease in the levels of BDNF and Bcl-2 as well as decreased cell viability. In the impaired neurons, we found a reverse change in the expression of total CREB and AMPK. Then, combined with the AMPK overexpression study, we initially identified that AMPK decreased the total CREB level through upregulating SIRT1 and CREB-targeting miR-134. In addition, in one of our previous studies, we had proven that miR-134 caused ischemia/reperfusion injury-induced neuronal cell death by directly targeting and negatively regulating CREB/BDNF pathway (Huang et al. 2014). It has been reported that AMPK is a gene upstream of SIRT1 in multiple metabolic processes in non-nervous tissues or cells (Cantó et al. 2009; Chau et al. 2010). Our results here reveal that AMPK also positively regulates SIRT1 in mouse nervous system cells and has some relationship with hippocampal neuronal damage.
AMPK is a well-studied phosphorylated kinase that controls aging through multiple metabolic processes, such as autophagy, oxidation stress, apoptosis resistance, and inflammation (Salminen and Kaarniranta 2012). The most recent research has shown that activating AMPK slows aging in the intestine, mediated by CREB-regulated transcriptional coactivator 1 (CRTC-1), and CREB (Mair et al. 2011). In fact, CREB is also a substrate of AMPK, and activation of CREB depends on phosphorylation by AMPK. Here, in mouse primary hippocampal neurons, we found that phosphorylation of CREB was severely affected in the absence of AMPK, resulting in the downregulation of BDNF and reduced cell viability. These results indicate that AMPK is very important for the phosphorylation of CREB in hippocampal neurons.
CREB is necessary for the late stage of long-term potentiation and is also important for the survival of neurons. Whole body knockout of CREB in the mouse causes immediate death after birth, highlighting the vital role of CREB (Arthur and Cohen 2000). In the canonical cAMP-CREB signaling pathway (Al‐Wadei et al. 2006; Beyer and Karolczak 2000; Chen et al. 2012; Vitolo et al. 2002), the cAMP responding element modulates the effects of PKA and then regulates the phosphorylation-activation of CREB. Sometimes, phosphorylation-activation of CREB is mediated by MAPK or AMPK (Al‐Wadei et al. 2006; Delghandi et al. 2005; Mair et al. 2011). Our data indicate that, in hippocampal neurons, AMPK is very important for the phosphorylation of CREB. However, the regulatory effects of AMPK on CREB involve not only phosphorylation, but also SIRT1/miR-134-mediated posttranscriptional suppression. Thus, hippocampal neurons are affected by the balance of posttranscriptional suppression and phosphorylation-activation. The absence of AMPK destroys this balance, and thereby induces dysregulation in the downstream genes of CREB and influences neuronal viability.
Collectively, we found that AMPK plays a dual role in the regulation of CREB in mouse primary hippocampal cells: negative in terms of total CREB expression through elevating SIRT1/miR-134 and positive in terms of activity via phosphorylation.
References
Aggarwal S, Kim S-W, Ryu S-H, Chung W-C, Koo JS (2008) Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein. Cancer Res 68:981–988
Al‐Wadei HA, Takahashi T, Schuller HM (2006) Growth stimulation of human pulmonary adenocarcinoma cells and small airway epithelial cells by β‐carotene via activation of cAMP, PKA, CREB and ERK1/2. Int J Cancer 118:1370–1380
Arthur JSC, Cohen P (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett 482:44–48
Beyer C, Karolczak M (2000) Estrogenic stimulation of neurite growth in midbrain dopaminergic neurons depends on cAMP/protein kinase A signalling. J Neurosci Res 59:107–116
Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC et al (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060
Chau MD, Gao J, Yang Q, Wu Z, Gromada J (2010) Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc Natl Acad Sci 107:12553–12558
Chen Y, Huang X, Y-w Z, Rockenstein E, Bu G, Golde TE et al (2012) Alzheimer’s β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of β-amyloid. J Neurosci 32:11390–11395
Chun H, Hao W, Honghai Z, Ning L, Yasong W, Chen D (2009) CCL3L1 prevents gp120-induced neuron death via the CREB cell signaling pathway. Brain Res 1257:75–88
Delghandi MP, Johannessen M, Moens U (2005) The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal 17:1343–1351
Fryer LG, Parbu-Patel A, Carling D (2002) The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277:25226–25232
Gao J, Wang W-Y, Mao Y-W, Gräff J, Guan J-S, Pan L et al (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466:1105–1109
Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785
Huang W, Liu X, Cao J, Meng F, Li M, Chen B et al (2014) miR-134 regulates ischemia/reperfusion injury-induced neuronal cell death by regulating CREB signaling. J Mol Neurosci 1-9
Ignácio ZM, Réus GZ, Abelaira HM, Quevedo J (2014) Epigenetic and epistatic interactions between serotonin transporter and brain-derived neurotrophic factor genetic polymorphism: insights in depression. Neuroscience
Kim H-W, Chang YC, Chen M, Rapoport SI, Rao JS (2009) Chronic NMDA administration to rats increases brain pro-apoptotic factors while decreasing anti-apoptotic factors and causes cell death. BMC Neurosci 10:123
Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM (2009) Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids 74:555–561
Li N, Liu G-t (2010) The novel squamosamide derivative FLZ enhances BDNF/TrkB/CREB signaling and inhibits neuronal apoptosis in APP/PS1 mice. Acta Pharmacol Sin 31:265–272
Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ et al (2011) Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470:404–408
Min A, Liu F, Yang X, Chen M (2014) Benzyl butyl phthalate exposure impairs learning and memory and attenuates neurotransmission and CREB phosphorylation in mice. Food Chem Toxicol 71:81–89
Özgen N, Guo J, Gertsberg Z, Danilo P, Rosen MR, Steinberg SF (2009) Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol Pharmacol 76:896–902
Salminen A, Kaarniranta K (2012) AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11:230–241
Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci 99:13217–13221
Zhang L, Jin C, Lu X, Yang J, Wu S, Liu Q et al (2014) Aluminium chloride impairs long-term memory and downregulates cAMP-PKA-CREB signalling in rats. Toxicology
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ et al (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci 99:15983–15987
Author information
Authors and Affiliations
Corresponding author
Additional information
Weidong Huang and Jie Cao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Huang, W., Cao, J., Liu, X. et al. AMPK Plays a Dual Role in Regulation of CREB/BDNF Pathway in Mouse Primary Hippocampal Cells. J Mol Neurosci 56, 782–788 (2015). https://doi.org/10.1007/s12031-015-0500-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0500-2