
Abstract
Cardiac ischemia–reperfusion (I/R) injury results mainly from mitochondrial dysfunction and cardiomyocyte death. Mitophagy sustains mitochondrial function and exerts a pro-survival effect on the reperfused heart tissue. Mammalian STE20-like kinase 1 (Mst1) regulates chronic cardiac metabolic damage and autophagic activity, but its role in acute cardiac I/R injury, especially its effect on mitophagy, is unknown. The aim of this study is to explore whether Mst1 is involved in reperfusion-mediated cardiomyocyte death via modulation of FUN14 domain containing 1 (FUNDC1)-related mitophagy. Our data indicated that Mst1 was markedly increased in reperfused hearts. However, genetic ablation of Mst1 in Mst1-knockout (Mst1-KO) mice significantly reduced the expansion of the cardiac infarction area, maintained myocardial function and abolished I/R-mediated cardiomyocyte death. At the molecular level, upregulation of Mst1 promoted ROS production, reduced mitochondrial membrane potential, facilitated the leakage of mitochondrial pro-apoptotic factors into the nucleus, and activated the caspase-9-related apoptotic pathway in reperfused cardiomyocytes. Mechanistically, Mst1 activation repressed FUNDC1 expression and consequently inhibited mitophagy. However, deletion of Mst1 was able to reverse FUNDC1 expression and thus re-activate protective mitophagy, effectively sustaining mitochondrial homeostasis and blocking mitochondrial apoptosis in reperfused cardiomyocytes. Finally, we demonstrated that Mst1 regulated FUNDC1 expression via the MAPK/ERK-CREB pathway. Inhibition of the MAPK/ERK-CREB pathway prevented FUNDC1 activation caused by Mst1 deletion. Altogether, our data confirm that Mst1 deficiency sends a pro-survival signal for the reperfused heart by reversing FUNDC1-related mitophagy and thus reducing cardiomyocyte mitochondrial apoptosis, which identifies Mst1 as a novel regulator for cardiac reperfusion injury via modulation of mitochondrial homeostasis.
Similar content being viewed by others


Introduction
Acute myocardial infarction, which is caused by a sudden embolism of the coronary arteries, is a leading cause of mortality [1, 2]. Timely myocardial reperfusion by reopening occluded vessels is the primary clinical therapy for treatment of myocardial infarction. Unfortunately, the reperfusion strategy also causes ischemia–reperfusion (I/R) injury to the heart [3, 4]. Cardiac I/R injury is caused mainly by cardiomyocyte and endothelial apoptosis, which induce malignant arrhythmia, constriction dysfunction, microvascular spasms and capillary obstruction following reinstitution of epicardial blood flow [5, 6]. Mitochondrial dysfunction, such as mitochondrial fission [7], mitochondrial oxidative stress [8], and mitochondrial calcium overload [9], is known to be intimately involved in the processes that contribute to reperfusion injury. Mechanistically, aberrant mitochondria fail to produce enough energy to meet the requirements of the heart tissue [10, 11]. In addition, damaged mitochondria generate excessive ROS that cause cardiomyocyte senescence. More severely damaged mitochondria would liberate pro-apoptotic factors and initiate programmed cell death, promoting the loss of functional cardiomyocytes [12, 13]. Accordingly, recovery of mitochondrial homeostasis is vital to reduce cardiac I/R injury.
Based on several reports, selective mitochondrial autophagy (mitophagy) forms the essential axis of mitochondrial quality control in response to mitochondrial damage [14]. The decisive role of mitophagy in the removal of faulty mitochondria determines its protective effects on mitochondria homeostasis and myocardial apoptosis [12, 15]. As to mitophagy, FUN14 domain containing 1 (FUNDC1), a novel mitophagy receptor, has been reported to be involved in cardioprotection in the context of cardiac disorder [12]. Activated FUNDC1 elevates mitophagy activity in the reperfused heart and prevents mitochondrial apoptosis activation. Moreover, FUNDC1-related mitophagy also attenuates cardiomyocyte oxidative stress, alleviates mitochondrial fission, sustains mitochondrial membrane potential, promotes mitochondrial biosynthesis, maintains ATP production, and sends pro-survival signals to cardiomyocytes in the context of I/R injury [12, 13, 16]. However, based on recent findings, FUNDC1-related mitophagy is unfortunately inactivated at the stage of reperfusion [12, 17], and the upstream molecular signals controlling FUNDC1-mediated mitophagy in I/R injury have not been adequately explored. Although previous studies have suggested that FUNDC1-mediated mitophagy is primarily regulated by the ERK-CREB pathway in the post-infarct heart [18, 19], the role of ERK-CREB axis in cardiac I/R injury has not been fully described.
Mammalian STE20-like kinase 1 (Mst1) is the major effector in the classical Hippo pathway. Early research has reported that cardiac-specific knockout of Mst1 blocks the Hippo pathway, leading to an enlarged heart without alterations in cardiomyocyte size [20]. By comparison, the increased Mst1 expression promotes apoptosis in cardiomyocytes [21]. This conclusion is further supported by a careful study in a diabetic cardiomyopathy model, which demonstrates that Mst1 deletion enhances the interaction between Bcl-2 and Bax, reducing cardiomyocyte mitochondrial apoptosis [22]. Subsequent research further reveals that upregulated Mst1 phosphorylates Bcl-XL at Ser14, promoting Bax translocation from the cytoplasm to the mitochondria. In addition, the Mst1-Hippo pathway also participates in several physiological and pathological processes in the heart ranging from heart development, hypertrophy, and angiogenesis to cardiomyocyte regeneration [23]. These pieces of evidence illustrate a strong correlation between Mst1 activation and mitochondrial damage in cardiomyocytes. However, the contributory action of Mst1 in FUNDC1-related mitophagy in cardiac I/R injury is poorly understood. Notably, accumulating evidence is available to confirm the inhibitory role of Mst1 in autophagic flux [24]. Mst1 activation is associated with an increase in Beclin1 phosphorylation at Thr108, disrupting the Beclin1-PI3 K interaction and consequently reducing autophagy activity [25]. Furthermore, Mst1 has been well recognized as a major transcription factor in inhibiting Parkin expression and mitophagy activity [26]. However, whether Mst1 is involved in cardiomyocyte viability and mitochondrial homeostasis under I/R injury via FUNDC1-related mitophagy is far less clear. In the present study, we found that Mst1 was upregulated by cardiac I/R injury and that activated Mst1 promoted myocardial damage via repressing FUNDC1-related mitophagy. Mechanistically, Mst1 inhibited the ERK-CREB pathway and thus repressed FUNDC1 expression. However, genetic ablation of Mst1 normalized the ERK-CREB pathway and consequently reversed FUNDC1-mediated mitophagy, sustaining mitochondrial homeostasis and cardiac function in the context of cardiac I/R injury.
Methods
Animal experiments
To examine the physiological role of Mst1 in mammals, we purchased Mst1-knockout (Mst1-KO) mice and wild-type (WT) mice on a C57BL/6 background from K&D Gene Technology (WuHan) as in a previous study [26]. In the cardiac I/R model, the above mice were anesthetized and then the hearts were exposed through the left thoracotomy. Subsequently, the left anterior descending coronary artery was tightened around using a 5.0 Prolene suture. The hearts were subjected to 30 min of ischemia. Then the ligature was released and reperfusion was achieved for 2 h according to a previous study [12]. After reperfusion, the hearts were excised and the ventricles were sliced. Then, 1% triphenyltetrazolium chloride (TTC) and 2% Evans blue stain were used to stain the infarcted area at 37 °C for 15 min. Cardiac function was assessed via transthoracic echocardiographic analysis as in a previous study. In addition, the blood was collected and several cardiac damage markers such as CK-MB, troponin T and LDH were analyzed via ELISA.
Cardiomyocyte culture and I/R injury in vitro
In vitro, cardiomyocytes were isolated from WT mice and Mst1-KO mice using the enzyme dissociation method according to a previous study [12]. The primary cardiomyocytes were cultured at 37 °C/5% CO2. The I/R injury was performed in vitro using hypoxia and reoxygenation [27, 28]. Hypoxia was induced under 95% N2 and 5% CO2 for 30 min. Then, the cardiomyocytes were cultured with fresh DMEM with 20% FBS at 37 °C/5% CO2 for 2 h. To inhibit the activity of ERK, we applied PD98059 to cardiomyocytes for 2 h.
RNAi knockdown assay
The small interfering RNA (siRNA) against FUNDC1 was designed and purchased from Shanghai Gene-Pharma Co. (Shanghai, China). The sequences for RNAi were as follows: siRNA-FUNDC1: 5′-GAGTCGCATGACTTAGCTATG-3′; anti-sense: 5′-UUAUCUTAUCGUAGUCTUGUC-3′, siRNA-Ctrl: 5′-UCUAGUUCUGATUUGUCUGCA-3′; anti-sense: 5′-CUGGAUGGTUCUAUGUGCUUA-3′. The siRNA (1 µg/ml) was transfected into cardiomyocytes using Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) in serum-free Opti-MEM (Invitrogen, USA) according to the manufacturer’s instructions [29]. After 48–72 h, the cardiomyocytes were isolated, and the knockdown efficiency was confirmed via western blotting [30].
Electron microscopy (EM)
EM was used to observe the ultrastructure of the myocardium. After I/R injury, hearts were washed with cold PBS 3 times and then fixed in 3% glutaraldehyde at 4 °C for 2 h [31]. Then, the sample was treated with 1% cacodylate-buffer osmium tetroxide at 4 °C for 4 h, followed by dehydration using graded ethanol (50, 70, 90 and 100% for 30 min each). Then, 60-nm sections were cut via a Leica EM UC6rt (Leica Microsystems GmbH, Wetzlar, Germany), and then stained using uranyl acetate and lead citrate. Finally, the samples were observed under a Hitachi H7650 transmission electron microscope (TEM; Hitachi, Ltd., Tokyo, Japan). At least three randomly selected areas were obtained and the EM assay was repeated 3 times [32].
Immunofluorescence staining
In the immunofluorescence assay, cells were first fixed with 3.7% paraformaldehyde at room temperature for 10 min, followed by permeabilization using 0.05% Triton X-100 at room temperature for 30 min [33]. Subsequently, the samples were washed with cold PBS 3 times at the room temperature, and then 10% goat serum albumin (Invitrogen, USA) was used as a blocking agent. After washing with PBS 3 times at room temperature, samples were incubated with primary antibodies at 4 °C overnight. After being extensively washed with PBS, samples were treated with Alexa-Fluor 488 donkey anti-rabbit secondary antibody (1:1000; cat. no. A-21206; Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for 45 min. Finally, DAPI (5 mg/ml; Sigma-Aldrich; Merck KGaA) was used to tag the nucleus at 37 °C for 3 min. The samples were observed under an Olympus BX 50 microscope [34]. The primary antibodies used in the study were: Troponin T (1:1000, Abcam, #ab8295), p-ERK (1:500, Abcam, #ab176660), FUNDC1 (1:500, Abcam #ab224722), Tom20 (mitochondrial marker; 1:1000, Abcam, #ab186735), cyt-c (1:500; Abcam; #ab90529), and LAMP1 (lysosome marker; 1:500, Abcam, #ab24170).
Western blot analysis
Heart tissue and cardiomyocytes were homogenized in a lysis buffer with a protease inhibitor cocktail [35]. The primary antibodies used for immunoblotting were as follows: pro-caspase-3 (1:1000, Cell Signaling Technology, #9662), cleaved caspase-3 (1:1000, Cell Signaling Technology, #9664), Bax monoclonal antibody (6A7) (1:1000, Thermo Fisher Scientific, #14-6997-81), Bcl2 (1:1000, Cell Signaling Technology, #3498), caspase-9 (1:1000, Abcam #ab32539), LC3II (1:1000, Cell Signaling Technology, #3868), LC3I (1:1000, Cell Signaling Technology, #4599), Beclin1 (1:1000, Cell Signaling Technology, #3495), Atg5 (1:1000, Cell Signaling Technology, #12994), p-ERK (1:1000, Abcam, #ab176660), t-ERK (1:1000, Abcam #ab54230), FUNDC1 (1:1000, Abcam #ab224722), cyt-c (1:1000; Abcam; #ab90529), TNFα (1:1000, Abcam, #ab6671), MMP9 (1:1000, Abcam, #ab38898), IL-8 (1:1000, Abcam, #ab7747), c-IAP (1:1000, Cell Signaling Technology, #4952), Mst1 (1:1000, Cell Signaling Technology, #3682), survivin (1:1000, Cell Signaling Technology, #2808), t-CREB (1:1000, Cell Signaling Technology, #9197), and p-CREB (1:1000, Cell Signaling Technology, #9198). The membranes were incubated with horseradish peroxidase (HRP)-coupled secondary antibodies (1:2000, Cell Signaling Technology, #7074 and #7076). The intensity of immunoblot bands was normalized to that of GAPDH (1:1000, Cell Signaling Technology, #5174) and/or β-actin (1:1000, Cell Signaling Technology, #4970) [36].
Mitochondrial function assessment
Mitochondrial function was evaluated via mitochondrial potential observation and ROS staining. Mitochondrial potential was observed via JC-1 probe according to a previous study [37]. After I/R injury, cardiomyocytes were treated with 5 mg/ml JC-1 probe at 37 °C for 15 min. Then, PBS was used to wash cells to remove the free JC-1 probe. After being stained with DAPI, cardiomyocytes were observed under an Olympus BX 50 microscope. Images were analyzed with Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD) to obtain the mean densities of the region of interest, which was normalized to that of the control group. Cellular ROS was assessed via ROS probe DHE. After I/R injury, cardiomyocytes were washed with PBS and then incubated with DHE probe at 37 °C for 10 min. Then, PBS was used again to wash cardiomyocytes to remove the free ROS probe. Subsequently, ROS were detected via flow cytometry based on a recent report [38].
Cell shortening/relengthening assay and cardiomyocyte calcium mapping
The mechanical properties of primary cardiomyocytes after I/R injury were assessed using a SoftEdge MyoCam system (IonOptix, Milton, MA). Cardiomyocytes were isolated from reperfused hearts and then plated in a chamber on the stage of an inverted microscope. Cell shortening and relengthening were assessed, including PS, TPS, TR90, and ± dL/dt according to previous studies [7, 39]. +dL/dt is the maximal velocity of shortening. −dL/dt is the maximal velocity of relengthening. TPS is the time to peak shortening. TR90 is the time to 90% relengthening. Cardiomyocyte calcium mapping was conducted using Fluo-2-AM (Molecular Probes). After I/R injury, cardiomyocytes were washed with PBS 3 times and then incubated with a Fluo-2 probe at 37 °C for 10 min. Then, the cellular calcium was quantified via evaluating the excitation wavelengths of 340 nm and emission wavelengths of 380 nm based on a previous study [40].
Cellular viability evaluation and MMP9 activity detection
Cellular viability was assessed via analyzing caspase-3/9 activity and LDH release. caspase-9 activation is the early hallmark of mitochondrial apoptosis, which promotes caspase-3 activation. The caspase-3 and caspase-9 activity assays were performed according to the manufacturer’s instructions (Beyotime Institute of Biotechnology, China) [41]. The relative protein activity was calculated from the ratio of I/R-treated group to control group. In the later stage of cell death, the breakage of the cellular membrane induces LDH release into the medium. Accordingly, the content of LDH in the medium was another marker of cellular viability. LDH release was examined using an LDH release kit (Beyotime Institute of Biotechnology, China) according to a previous study [42]. An MMP9 activity kit (Beyotime Institute of Biotechnology) was used according to the manufacturer’s protocols. The relative MMP9 activity was calculated from the ratio of treated cells to control cells. The assays were repeated 3 times.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) staining
Reperfused myocardial tissues and cardiomyocytes were collected and immediately fixed by 3.7% paraformaldehyde. The TUNEL kit (Roche Apoptosis Detection Kit, Roche, Mannheim, Germany) was used on the sections according to the instructions [43, 44]. DAPI (5 mg/ml; Sigma-Aldrich; Merck KGaA) was used to tag the nucleus. The samples were observed under an Olympus BX 50 microscope. Finally, the apoptotic index (AI) was the proportion of apoptotic cells to total cells [45].
Statistical analysis
All data were analyzed using SPSS 20.0 software (IBM Corporation, Armonk, NY, USA). The experiments were repeated at least 3 times. The data are presented as the mean ± SEM and statistical significance was assessed by a one-way analysis of variance followed by Tukeyʼs test for the post hoc analysis. P < 0.05 indicates a statistically significant difference.
Results
Mst1 is upregulated in response to I/R injury
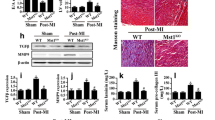
First, we evaluated Mst1 expression levels before and after cardiovascular I/R injury in WT mice. Relative to the baseline, Mst1 expression tended to be increased in the heart after I/R (Fig. 1a, b). Furthermore, to gain the functional role of Mst1 in cardiac I/R injury, Mst1 knockout (Mst1-KO) mice were subjected to I/R injury, and the degree of infarction was quantified. Genetic ablation of Mst1 reduced the contents of Mst1 in the infarcted heart under I/R injury (Fig. 1a–b) and subsequently reduced the infarcted area when compared with WT mice (Fig. 1c, e). Moreover, TUNEL assay hinted that more cell death appeared in I/R-treated mice when compared to the sham group (Fig. 1d, f). However, Mst1 deletion alleviated the number of dead cells (Fig. 1d, f). In addition to functional alterations, we also observed a change of cardiac structure via electron microscope (EM). Compared to the sham group, I/R injury caused myocardium dissolution and muscle fiber breakage (Fig. 1g); this structural change was reversed by Mst1 deletion. Finally, we evaluated the impact of Mst1 deletion on myocardial inflammation induced by cardiac I/R injury. As shown in Fig. 1h–k, I/R injury elevated the expression of MMP9, TNFα, and IL-8, and these effects were rescued by Mst1 deletion. Meanwhile, we also found that MMP9 activity was increased in response to I/R injury and was reduced with Mst1 deletion (Fig. 1l). Altogether, the present data indicated that upregulated Mst1 mediated by I/R injury contributed to cardiomyocyte death and myocardial inflammation.

Mst1 is upregulated by cardiac I/R injury and contributes to myocardial damage and expansion of the infarct area. a, b Cardiac I/R injury was induced via 30 min of ischemia and 2 h of reperfusion in WT mice and Mst1-knockout (Mst1-KO) mice. Then, heart tissues were isolated and western blotting was used to analyze the Mst1 expression before and after cardiac I/R injury. c and e TTC and Evans blue staining were used to analyze the infarction area. The white area in the heart section indicates the infarcted zone. d, f After cardiac I/R injury, a TUNEL assay was carried out to observe cardiomyocyte death. The TUNEL-positive cells detected by TUNEL manifesting brown nuclei were apoptotic cells. g An electron microscope was used to observe the ultrastructure of infarcted hearts. h–k After cardiac I/R injury, heart tissues were isolated and western blotting was performed to detect the levels of inflammation. MMP9, TNFα and IL-8 were increased in response to I/R injury and were reduced to near-normal levels with Mst1 deletion. l The activity of MMP9 was measured via ELISA. MMP9 matrix metalloproteinase 9, TNFα tumor necrosis factor alpha, IL-8 interleukin 8. Data are presented as the mean ± SEM (n = 6, for each group). I/R injury ischemia–reperfusion injury. *P < 0.05
Loss of Mst1 sustains cardiac function
The echocardiogram was used to evaluate the change of left ventricular function after I/R injury (Fig. 2a–c). Left ventricular ejection fraction (LVEF) and left ventricular fractional shorting (LVFS) significantly declined whereas left ventricular diastolic dimension (LVDd) was increased in the I/R-treated mice, when compared with the sham group (Fig. 2a–c). However, genetic ablation of Mst1 improved the cardiac function parameters (Fig. 2a–c). Moreover, compared with the sham group, I/R injury also elevated the concentration of LDH, Troponin T, and CK-MB (Fig. 2d–f); this effect was attenuated by Mst1 deletion (Fig. 2d–f). These data suggest that Mst1 deficiency sustained the cardiac function and alleviated cardiac damage under I/R injury.

Mst1 deletion improves cardiac function in the context of cardiac I/R injury. a–c Cardiac function was measured via echocardiography. FS fractional shortening, LVDd left ventricular diastolic dimension, LVEF left ventricular ejection fraction. d–f After cardiac I/R injury, blood was collected and the concentrations of cardiac damage markers were detected via ELISA. LDH lactate dehydrogenase; CK-MB: creatine kinase-MB. g–l The contractile properties of cardiomyocytes in WT mice and Mst1-KO mice in the context of IR injury. +dL/dt is the maximal velocity of shortening. −dL/dt is the maximal velocity of relengthening. TPS is the time to peak shortening. TR90 is the time to 90% relengthening. m–o Cardiomyocyte calcium mapping via the Fluo-2 probe. Representative tracings of F340/F380 fluorescence ratio. The calcium transient baseline and the amplitude of calcium transients were measured. Data are presented as the mean ± SEM (n = 6, for each group). I/R injury ischemia–reperfusion injury. *P < 0.05
Furthermore, we isolated the cardiomyocytes from the mice and then observed the mechanical parameters of cardiomyocytes in response to Mst1 deletion. Compared to the sham group, neither I/R injury nor Mst1 deletion had influences on the resting cell length in cardiomyocytes (Fig. 2g). However, I/R injury obviously repressed PS and ± dL/dt (Fig. 2h–j); this effect was rescued by Mst1 deletion. In addition, TPS and TR90 were also increased in response to I/R injury and were reduced to normal levels with Mst1 deletion (Fig. 2k, l).
Moreover, we also observed the change of intracellular Ca2+ transients in isolated cardiomyocytes via Fura-2 staining. As shown in Fig. 2m–o, compared to the sham group, I/R injury significantly increased the intracellular resting calcium, indicative of baseline calcium overload. However, Mst1 deletion could eliminate I/R-mediated cardiomyocyte calcium overload. Moreover, the intracellular calcium transient amplitude was decreased by I/R injury and was increased by Mst1 deletion (Fig. 2m–o). Altogether, these data confirmed that Mst1 deficiency sustained myocardial contractile function in the context of cardiac I/R injury.
Mst1 promotes cardiomyocyte death via mitochondrial apoptosis
Given that upregulated Mst1 played an unfavorable role in mitochondrial homeostasis in diabetic cardiomyopathy [26] and infarcted heart [22, 46], we wanted to know whether Mst1 also modulated mitochondrial function in cardiac I/R injury. To validate our speculation, we isolated cardiomyocytes from WT and Mst1-KO mice. Then, a treatment consisting of 30 min of hypoxia with serum starvation and 2 h of reoxygenation (H/R injury) was used to mimic I/R injury in vitro. Mitochondrial function was assessed via ROS production. As shown in Fig. 3a, b, H/R injury obviously increased the ROS production in the cardiomyocytes, and this effect was blocked by Mst1 deletion. Excessive ROS production causes damage to the mitochondrial membrane, leading to potential collapse of the mitochondrial membrane. Accordingly, JC-1 staining was used to observe the alterations in mitochondrial membrane potential. As shown in Fig. 3c, d, mitochondrial membrane potential was reduced by H/R stress and was reversed to near-normal levels with Mst1 deletion.

Mst1 deletion protects mitochondria against reperfusion-mediated damage. Cardiomyocytes were isolated from WT mice and Mst1-deleted mice. Then, hypoxia for 30 min and reoxygenation for 2 h were applied in vitro to mimic cardiac I/R injury. Subsequently, mitochondrial function and mitochondrial apoptosis were measured. a, b ROS production was detected via flow cytometry. c, d Mitochondrial potential was observed via JC-1 staining; the normal mitochondria displayed red fluorescence, whereas the damaged mitochondria exhibited green fluorescence. e, f The cellular location of cyt-c was observed via immunofluorescence assay. The nuclear expression of cyt-c was recorded. g–m After H/R injury in vitro, proteins were isolated and western blotting was performed to analyze the expression of proteins related to mitochondrial apoptosis. Data are presented as the mean ± SEM. H/R injury hypoxia–reoxygenation injury, WT-cell cardiomyocytes isolated from WT mice, Mst1-KO cell cardiomyocytes isolated from Mst1 knockout mice. *P < 0.05
The mitochondrial membrane potential reduction is an early hallmark of mitochondrial apoptosis which is characterized by cyt-c liberation into the nucleus and caspase-9 activation. Based on this information, immunofluorescence was used to observe cyt-c location. In the H/R-treated cardiomyocytes, cyt-c was liberated into the nucleus, and this conformational change was reversed by Mst1 deletion (Fig. 3e, f). This finding was further supported via western blotting analysis (Fig. 3g, h). As the consequence of cyt-c liberation, mitochondrial pro-apoptotic proteins, such as caspase-3, caspase-9 and Bax, were upregulated in response to H/R injury (Fig. 3g–m) and were reduced by Mst1 deletion. By comparison, anti-apoptotic factors, including Bcl2 and c-IAP, were repressed by H/R injury and were reversed to near-normal levels in Mst1-deleted cardiomyocytes (Fig. 3g–m). Altogether, the above data indicated that I/R injury induced cardiomyocyte death via mitochondrial apoptosis and Mst1 deletion had the ability to block mitochondrial apoptosis activation, promoting cardiomyocyte survival and sustaining mitochondrial function.
Mst1 represses FUNDC1-mediated mitophagy
Mitophagy is another complementary system for preserving mitochondrial homeostasis. To examine whether Mst1 regulates mitochondrial function via mitophagy, we used western blotting to analyze the activity of mitophagy in response to Mst1 deletion under H/R injury. Compared to the control group, H/R treatment reduced the LC3II expression and increased the LC3I content (Fig. 4a–g), suggestive of an autophagosome synthesis defect. Subsequently, we isolated the mitochondria and analyzed the expression of mitochondrial LC3II (mito-LC3II) expression. Similarly, H/R treatment significantly reduced the expression of mito-LC3II (Fig. 4a–g). Moreover, the proteins related to autophagy (ATG5, Beclin1 and Vps34) were also repressed by H/R treatment, when compared to control group (Fig. 4a–g). Interestingly, loss of Mst1 reversed mitochondrial autophagy activity in the presence of H/R stress (Fig. 4a–g), as evidenced by increased expression of mito-LC3II, ATG5, Vps34 and Beclin1. These data highlighted that mitophagy was drastically suppressed by H/R injury via Mst1.

Mst1 deletion reverses FUNDC1-related mitophagy in the context of cardiac I/R injury. a–g After H/R injury in vitro, proteins were isolated and mitophagy activity was monitored via western blotting. Moreover, to verify whether mitophagy activity was predominately regulated by FUNDC1, we transfected an siRNA into Mst1-deleted cardiomyocytes. The transfection efficiency was confirmed via western blotting. After loss of FUNDC1 in Mst1-deleted mice, mitophagy activity was re-examined via western blotting. h, i Mitophagic activity was further validated via immunofluorescent staining of mitochondria and lysosomes. The fusion of mitochondria (green fluorescence) and lysosomes (red fluorescence) indicates mitophagy, which is indicated by orange fluorescence. Data are presented as the mean ± SEM. H/R injury hypoxia–reoxygenation injury, WT-cell cardiomyocytes isolated from WT mice, Mst1-KO cell cardiomyocytes isolated from Mst1 knockout mice. *P < 0.05
Previous studies have found that cardiomyocyte mitophagy is regulated primarily by FUNDC1 in the context of H/R injury [12, 13]. In the present study, we also found that FUNDC1 expression was significantly downregulated by H/R treatment (Fig. 4a, b). Interestingly, Mst1 deletion has the ability to reverse FUNDC1 expression despite H/R treatment (Fig. 4a, b). This information indicated that Mst1 could be considered the upstream inhibitor of FUNDC1. To verify whether FUNDC1 is required for Mst1-modulated mitophagy, we knocked down FUNDC1 in Mst1-deleted cardiomyocytes. The knockdown efficiency was confirmed via western blotting in Fig. 4a, b. Then, mitophagy parameters were assessed again. The results shown in Fig. 4a–g demonstrated that loss of FUNDC1 re-inhibited mitophagy activity in Mst1-deleted cells. This information illustrated that Mst1 modified mitophagy activity via FUNDC1 in the context of H/R injury.
Subsequently, mitophagy activity was further evaluated via observing the co-location of mitochondria and lysosome. Compared to the H/R-treated cardiomyocytes, Mst1 deletion increased the number of mitochondria that were swallowed by lysosomes (Fig. 4h, i), and this effect was abrogated by FUNDC1 deletion. Altogether, this information revealed that Mst1 deletion sustained FUNDC1-related mitophagy activity under H/R injury.
FUNDC1-related mitophagy maintains mitochondrial function and reduces H/R-mediated cardiomyocyte death
Next, to explain the role of FUNDC1-mediated mitophagy in cardiomyocyte damage, we assessed mitochondrial function again. First, ATP production was decreased in response to H/R treatment (Fig. 5a) and was reversed to near-normal levels by Mst1 deletion in a FUNDC1-dependent manner (Fig. 5a). In addition, H/R-mediated caspase-3 activation was also inhibited in Mst1-deleted cardiomyocytes via reversing FUNDC1 mitophagy (Fig. 5b). Moreover, Mst1 deletion repressed the expression of pro-apoptotic proteins and upregulated the content of anti-apoptotic factors under H/R stimulus, and this effect was abrogated by FUNDC1 deficiency (Fig. 5c–h). Finally, a TUNEL assay was performed to analyze the cell apoptotic index. Compared to the control group, H/R increased the number of TUNEL-positive cells (Fig. 5i, j); this effect was strongly inhibited by Mst1 deletion. However, loss of FUNDC1 attenuated the anti-apoptotic property of Mst1 deficiency in H/R-treated cardiomyocytes (Fig. 5i, j). Altogether, this information indicated that Mst1 deletion maintained mitochondrial function and cardiomyocyte survival via reversing FUNDC1-related mitophagy.

FUNDC1-related mitophagy sustains mitochondrial dysfunction and blocks I/R-mediated mitochondrial apoptosis. a ATP production was measured via ELISA in response to FUNDC1 deficiency and Mst1 deletion. b Caspase-3 activity was measured after I/R injury in cardiomyocytes transfected with FUNDC1 siRNA. c–h After H/R injury in vitro, proteins were isolated and western blotting was performed to analyze the expression of proteins related to mitochondria. i, j TUNEL assay for cellular apoptosis. Green nuclei indicate apoptotic cells, and the number of TUNEL-positive cells was quantified. Data are presented as the mean ± SEM. H/R injury hypoxia–reoxygenation injury, WT-cell cardiomyocytes isolated from WT mice, Mst1-KO cell cardiomyocytes isolated from Mst1 knockout mice. *P < 0.05
Mst1 regulates FUNDC1 via the MAPK/ERK-CREB pathway
Finally, we want to know the underlying mechanism by which Mst1 modulated FUNDC1-related mitophagy. Recent studies have suggested that FUNDC1 mitophagy activity is highly regulated by the ERK-CREB pathway in the infarcted heart [18]. In the present study, our data demonstrated that both ERK and CREB were inhibited by H/R injury (Fig. 6a–d). However, loss of Mst1 reversed the ERK and CREB activity, as evidenced by increased ERK and CREB phosphorylation. This finding was further supported by immunofluorescence assay (Fig. 6e). To explain whether the ERK-CREB pathway is required for Mst1-modulated FUNDC1 expression, we blocked the pathway and then measured FUNDC1 expression again. As shown in Fig. 6a–d, Mst1 deletion reversed H/R-inhibited FUNDC1 expression, and this effect was abrogated by ERK inhibitor, PD98059. These results were also validated via immunofluorescence assay (Fig. 6e). Accordingly, our data indicated that Mst1 regulated FUNDC1 via the MAPK/ERK-CREB pathway.

Mst1 modulates FUNDC1 via the ERK-CREB pathway. a–d Western blotting was used to analyze the effect of Mst1 deletion on ERK phosphorylation and CREB phosphorylation. Subsequently, to inhibit the ERK activity in Mst1-deleted cardiomyocytes, we added PD98059 to Mst1-deleted cells. Then, the FUNDC1 expression was detected to establish the regulatory effect of ERK on FUNDC1 activation. e Immunofluorescence assay for p-CREB and FUNDC1. Both p-CREB and FUNDC1 expression were significantly inhibited by H/R injury and were reversed to near-normal levels with Mst1 deletion. f, g Caspase-9 activity and LDH release assays were measured to confirm the role of the ERK-CREB pathway in mitochondrial apoptosis. Data are presented as the mean ± SEM. H/R injury hypoxia–reoxygenation injury, WT-cell cardiomyocytes isolated from WT mice, Mst1-KO cell cardiomyocytes isolated from Mst1 knockout mice. *P < 0.05
Furthermore, LDH release and caspase-9 activity assays were performed to validate whether the MAPK/ERK-CREB pathway was also involved in Mst1-related cardiomyocyte death under H/R stress. As shown in Fig. 6f, g, H/R-mediated LDH release and caspase-9 activation were effectively inhibited by Mst1 deletion. However, with blockade of the MAPK/ERK-CREB pathway in Mst1-deleted cardiomyocytes, LDH release and caspase-9 activity were increased again (Fig. 6f, g).
Discussion
Although it has been over a decade since the discovery of Mst1, its precise functions, especially its role in cardiac I/R injury, have not been determined yet. In the study, we found that (1) I/R injury induced intracellular Mst1 upregulation, which was the pathogenesis for the cardiac I/R injury because loss of Mst1 reduced the infarction area, improved myocardial function, and promoted cardiomyocyte survival; (2) mechanistically, Mst1 inactivated the MAPK/ERK-CREB pathway and consequently repressed FUNDC1 expression; (3) depleted FUNDC1 was unable to launch protective mitophagy, ultimately leading to mitophagy delay; and (4) defective mitophagy promoted reperfusion-mediated mitochondrial damage, evoking oxidative stress, energy shortage, and cardiomyocyte apoptosis activation. To the best of our knowledge, this is the first investigation to establish the functional role of Mst1 in cardiac I/R injury via regulating mitophagy activity and the MAPK/ERK-CREB pathway.
Mst1, also known as STK4 or KRS2, is a type of serine/threonine kinase. Mst1 belongs to the Hippo pathway and was originally found to be associated with cancer proliferation, survival and stress response. Shreds of evidence are currently accumulating to establish the pro-apoptotic action of Mst1 on several kinds of cancer. For example, Mst1 deficiency promotes endometriosis survival and migration via modulating mitochondrial homeostasis [47, 48]. In breast and lung cancer, Mst1 deletion enhances cancer survival and invasion [49]. In addition, Mst1 expression is positively associated with the activity of caspase-9 in pancreatic cancer [50]. In colorectal cancer, Mst1 expression can be considered a potential early biomarker for cancer progression and metastasis [51]. This information illustrates that Mst1 is a critical molecule in the management of cell death. However, no data are available to describe the role of Mst1 reperfusion-mediated cardiomyocyte death. Our present study demonstrated that Mst1 was dramatically increased by cardiac reperfusion injury and higher Mst1 expression was correlated to more cardiomyocyte death. However, genetic ablation of Mst1 reduced the cardiomyocyte death and therefore alleviated cardiac I/R injury. Our data, combined with the previous evidence, define Mst1 as a novel causative factor of cardiac I/R injury that acts by regulating cardiomyocyte viability, with potential implications for new approaches to myocardial I/R injury.
The key molecular machinery to appropriately control mitochondrial homeostasis is mitophagy, a selective form of autophagy that removes malfunctioning mitochondria. In the past few decades, several essential factors for mitophagy activation have been identified and characterized, such as Bnip3 [52], FUNDC1 [14], Mfn2 [53] and Parkin. Notably, different mitophagy activators in various disease models play distinct roles in modulating mitochondrial homeostasis. For example, in fatty liver disease, Bnip3-related mitophagy is beneficial for lipid metabolism and therefore retards the progression of cirrhosis [52]. By comparison, in high-fat-associated atherosclerosis, Parkin-mediated mitophagy is deleterious to endothelial function and promotes atherosclerosis formation [54]. This information indicates that mitophagy modulators have a decisive role in mitophagy function. In cardiac I/R injury, FUNDC1 has been tested in several animal studies [13, 15], and ample evidence has confirmed its protective action on myocardial viability via reducing mitochondria oxidative stress, blocking mitochondrial fission, attenuating mitochondrial calcium overload, promoting mitochondrial energy production, and preventing mitochondrial apoptosis activation. Unfortunately, FUNDC1-related mitophagy is prone to be repressed by reperfusion injury, and the upstream mediator has not been elucidated [12]. In the present study, we found that FUNDC1 expression and mitophagy activity are primarily regulated by Mst1. Loss of Mst1 reverts FUNDC1 content to normal and therefore enhances protective mitophagy activity in the reperfused heart. Moreover, consistent with the previous study, our data reconfirmed that FUNDC1-related mitophagy could reverse ATP production, stabilize mitochondrial membrane potential and block I/R-activated mitochondrial apoptosis. In this study, our findings help fill in the gaps that Mst1 could be considered as the upstream regulator for FUNDC1-related mitophagy in the context of cardiac I/R injury. In addition, Mst1 has also been found to be associated with other mitophagy mediators. For example, in colorectal cancer, Mst1 inhibits Bnip3 expression and further represses mitophagy activity via the JNK/p53 pathway [55]. In endometriosis, Mst1 regulates cellular apoptosis and migration via Parkin-related mitophagy [56]. These pieces of evidence suggest that Mst1 effectively manages mitophagy activity by controlling the expression of multiple mitophagy receptors, which firmly establishes a central role of Mst1 in mitophagy modification.
Finally, we explored the mechanism by which Mst1 controlled FUNDC1. A previous study has suggested that FUNDC1 expression is regulated by the transcriptional factor, CREB [18]. In agreement with the findings of that study, we also demonstrated that the ERK-CREB pathway was significantly inactivated by Mst1, contributing to FUNDC1 downregulation. However, genetic deletion of Mst1 re-activated the ERK-CREB pathway and resultantly elevated FUNDC1 expression. These findings suggest that Mst1 inhibits FUNDC1-related mitophagy that occurs, at least in part, through the ERK-CREB pathway in cardiac I/R injury. In accordance with our finding, Bnip3-related mitophagy is also regulated by the MAPK-ERK pathway in the nervous system [57]. Similarly, in β-cells, ERK activation is associated with mitophagy initiation [58]. In liver tissue, ERK inhibition interrupts mitochondrial degradation and reduces glucose production [59]. This information highlights the relevance of the ERK pathway in the regulation of mitophagy activity.
Collectively, our data reported that cardiac I/R injury is associated with Mst1 upregulation which blunts FUNDC1-related mitophagy via repression of the MAPK/ERK-CREB pathway. Genetic ablation of Mst1 boosted FUNDC1-mediated mitophagy, effectively alleviating mitochondrial damage and reducing cardiomyocyte death. Our results in vivo and in vitro lay the foundation for a detailed study of the molecular mechanisms of mitophagy regulation, mitochondrial homeostasis and cardiomyocyte death in response to acute cardiac I/R injury.
References
Hu SY, Zhang Y, Zhu PJ, Zhou H, Chen YD (2017) Liraglutide directly protects cardiomyocytes against reperfusion injury possibly via modulation of intracellular calcium homeostasis. J Geriatr Cardiol 14:57–66
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y (2018) Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res 64(3):e12471
Garcia-Nino WR, Correa F, Rodriguez-Barrena JI, Leon-Contreras JC, Buelna-Chontal M, Soria-Castro E, Hernandez-Pando R, Pedraza-Chaverri J, Zazueta C (2017) Cardioprotective kinase signaling to subsarcolemmal and interfibrillar mitochondria is mediated by caveolar structures. Basic Res Cardiol 112:15
Reinthaler M, Braune S, Lendlein A, Landmesser U, Jung F (2016) Platelets and coronary artery disease: interactions with the blood vessel wall and cardiovascular devices. Biointerphases 11:029702
Jovancevic N, Dendorfer A, Matzkies M, Kovarova M, Heckmann JC, Osterloh M, Boehm M, Weber L, Nguemo F, Semmler J, Hescheler J, Milting H, Schleicher E, Gelis L, Hatt H (2017) Medium-chain fatty acids modulate myocardial function via a cardiac odorant receptor. Basic Res Cardiol 112:13
Zhai M, Li B, Duan W, Jing L, Zhang B, Zhang M, Yu L, Liu Z, Yu B, Ren K, Gao E, Yang Y, Liang H, Jin Z, Yu S (2017) Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 63(2):e12419
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H (2018) DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol 14:576–587
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y (2016) Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3 K/Akt/survivin pathways. Free Radic Biol Med 95:278–292
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y (2018) Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 23:101–113
Fuhrmann DC, Brune B (2017) Mitochondrial composition and function under the control of hypoxia. Redox Biol 12:208–215
Xu J, Wu Y, Lu G, Xie S, Ma Z, Chen Z, Shen HM, Xia D (2017) Importance of ROS-mediated autophagy in determining apoptotic cell death induced by physapubescin B. Redox Biol 12:198–207
Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y (2018) Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ 25:1080–1093
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y (2018) NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol 113:23
Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q, Du L, Zheng M, Zhang C, Liu J, Chen Q (2016) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5:e21407
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y (2017) Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol 13:498–507
Zhou H, Shi C, Hu S, Zhu H, Ren J, Chen Y (2018) BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. https://doi.org/10.1007/s10456-018-9611-z
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y (2017) Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res 63:e12438
Lee H J, Jung Y H, Choi G E, Ko S H, Lee S J, Lee S H, Han H J (2017) BNIP3 induction by hypoxia stimulates FASN-dependent free fatty acid production enhancing therapeutic potential of umbilical cord blood-derived human mesenchymal stem cells. Redox Biol 13:426–443
Zhang W, Siraj S, Zhang R, Chen Q (2017) Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 13:1080–1081
Zhou H, Wang J, Zhu P, Hu S, Ren J (2018) Ripk3 regulates cardiac microvascular reperfusion injury: the role of IP3R-dependent calcium overload, XO-mediated oxidative stress and F-action/filopodia-based cellular migration. Cell Signal 45:12–22
Rossello X, Riquelme JA, He Z, Taferner S, Vanhaesebroeck B, Davidson SM, Yellon DM (2017) The role of PI3K alpha isoform in cardioprotection. Basic Res Cardiol 112:66
Nunez-Gomez E, Pericacho M, Ollauri-Ibanez C, Bernabeu C, Lopez-Novoa JM (2017) The role of endoglin in post-ischemic revascularization. Angiogenesis 20:1–24
Yang Y, Wang H, Ma Z, Hu W, Sun D (2018) Understanding the role of mammalian sterile 20-like kinase 1 (MST1) in cardiovascular disorders. J Mol Cell Cardiol 114:141–149
Thirusangu P, Vigneshwaran V, Prashanth T, Vijay Avin B R, Malojirao V H, Rakesh H, Khanum S A, Mahmood R, Prabhakar B T (2017) BP-1T, an antiangiogenic benzophenone-thiazole pharmacophore, counteracts HIF-1 signalling through p53/MDM2-mediated HIF-1alpha proteasomal degradation. Angiogenesis 20:55–71
Zhou H, Li D, Zhu P, Ma Q, Sam T, Wang J, Hu S, Chen Y, Zhang Y (2018) Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia reperfusion injury. J Pineal Res 65:e12503
Kingery J R, Hamid T, Lewis R K, Ismahil M A, Bansal S S, Rokosh G, Townes T M, Ildstad S T, Jones S P, Prabhu S D (2017) Leukocyte iNOS is required for inflammation and pathological remodeling in ischemic heart failure. Basic Res Cardiol 112:19
Pickard JM, Burke N, Davidson SM, Yellon DM (2017) Intrinsic cardiac ganglia and acetylcholine are important in the mechanism of ischaemic preconditioning. Basic Res Cardiol 112:11
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc 6:e005328
Alghanem AF, Wilkinson EL, Emmett MS, Aljasir MA, Holmes K, Rothermel BA, Simms VA, Heath VL, Cross MJ (2017) RCAN1.4 regulates VEGFR-2 internalisation, cell polarity and migration in human microvascular endothelial cells. Angiogenesis 20:341–358
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S, Sun C (2017) Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal Res 63:e12414
Banerjee K, Keasey MP, Razskazovskiy V, Visavadiya NP, Jia C, Hagg T (2017) Reduced FAK-STAT3 signaling contributes to ER stress-induced mitochondrial dysfunction and death in endothelial cells. Cell Signal 36:154–162
Dong X, Fu J, Yin X, Qu C, Yang C, He H, Ni J (2017) Induction of apoptosis in HepaRG cell line by aloe-emodin through generation of reactive oxygen species and the mitochondrial pathway. Cell Physiol Biochem 42:685–696
Daiber A, Oelze M, Steven S, Kroller-Schon S, Munzel T (2017) Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol 12:35–49
Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D, Reiter RJ, Roy SS (2017) Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J Pineal Res 62:e12404
Couto JA, Ayturk UM, Konczyk DJ, Goss JA, Huang AY, Hann S, Reeve JL, Liang MG, Bischoff J, Warman ML, Greene AK (2017) A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis 20:303–306
Brasacchio D, Alsop AE, Noori T, Lufti M, Iyer S, Simpson KJ, Bird PI, Kluck RM, Johnstone RW, Trapani JA (2017) Epigenetic control of mitochondrial cell death through PACS1-mediated regulation of BAX/BAK oligomerization. Cell Death Differ 24:961–970
Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang Z, Li Z, Feng X, Hao J, Zhang K, Xiao B, Chen M, Huang W, Xiong S, Wu X, Deng W (2017) Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3 K/AKT and NF-kappaB/iNOS signaling pathways. J Pineal Res 62:e12380
Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, Wang J, Qin Y, Liu Y, Tang C, He L, Greka A, Zhou Z, Liu F, Dong Z, Sun L (2017) The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 11:297–311
Vargas LA, Velasquez FC, Alvarez BV (2017) Compensatory role of the NBCn1 sodium/bicarbonate cotransporter on Ca(2+)-induced mitochondrial swelling in hypertrophic hearts. Basic Res Cardiol 112:14
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y (2018) Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol 16:157–168
Murphy PS, Wang J, Bhagwat SP, Munger JC, Janssen WJ, Wright TW, Elliott MR (2017) CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ 24:559–570
Randriamboavonjy V, Kyselova A, Elgheznawy A, Zukunft S, Wittig I, Fleming I (2017) Calpain 1 cleaves and inactivates prostacyclin synthase in mesenteric arteries from diabetic mice. Basic Res Cardiol 112:10
Ligeza J, Marona P, Gach N, Lipert B, Miekus K, Wilk W, Jaszczynski J, Stelmach A, Loboda A, Dulak J, Branicki W, Rys J, Jura J (2017) MCPIP1 contributes to clear cell renal cell carcinomas development. Angiogenesis 20:325–340
Akin S, Naito H, Ogura Y, Ichinoseki-Sekine N, Kurosaka M, Kakigi R, Demirel HA (2017) Short-term treadmill exercise in a cold environment does not induce adrenal Hsp72 and Hsp25 expression. J Physiol Sci 67:407–413
Merjaneh M, Langlois A, Larochelle S, Cloutier CB, Ricard-Blum S, Moulin VJ (2017) Pro-angiogenic capacities of microvesicles produced by skin wound myofibroblasts. Angiogenesis 20:385–398
Wang X, Song Q (2018) Mst1 regulates post-infarction cardiac injury through the JNK-Drp1-mitochondrial fission pathway. Cell Mol Biol Lett 23:21
Zhou H, Yue Y, Wang J, Ma Q, Chen Y (2018) Melatonin therapy for diabetic cardiomyopathy: a mechanism involving Syk-mitochondrial complex I-SERCA pathway. Cell Signal 47:88–100
Oyama Y, Iigaya K, Minoura Y, Okabe T, Izumizaki M, Onimaru H (2017) An in vitro experimental model for analysis of central control of sympathetic nerve activity. J Physiol Sci 67:629–635
Tobisawa T, Yano T, Tanno M, Miki T, Kuno A, Kimura Y, Ishikawa S, Kouzu H, Nishizawa K, Yoshida H, Miura T (2017) Insufficient activation of Akt upon reperfusion because of its novel modification by reduced PP2A-B55alpha contributes to enlargement of infarct size by chronic kidney disease. Basic Res Cardiol 112:31
Torres-Estay V, Carreno DV, Fuenzalida P, Watts A, San Francisco IF, Montecinos VP, Sotomayor PC, Ebos J, Smith GJ, Godoy AS (2017) Androgens modulate male-derived endothelial cell homeostasis using androgen receptor-dependent and receptor-independent mechanisms. Angiogenesis 20:25–38
Lee MS, Yin TC, Sung PH, Chiang JY, Sun CK, Yip HK (2017) Melatonin enhances survival and preserves functional integrity of stem cells: a review. J Pineal Res 62:e12372
Zhou H, Du W, Li Y, Shi C, Hu N, Ma S, Wang W, Ren J (2018) Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 64:e12450
Sarkar C, Ganju R K, Pompili V J, Chakroborty D (2017) Enhanced peripheral dopamine impairs post-ischemic healing by suppressing angiotensin receptor type 1 expression in endothelial cells and inhibiting angiogenesis. Angiogenesis 20:97–107
Takeya M, Okumura Y, Nikawa T (2017) Modulation of cutaneous extracellular collagen contraction by phosphorylation status of p130Cas. J Physiol Sci 67:613–622
Kang P T, Chen C L, Lin P, Chilian W M, Chen Y R (2017) Impairment of pH gradient and membrane potential mediates redox dysfunction in the mitochondria of the post-ischemic heart. Basic Res Cardiol 112:36
Zhao Q, Ye M, Yang W, Wang M, Li M, Gu C, Zhao L, Zhang Z, Han W, Fan W, Meng Y (2018) Effect of Mst1 on endometriosis apoptosis and migration: role of Drp1-related mitochondrial fission and Parkin-required mitophagy. Cell Physiol Biochem 45:1172–1190
Lei Q, Tan J, Yi S, Wu N, Wang Y, Wu H (2018) Mitochonic acid 5 activates the MAPK-ERK-yap signaling pathways to protect mouse microglial BV-2 cells against TNFalpha-induced apoptosis via increased Bnip3-related mitophagy. Cell Mol Biol Lett 23:14
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H (2018) Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol 14:59–71
Maezawa T, Tanaka M, Kanazashi M, Maeshige N, Kondo H, Ishihara A, Fujino H (2017) Astaxanthin supplementation attenuates immobilization-induced skeletal muscle fibrosis via suppression of oxidative stress. J Physiol Sci 67:603–611
Funding
This study was supported by the Natural Science Foundation of Shandong Province of China (No. ZR2013HM056), Shandong Province Science and Technology Development Plan (2014GH218016).
Author information
Authors and Affiliations
Contributions
WCY, MX, and TZ conceived the research; QZ and CWZ performed the experiments; all authors participated in discussing and revising the manuscript.
Corresponding author
Ethics declarations
Conflict of interest statement
The authors have declared that they have no conflicts of interest.
Ethics approval and consent to participate
The animal study was performed in accordance with the Declaration of Helsinki. All experimental protocols were approved by the Ethics Committee of the Department of Cardiac Surgery, Provincial Hospital Affiliated to Shandong University. The ethics reference number is SDQL33SKT1.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
About this article

Cite this article
Yu, W., Xu, M., Zhang, T. et al. Mst1 promotes cardiac ischemia–reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J Physiol Sci 69, 113–127 (2019). https://doi.org/10.1007/s12576-018-0627-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-018-0627-3