
Abstract
The mitochondrial electrochemical gradient (Δp), which comprises the pH gradient (ΔpH) and the membrane potential (ΔΨ), is crucial in controlling energy transduction. During myocardial ischemia and reperfusion (IR), mitochondrial dysfunction mediates superoxide (·O2 −) and H2O2 overproduction leading to oxidative injury. However, the role of ΔpH and ΔΨ in post-ischemic injury is not fully established. Here we studied mitochondria from the risk region of rat hearts subjected to 30 min of coronary ligation and 24 h of reperfusion in vivo. In the presence of glutamate, malate and ADP, normal mitochondria (mitochondria of non-ischemic region, NR) exhibited a heightened state 3 oxygen consumption rate (OCR) and reduced ·O2 − and H2O2 production when compared to state 2 conditions. Oligomycin (increases ΔpH by inhibiting ATP synthase) increased ·O2 − and H2O2 production in normal mitochondria, but not significantly in the mitochondria of the risk region (IR mitochondria or post-ischemic mitochondria), indicating that normal mitochondrial ·O2 − and H2O2 generation is dependent on ΔpH and that IR impaired the ΔpH of normal mitochondria. Conversely, nigericin (dissipates ΔpH) dramatically reduced ·O2 − and H2O2 generation by normal mitochondria under state 4 conditions, and this nigericin quenching effect was less pronounced in IR mitochondria. Nigericin also increased mitochondrial OCR, and predisposed normal mitochondria to a more oxidized redox status assessed by increased oxidation of cyclic hydroxylamine, CM-H. IR mitochondria, although more oxidized than normal mitochondria, were not responsive to nigericin-induced CM-H oxidation, which is consistent with the result that IR induced ΔpH impairment in normal mitochondria. Valinomycin, a K+ ionophore used to dissipate ΔΨ, drastically diminished ·O2 − and H2O2 generation by normal mitochondria, but less pronounced effect on IR mitochondria under state 4 conditions, indicating that ΔΨ also contributed to ·O2 − generation by normal mitochondria and that IR mediated ΔΨ impairment. However, there was no significant difference in valinomycin-induced CM-H oxidation between normal and IR mitochondria. In conclusion, under normal conditions the proton backpressure imposed by ΔpH restricts electron flow, controls a limited amount of ·O2 − generation, and results in a more reduced myocardium; however, IR causes ΔpH impairment and prompts a more oxidized myocardium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondrial dysfunction is the major contributor to myocardial ischemia/reperfusion injury. The investigations into reactive oxygen species (ROS) generation by mitochondria during ischemia and reperfusion have been extensively reviewed [1, 2]. Cardiac ischemia puts the myocardium into the physiological conditions of hypoxia, substrate deprivation, and acidosis. Mitochondria are thus situated in a highly reductive environment with low pO2 (oxygen tension) and low ADP. In the ischemic heart, oxygen delivery to the myocardium is not sufficient to meet the needs of mitochondrial oxidative metabolism. Re-introduction of oxygen upon reperfusion induces marked hyperoxygenation in the myocardium [3], and greatly increases electron leakage from the mitochondrial electron transport chain (ETC), leading to overproduction of ·O2 − and ·O2 −-derived oxidants in the mitochondria [4, 5].
The proton motive force (Δp, or electrochemical gradient) across the inner mitochondrial membrane is generated from proton pumping and represents the potential energy driving proton re-entry into the matrix for ATP synthesis. The Δp consists of a proton gradient (ΔpH) and a membrane potential (ΔΨ), which potentially contributes to ·O2 − production mediated by mitochondria. In vitro studies have firmly established the effect of ΔΨ on ·O2 − production by mitochondrial complex III, and this result led to the suggestion that cells modulate the magnitude of mitochondrial proton motive force to protect the mitochondria from excess production of ·O2 − [6].
Earlier studies with isolated mitochondria of rat and murine hearts indicated that mitochondria produce large amount of ·O2 − under oligomycin-induced state 4 conditions, but ·O2 − generation is diminished under conditions of ADP-mediated state 3 and FCCP-mediated uncoupling [7, 8]. The results thus lend support to the hypothesis that the proton motive force is high, and substantially contributes to ·O2 − production when the activity of F1F0 ATP synthase (F1F0 ATPase) is low (state 2 conditions) or F1F0 ATPase is inhibited (state 4 conditions). Restoration of ATP synthesis (state 3 conditions) via oxidative phosphorylation diminishes the ΔpH and ΔΨ, thus greatly reducing ·O2 − generation.
Studies with mouse hearts have indicated that ischemia can depolarize ΔΨ, which is directly correlated to the decreased reverse electron flow-induced ROS production by ischemia-damaged mitochondria, and indirectly supports the role of ΔΨ in complex I-mediated ·O2 − generation [9]. In the disease model of myocardial ischemia and reperfusion injury, oxidative damage to the ETC is marked in the region of myocardial infarction [10, 11]. Studies have further demonstrated that redox-dependent alterations of ETC and F1F0 ATPase in the post-ischemic myocardium are involved in oxidative post-translational modifications, including increasing protein tyrosine nitration of complex I, complex III, and F1F0 ATPase [12], increasing protein tyrosine nitration of complex II [13], increasing protein S-glutathionylation of complex I [14], and increasing protein sulfonation and disulfide formation of complex II [15], suggesting a redox-dependent dysfunction in the mitochondria of the post-ischemic heart. The results logically lend support to the hypothesis that oxidative injury of ETC and F1F0 ATPase can weaken the proton motive force, which further mediates the redox dysfunction in the post-ischemic heart. In the current study, we attempt to establish the roles of ΔpH and ΔΨ in post-ischemic injury by isolated mitochondria from a disease model of in vivo myocardial ischemia and reperfusion, followed by determining the rate of ·O2 − production, H2O2 generation, and redox activity using EPR spin probes. Our findings indicated that the proton backpressure imposed by mitochondrial ΔpH and ΔΨ can restrict electron flow and result in a more reduced myocardium environment, which modulates electron leakage for ·O2 − and H2O2 generation (which functions as a physiological signaling molecule). Conversely, ischemia and reperfusion injury (IR) causes Δp impairment, destroys physiological control of electron leakage by the ETC, and prompts the myocardium to become a more oxidized environment.
Materials and methods
Animals
Male Sprague–Dawley rats were purchased from Harlan (Indianapolis, IN, USA; ~300 g and 8 weeks old), rats were quarantined for one week prior to the surgical procedure of in vivo occlusion. The SOD2-tg mice (~27–33 g and 12 weeks old) were obtained from the Jackson Laboratory. All procedures were performed with the approval (Protocol No. 15-028) of the Institutional Animal Care and Use Committee (IACUC) at Northeast Ohio Medical University (Rootstown, OH, USA) and conformed to the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the NIH.
Reagents
Ammonium sulfate, diethylenetriaminepentaacetic acid (DTPA), ubiquinone-2 (Q2), deoxycholic acid, FCCP (carbonylcyanide-p-trifluoromethoxyphenylhydrazone), oligomycin, nigericin, valinomycin, gramicidin (a polypeptide antibiotic of channel-forming ionophores that conduct ions through diffusion and destroy the ΔΨ derived from the ion gradient), and polyethylene glycol superoxide dismutase (PEG-SOD) were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA) and used as received. The spin probe of CM-H, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl, was purchased from Enzo Life Sciences Inc. (Farmingdale, NY, USA). The DMPO spin trap was purchased from Dojindo Molecular Technologies, Inc. (Rockville, MD, USA), and stored under argon at −80 °C until needed.
Myocardial infarction model
The procedure to produce a myocardial infarct involved an in vivo ischemia–reperfusion rat heart model [3, 13, 16,17,18]. Sprague–Dawley rats (~300–350 g and 9–10 weeks old) were anesthetized with isoflurane ((2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane)) via inhalation. After the rats were fully anesthetized, they were intubated and then ventilated with room air (1.0 mL/100 g/min, rate of 75–85 breaths/min) using a mechanical ventilator Model 683 (Harvard Apparatus, Holliston, MA, USA). The rats then underwent a left lateral thoracotomy, the pericardium was opened, and a pericardial cradle formed to allow adequate exposure of the heart surface. The left anterior descending coronary artery (LAD) was then occluded by placing a suture (6.0 nylon) around the origin of the LAD (illustrated in the Scheme 1a of enclosed box inset).

a The approach to construct the model of in vivo myocardial ischemia and reperfusion rat heart as described in the “Materials and Methods”. b TCC staining of the infarct area or the risk region in the post-ischemic myocardium. c Strategic design to assess the ΔpH and ΔΨ of mitochondria. Mitochondria were isolated from the myocardium of non-ischemic region (NR or normal mitochondria) and infarct region (IR or post-ischemic mitochondria). The substrate of glutamate plus malate was used to induce state 2 NADH-linked oxygen consumption. The electrochemical gradient, Δp, was increased because ΔpH was established and proton circuit is not completed by H+ re-entry through the ATPase (ATP synthase). The addition of ADP was used to induce state 3 oxygen consumption, allowing the ATPase to synthesize ATP coupled to H+ re-entry across the membrane through the proton channel of ATPase, Δp was thus decreased. Oligomycin was used to block the proton channel of ATPase and induce state 4 oxygen consumption. Δp was thus enhanced due to re-establishment of the proton gradient. Proton ionophore, FCCP, was used to dissipate protein gradient and membrane potential, allowing Δp diminishment. Nigericine was used to enhance proton influx and dissipate ΔpH. Potassium ionophore, valinomycin, was used to dissipate the membrane potential
After 30 min of ischemia, the ligature around the coronary artery was removed, allowing reperfusion. Following the reperfusion, all wounds were closed and injected with buprenorphine (0.05 mg/kg, SQ). The muscular layers and skin incisions were closed with 4.0 nylon sutures.
Upon spontaneous breathing, the animal was allowed to recover and a physiological assessment was performed. During the recovery period the animals received supportive post-operative care as needed. Body temperature was maintained at 37 °C by a thermal heating pad. By 6-h post-operation, the animals had recovered sufficiently to eat and drink independently.
At 24-h post-infarction, the rats were placed under deep anesthesia with isoflurane. The rats were then killed, and the hearts excised and placed in the PBS buffer. The infarct area was identified by TTC (2,3,5-triphenyltetrazolium chloride) staining (illustrated in the Scheme 1b of the enclosed box inset). The risk region of myocardial tissue without TTC staining was excised and subjected to mitochondrial preparations and biochemical analysis (denoted by IR in the Scheme 1b). The remaining tissue as non-ischemic region was served as the control or normal region (denoted by NR in the Scheme 1b). The ratio (w/w) of risk region tissue to non-ischemic region tissue is 37.4 ± 11.9% (n = 19).
Analytical methods
Optical spectra were measured on a Shimadzu 2600 UV/VIS recording spectrophotometer. The protein concentrations of mitochondrial preparations were determined by the Lowry method using BSA as a standard. The concentrations of Q 1 and Q 2 were determined by absorbance spectra from NaBH4 reduction using a millimolar extinction coefficient of ε (275–290 nm) = 12.25 mM−1 cm−1. Optical spectra were measured on a Shimadzu UV2600 recording spectrophotometer. The heme b and aa 3 concentrations of isolated mitochondria were calculated from the differential spectrum between dithionite reduction and ferricyanide oxidation.
Mitochondria were prepared from rat hearts by differential centrifugation as described in the published method [8, 11, 16]. Mitochondria were precipitated by centrifugation at 20,000×g for 10 min in the final step and re-suspended in media (M-buffer) containing the following agents (in mM): mannitol (230), sucrose (70), EDTA (1), and Trizma (1), pH 7.4. Mitochondrial respiration (or NADH-lined OCR) was measured by the polarographic method using a Clark-type oxygen electrode (Oxytherm, Hansatech Instruments, Norfolk, England) at 30 °C by following the same protocol as detailed in a previous publication [8]. The electron transfer activities of Complexes I–IV from the heart mitochondrial preparations were assayed by a published method [11]. The recovery of mitochondria from the post-ischemic rat heart is 24.3 ± 2.6% for non-ischemic region (n = 3) and 20.0 ± 0.4% for risk region (n = 3) based on measuring percent recovery of citrate synthase activity in isolated mitochondria and tissue homogenate. The mitochondria as prepared contain 474.6 ± 18.2 nmol heme b/mg protein and 596.9 ± 14.7 nmol aa 3/mg protein for non-ischemic region, 456.3 ± 10.9 nmol heme b/mg protein and 511.4 ± 21.0 nmol aa 3/mg protein for the infarct tissue, and no detectable contamination of the mitochondrial preparation with nuclear (probed with histone) or cytosolic proteins (probed with GAPDH).
Measurement of the mitochondrial State 3 ATP generation flux was conducted by combining oxygen electrode and ATP assay to simultaneously monitor mitochondrial state 3 ATP generation flux and OCR as reported in the literature [19, 20]. The mitochondrial ATP flux (accumulated ATP within 1 min) was measured using ATP Bioluminescent Assay Kit (Sigma-Aldrich, St. Louis, MO, USA) following manufacturer’s protocol. The mitochondrial state 3 OCR was simultaneously measured on the Oxytherm under the conditions as previously described. Briefly, the mitochondrial preparation (to a final of 0.4 mg/mL) was added into NADH-linked respiration buffer containing Ap5A (adenylate kinase inhibitor, 20 µM), to which ADP (to a final of 1 mM) was added. 5 µL of reaction mixture was withdrawn from the reaction chamber and immediately added into 495 µL of pre-heated ATP-assay buffer (70 °C, 10 min) as initial ATP concentration. Another 5 µL of reaction mixture was sampled 20 and 40 s after the initial sampling as the final ATP concentration. The linear OCR during the ATP sampling time span was ensured by the simultaneous Oxytherm polarography monitoring. The bioluminescent signal was recorded on the OrionL microplate luminometer (Titertek-Berthold Detection Systems GmbH, Pforzheim, Germany).
Measurements of mitochondrial ·O2 − production by EPR spin trapping were carried out on a Bruker EMX Micro spectrometer operating at 9.43 GHz with 100 kHz modulation frequency at room temperature [8]. The reaction mixture containing the NADH-linked respiration buffer supplemented with DTPA (1 mM)/DMPO (90 mM) was mixed with a mitochondrial preparation (to a final concentration of 0.5 mg protein/mL) at 30 °C for 4 min. The reaction mixture was then transferred to a 50-µL capillary tube (Drummond Wiretrol, Broomall, PA, USA), loaded into the EPR resonator (HS cavity, Bruker Instrument, Billerica, MA, USA), equilibrated to 298°K, and tuned within 2 min. The scan of EPR spectra was started at exactly 6 min after the initial reaction. Parameters: center field 3360 G, sweep width 100 G, power 20 mW, receiver gain 1 × 105, modulation amplitude 1 G, conversion time 40.96 ms, time constant 163.84 ms, and number of scans 5. The spectral simulations were performed using the WinSim program developed at NIEHS by Duling [21].
The redox activities of mitochondria or tissue homogenates from the myocardium were measured by EPR with a redox probe of CM-H [7, 20]. The mitochondrial preparation or tissue homogenates in M-buffer were energized by glutamate and malate in the presence of CM-H and then subjected to EPR measurement at 298°K. The instrumental settings used for detecting the three-line spectrum of the nitroxide were: center field, 3360.3 G; sweep width, 60 G; microwave frequency, 9.43 GHz; power, 20 mW; receiver gain, 5.02 × 103; modulation frequency, 100 kHz; modulation amplitude, 1 G; time constant, 163.84 ms; conversion time, 41 ms; sweep time, 42 s; number of X-scans, 1. The parameters for the kinetic mode were: static field, 3360.3 G; receiver gain, 2 × 103; time constant, 2624.44 ms; conversion time, 1000 ms; sweep time, 300 s; number of scans, 1.
The enzymatic activity of aconitase in mitochondria was assayed by measuring the conversion of citrate to α-ketoglutarate in the presence of isocitrate dehydrogenase by the absorbance increase at 340 nm at 37 °C as described in a previous publication [7].
The enzymatic activity of glutathione reductase (GR) was assayed by measuring GSSG-mediated NADPH consumption by the absorbance decrease at 340 nm at 25 °C as described in a previous publication [8].
The redox status of NAD+ and NADH in the as-isolated mitochondria was determined by NAD+ recycling with colorimetric method using NAD+/NADH Quantification Colorimetric Kits from Sigma-Aldrich (Catalog Number: MAK037) according to the manufacture’s instructions.
Direct measurements of mitochondrial ΔΨ were carried out using the fluorescent probe of TMRM (tetramethylrhodamine methyl ester). Isolated rat heart mitochondrial suspension (160 µg protein/mL) was equilibrated with TMRM (final 2 µM) in respiration buffer containing potassium glutamate (140 mM) and malate (5 mM) at 30 °C for 10 min, and the fluorescence was recorded by a 96-well microplate reader (excitation 528/20 nm, emission 620/40 nm). Various effectors (final concentration: ADP 5 mM, oligomycin 4 µg/mL, nigericin 5 µM, valinomycin 180 nM) were then injected into the specified wells, and the fluorescence was continuously monitored for additional 25 min. Heat-denatured mitochondria (70 °C for 10 min), M-buffer without mitochondria, as well as trace amount of ethanol (vehicle control of compounds oligomycin, nigericin or valinomycin) were served as background controls. Most wells started in state 2 conditions (containing the energetic respiration buffer, mitochondria, and TMRM) except the state 4 control contained additional ADP and oligomycin in the reaction mixture at the onset of the measurement.
Immunoblotting analysis
Western blotting with mitochondrial preparations was performed as described previously [7, 8, 11, 20]. Immunoblotting was carried out with one of: an anti-51 kDa antibody (against the FMN-binding subunit of complex I, generated in-house), an anti-75 kDa polyclonal antibody (against the 75 kDa subunit of complex I, generated in-house), an anti-ND1 (hydrophobic protein of complex I, from Santa Cruz Biotechnology, Dallas, TX, USA), an anti-70 kDa antibody (against the FAD-binding subunit of complex II, generated in-house), an anti-FeS antibody (monoclonal antibody against Rieske iron–sulfur protein of complex III), an anti-CoX I antibody (monoclonal antibody against the subunit 1 of complex IV, from Santa Cruz Biotechnology), or an anti-aconitase, or anti-glutathione reductase antibody (polyclonal antibodies from Santa Cruz Biotechnology).
Data analysis
Statistical analysis was performed using Origin 9.1 data analysis software. Results were presented as mean ± SEM, followed by group number (n), and p value. Comparisons between two groups (the data sets of Figs. 1, 2B, 3, 4b, 5, 6, 7, 8C, D) were assessed by Student’s t test. Comparisons among multiple groups (the data set of three groups in Fig. 5) were assessed by one-way ANOVA followed by Tukey’s post hoc tests. A probability value of p < 0.05 was used to establish statistical significance.

Mitochondria were prepared from the myocardia of non-ischemic (normal mitochondria, black bar) and risk regions (post-ischemic mitochondria, gray bar), and then subjected to measurement of the OCR by oxygen polarography at 30 °C as described under “Materials and Methods”. a, b State 3 (200 µM ADP used), state 4 (2 µg/mL oligomycin used), and FCCP (2.5 µM used)-mediated OCRs from the isolated mitochondria. c Respiratory control index obtained from the ratio of state 3 OCR to state 4 OCR. n = 6; **p < 0.01, assessed by Student’s t test for normal mitochondria (NR) vs. post-ischemic mitochondria (IR) in a and b. d ATP production flux mediated by normal and post-ischemic mitochondria, n = 4; **p < 0.01

The ·O2 − generation mediated by mitochondria in the presence of glutamate and malate (in mM, potassium glutamate (140) and malate (5), NADH-linked) was assessed by EPR spin trapping with DMPO according to our published methods [8]. A The EPR spectra of the SOD-sensitive DMPO/•OH adduct mediated by normal mitochondria under the conditions of state 2 (spectrum b), state 3 (spectrum c), state 4 (spectrum d), FCCP uncoupling (spectrum e), and the effect of PEG-SOD on the detected DMPO/•OH adducts (spectrum f). B Comparison between two groups of normal mitochondria (NR, gray bar) and post-ischemic mitochondria (IR, black bar) in the mediation of NADH-linked ·O2 − generation (state 2, state 3, state 4, and FCCP). Data were collected based on spin quantitation of DMPO/•OH. Data were analyzed by Student’s t test (n = 6, **p < 0.01)

Mitochondria were isolated from the non-ischemic region (NR) and the risk region (IR), permeabilized by alamethicin (40 mg protein:1 mg alamethicin ratio), and then subjected to assay. a The enzymatic activities of ETC components in the mitochondria were assayed as described previously [8, 11] (NR vs. IR, n = 4, *p < 0.05). b The enzymatic activity of aconitase was measured as described previously [7] (NR vs. IR, n = 4, ***p < 0.001). Mitochondrial preparation (100 µg protein per lane) was probed with a polyclonal antibody against aconitase (1:500) and the subunit I of complex IV (anti-Cox I, 1:4,000) was used as the loading control. Data were normalized to the signal intensity of mitochondrial complex IV

The mitochondria were isolated from the non-ischemic region (NR) and the risk region (IR), and then subjected to measurement of OCR using an oxygen polarograph at 30 °C. The mitochondrial preparation (0.5 mg/mL) was added to an NADH-linked respiration buffer containing malate plus glutamate to initiate oxygen consumption. The OCR was then enhanced by an uncoupler. a The OCRs of normal mitochondrial preparations (0.5 mg/mL) were enhanced by uncoupling with FCCP (2.5 µM), nigericin (5 µM), valinomycin (180 nM), and gramicidin (2 µg/mL) (n = 3). b Upper panel the chart of extracellular flux analysis showing measurements of the percentage changes of OCR of HL-1 cells after compound injections. After oligomycin (port A) inhibition of ATP-linked oxygen consumption, various uncouplers (nigericin, FCCP, valinomycin, or gramicidin; port B) were injected into the microplate to induce oxygen consumption. The enhanced OCRs were attributed to Δp, ΔpH, or ΔΨ according to the nature of the uncouplers. The final injection of antimycin A (port C) inhibited the relative mitochondrial oxygen consumption and resulted in the same percentage of non-mitochondrial OCR, confirming that the effects of the uncouplers targeted mitochondria directly, and that this effect could be abolished by antimycin A. Lower panel, the average %OCR of various uncouplers was calculated based on the measurement in the graph in the upper panel. In the control group (Ctrl), the % OCR remained above 90% after three mock injections of assay medium, which served as an additional control indicating that the HL-1 cells remained viable throughout the 2 h of the assay time. Nig. nigericin, Val. valinomycin, Gram. gramicidin; n = 6, ***p < 0.001

Effects of nigericin and valinomycin on the NADH-linked ·O2 − generation mediated by the mitochondria isolated from the non-ischemic region (NR) and the risk region (IR). The assay was conducted under state 2 (in a) and state 4 (in b) respiratory conditions. Mitochondria (0.5 mg/mL) isolated from the non-ischemic myocardium or from the risk region of the post-ischemic myocardium were pre-treated with nigericin (5 µM) or valinomycin (4 µg/mL) prior to EPR assay. The ·O2 − generation mediated by as-isolated or nigericin-treated or valinomycin-treated mitochondria in the presence of glutamate and malate (NADH-linked) was assessed by EPR spin trapping with DMPO according to our published methods [8]. Data were collected based on spin quantitation of DMPO/•OH adducts and by comparisons among three groups of NR or IR, nigericin treatment (+Nig.), and valinomycin treatment (+Val.). n = 4, ***p < 0.001 for NR vs. NR + Nig or NR + Val. (state 2), and NR vs. NR + Nig. (state 4); **p < 0.01 for NR vs. NR + Val. (state 4); *p < 0.05 for IR vs. IR + Nig or IR + Val. (state 2), and IR vs. IR + Nig. (state 4)

a, The rates of H2O2 generation mediated by the mitochondria of non-ischemic and risk regions in the presence of glutamate and malate (NADH-linked) under state 3, state 4, and FCCP-uncoupling conditions were assessed by an HRP/Amplex Red assay (n = 8). b–c, the effects of nigericin (Nig.) and valinomycin (Val.) on H2O2 generation rates mediated by the normal mitochondria and the post-ischemic mitochondria (n = 8, **p < 0.01; ***p < 0.001)

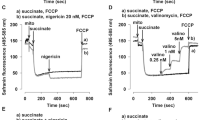
The mitochondria were isolated from the non-ischemic region (NR) and the risk region (IR), and then subjected to measurement of Δψ using a fluorescent probe, TMRM, as described in the experimental procedure. A Measurement of Δψ under state 3 conditions in the presence of ADP; B measurement of Δψ under state 4 conditions in the presence of oligomycin; C measurement of Δψ under state 4 conditions in the presence of nigericin; D measurement of Δψ under state 4 conditions in the presence of valinomycin. Each data point was collected from the average of three repeats (n = 3)

The effect of nigericin in the redox activity of CM-H oxidation mediated by the isolated mitochondria (in A–C) and tissue homogenate of post-ischemic myocardium (in D). A Mitochondria (0.4 mg/mL) isolated from the non-ischemic region (NR in a) and the risk region (IR in b) in respiration buffer (in mM, NaCl 10, MgCl2 1, EGTA 1, Trizma 1, phosphate 2.5, and cytochrome c 0.01; adjusted to pH 7.4) containing 1 mM DTPA were incubated with CM-H (1 mM) at room temperature. Oxidation of CM-H was immediately initiated by glutamate plus malate [(glu. plus mal.), 140 mM/5 mM] and then subjected to EPR measurement. To assess the effect of nigericin (Nig. in c and d), the mitochondrial preparation was pre-treated with nigericin (Nig., 5 µM) prior to CM-H incubation, activation of glutamate and malate, and EPR measurement. B Same as A, except that the redox activity of converting CM-H to a stable nitroxide is presented in a kinetic curve. C, same as A, except that the redox activity of converting CM-H to a stable nitroxide is presented in a bar graph (n = 5, **p < 0.01 and ***p < 0.001). D The tissue homogenates (0.25 mg/mL) of the non-ischemic region and the risk region excised from the post-ischemic myocardium in NADH-linked respiration buffer containing 1 mM DTPA, 140 mM glutamate, 5 mM malate, and 1 mM CM-H were subjected to EPR measurement for converting the hydroxylamine to a stable nitroxide (n = 5, **p < 0.01 and ***p < 0.001). The instrumental settings used for detecting the three-line spectrum of the nitroxide are: center field, 3360.3 G; sweep width, 60 G; microwave frequency, 9.43 GHz; power, 20 mW; receiver gain, 5.02 × 103; modulation frequency, 100 kHz; modulation amplitude, 1 G; time constant, 163.84 ms; conversion time, 41 ms; sweep time, 42 s; number of X-scans, 1
Results
Study design
Overall rationale and experimental design to assess states 2–4 OCR and the effect of ΔpH and ΔΨ on the mitochondria-mediated ·O2 − generation are illustrated in the Scheme 1c.
State 3 and state 4 OCR in the post-ischemic heart
It has been hypothesized that overproduction of oxygen free radicals during myocardial IR can depolarize mitochondrial membrane potentials and uncouple mitochondrial respiration [22, 23]. As indicated in Fig. 1a, b, NADH-linked and ADP-stimulated respiration of the mitochondria from the post-ischemic myocardium was significantly decreased. The ADP-independent OCR (state 4) was further deceased. The FCCP-induced uncoupled respiration was decreased, and the respiratory control index (the ratio of state 3 to state 4) was also decreased as indicated in Fig. 1c.
To evaluate the effect of IR on the efficacy of oxidative phosphorylation, we determined mitochondria-mediated ATP generation flux under the state 3 OCR in the presence of a higher ADP concentration (1 mM). The ATP production flux mediated by isolated mitochondria of the risk region was significantly diminished (Fig. 1d).
·O2 − generation mediated by the mitochondria of the non-ischemic myocardium
The NADH-linked ·O2 − production mediated by normal mitochondria at different respiratory states was induced by glutamate/malate (NADH-linked), and measured by EPR spin trapping with DMPO. A SOD 1 (Zn–Cu superoxide dismutase)-sensitive four-line spectrum of DMPO/•OH adduct was detected, indicating that ·O2 − generation was mediated by the normal mitochondria under state 2 conditions (Fig. 2A, b). Addition of ADP (state 3 respiration) significantly diminished mitochondria-mediated ·O2 − generation by 59.6 ± 7.7%, indicating that coupling of enhanced O2 consumption with oxidative phosphorylation for ATP synthesis decreased the e− leakage to molecular oxygen (Fig. 2A, c, B). In the presence of oligomycin (state 4 respiration), mitochondria-mediated ·O2 − generation induced by glutamate/malate was restored to the level of state 2 respiration (Fig. 2A, d, B). However, the addition of FCCP (1 μM) to uncouple the ∆Ψ and ∆pH significantly decreased the ·O2 − production to the level of state 3 conditions (Fig. 2A, e, B).
·O2 − generation mediated by the mitochondria of the post-ischemic heart
The measured NADH-linked ·O2 − production by mitochondria isolated from the post-ischemic myocardium was enhanced by 143.4 ± 33.1% under state 3 conditions (Fig. 2B). In the absence of ADP (state 2 in Fig. 2B) or in the presence of oligomycin (state 4 in Fig. 2B), the ·O2 − generation mediated by post-ischemic mitochondria was not significantly different from the level produced by the state 3 conditions, suggesting that ·O2 − generation by the post-ischemic mitochondria was less dependent on ∆pH. There was no significant difference in the ·O2 − production by the post-ischemic mitochondria and normal mitochondria under the oligomycin-induced state 4 conditions and the FCCP-induced uncoupling conditions (Fig. 2B).
Mitochondrial ETC and aconitase activities in the post-ischemic myocardium
Mitochondria were then subjected to analysis of ETC enzymatic activities. As indicated in Fig. 3a, a significant decrease in ETC activities was detected in the post-ischemic mitochondria compared to control, including complex I, complex II, complex III, and complex IV. The defect in the ETC activities was well correlated with the impairment of ADP-stimulated and FCCP-stimulated OCRs (Figs. 1a, b, 3a).
We used the redox sensor aconitase as another index of increased free radical production in IR mitochondria [24,25,26,27]. A severe decline in the enzymatic activity of aconitase to 57.5 ± 6.6% in the mitochondria of the risk region (Fig. 3b) was detected, which was corroborated by increased mitochondrial ·O2 − production in the post-ischemic myocardium. There was a modest decrease in protein expression of aconitase in the mitochondria of the post-ischemic myocardium (Fig. 3b).
NAD+ and NADH in the mitochondria of post-ischemic rat heart
The amount of NAD+ in the isolated mitochondria was decreased from 3.44 ± 0.06 (in nmol/mg protein) in the non-ischemic region to 1.42 ± 0.05 in the risk region (p < 0.001, n = 9) under non-energized conditions, indicating a depletion of NAD+ and NADH pool after myocardial I/R. However, the ratio of [NAD+]/[NADH] under energized conditions in the presence of glutamate plus malate was increased from 2.91 ± 0.08 in normal mitochondria to 8.12 ± 0.37 in post-ischemic mitochondria (p < 0.001, n = 8), implicating IR predisposed the mitochondria in a more oxidized physiological setting.
Mediation of uncoupling mitochondrial ΔpH and ΔΨ in the myocardium and HL-1 myocytes by nigericin, valinomycin, and gramicidin
To gain insights into the role of the electrochemical gradient in the post-ischemic injury, the specific inhibitors of ΔpH and ΔΨ were used, including nigericin, valinomycin, and gramicidin. The nature of their function as uncouplers was evaluated in the model system of isolated mitochondria with polarographic analysis, and in the HL-1 myocytes using the technique of extracellular flux analyzer.
Nigericin functions as an antiporter of H+ and K+, and thus dissipates the ΔpH and increases the ΔΨ via enhancing the K+ gradient across the inner membrane. Nigericin consistently partially uncoupled the normal mitochondria in the absence of ADP (Fig. 4a), suggesting that the proton backpressure of ΔpH negatively mediates electron transport for the OCR of mitochondria. The nature of nigericin functioning as an uncoupler was further evaluated in the model system of myocyte (HL-1 cell line). As indicated in Fig. 4b, nigericin partially uncoupled the OCR in HL-1, which resulted in a lower reserve bioenergetic capacity (~50%) compared with that of FCCP-induced uncoupling of the OCR in HL-1.
Valinomycin is a potassium-specific transporter and facilitates the movement of K+ across the membrane down an electrochemical potential gradient, leading to uncoupling oxidative phosphorylation in biological systems. Valinomycin partially uncoupled the normal mitochondria (Fig. 4a), indicating that the Δψ also negatively mediates electron transport for the OCR. However, it was observed that valinomycin completely uncoupled the OCR of HL-1 myocytes, exhibiting an uncoupling efficiency similar to that of FCCP (Fig. 4b).
Gramicidin partially uncoupled the OCR of normal mitochondria (Fig. 4a) via dissipating ΔΨ. In agreement with this result, gramicidin partially uncoupled the OCR of HL-1 myocytes as determined by extracellular flux analysis (Fig. 4b).
The effect of nigericin on NADH-linked ·O2 − production by the mitochondria of the post-ischemic myocardium
Nigericin abolished normal mitochondria-mediated ·O2 − generation under state 2 (Fig. 5a) and oligomycin-induced state 4 conditions, respectively (Fig. 5b). However, the quenching effect of nigericin on NADH-linked ·O2 − generation was less pronounced in the mitochondria isolated from the risk region (Fig. 5a, b).
The effect of valinomycin on NADH-linked ·O2 − production by the mitochondria of the post-ischemic heart
Valinomycin reduced ·O2 − generation by normal mitochondria under state 2 (Fig. 5a) and state 4 conditions (Fig. 5b). However, the quenching effect of valinomycin was less distinct in the post-ischemic mitochondria. The addition of valinomycin only modestly decreased NADH-linked ·O2 − generation under state 2 (Fig. 5a) and state 4 conditions (Fig. 5b).
Effect on the H2O2 emission by the mitochondria of the 24-h post-ischemic heart
The emission of NADH-linked H2O2 mediated by normal mitochondria and post-ischemic mitochondria was measured using Amplex Red/HRP according to previously published approach [10, 20, 28]. The levels of H2O2 emitted from normal and post-ischemic mitochondria were significantly higher in the state 4 conditions, and lower in the state 3 and FCCP-uncoupling conditions (n = 8, comparison of three groups in Fig. 6a), supporting the proton backpressure and membrane potential from electrochemical gradient as the endogenous source ROS. The amount of H2O2 emission by the post-ischemic mitochondria was significantly increased under state 3 and FCCP-uncoupling conditions (Fig. 6a), and more resistant to the quenching effect of nigericin (in Fig. 6b) and valinomycin (in Fig. 6c) as seen in the results of ·O2 − generation determined by EPR (Fig. 5). However, the capacity of a combination of nigericin and valinomycin to lower H2O2 emission by IR mitochondria is close to the effect of FCCP, and marginally higher than that of the single uncoupler used (data not shown).
Measurements of Δψ and ΔpH with the fluorescence probe of TMRM in the mitochondria of 24-h post-ischemic heart
TMRM is a commonly used fluorescent dye to monitor the mitochondrial Δψ. The lipophilic cationic probe is accumulated into the negatively charged mitochondrial matrix side by the polarized Δψ. The fluorescence of TMRM exhibits a red shift and the fluorescent intensity is quenched when the probe is accumulated by mitochondria [29]. The higher Δψ derived from polarized mitochondria would yield lower fluorescent intensity, and depolarized mitochondria would give higher fluorescent intensity.
As shown in Fig. 7a, the effect of ADP on normal mitochondria was highlighted. The ADP addition converted state 2 to state 3 conditions, and the fluorescent signals increased, confirming a shifting toward low Δψ status. When ATP was accumulated and ADP was exhausted over this time, the Δψ was gradually restored and the fluorescence returned to quenching state. The addition of oligomycin to normal mitochondria induced a transition to state 4 conditions as depicted in Fig. 7b, the fluorescence intensity gradually decreased over the course of 10-min (from 11th to 21st min), indicating the accumulation of Δψ resulted from buildup of the ΔpH. In contrast, ADP-stimulated fluorescent enhancement and oligomycin-induced quenching were subtle in the post-ischemic mitochondria, confirming a more depolarized status.
The effects of nigericin and valinomycin on the Δψ of normal mitochondria were highlighted in Fig. 7c, d. The addition of nigericin to normal mitochondria under state 4 conditions induced hyperpolarization as indicated by a rapid quenching of fluorescent intensity (time course of 10–15 min in Fig. 7c), confirming the nature of nigericin as the antiporter in dissipating the ΔpH and enlarging the Δψ. The addition of valinomycin to normal mitochondria in the presence of K+ significantly enhanced fluorescent intensity, thus indicating abolishment of mitochondrial Δψ (Fig. 7d). However, the post-ischemic mitochondria were much less sensitive to the quenching effect of nigericin and the enhancing effect of valinomycin (Fig. 7c, d).
Redox status in the mitochondria of the post-ischemic heart and the effect of nigericin
X-band EPR was employed to analyze the redox status of normal and post-ischemic mitochondria during the oxidation of CM-H to a stable nitroxide. It was observed that post-ischemic mitochondria exhibited a higher redox activity in mediating CM-H oxidation than normal mitochondria under basal or non-energized conditions (4.53 ± 0.30 nmol/min/mg protein vs. 3.63 ± 0.02 nmol/min/mg protein, n = 4 and p < 0.05).
Under the NADH-linked energized and oligomycin-induced state 4 conditions, the redox activity of post-ischemic mitochondria was significantly increased (Fig. 8A, B), supporting the conclusion of a more oxidized physiological setting in the post-ischemic myocardium. Nigericin treatment significantly enhanced the redox activity of normal mitochondria to the level of post-ischemic mitochondria, but did not impact the redox activity of post-ischemic mitochondria to a significant level (Fig. 8C), indicating that impairment of ΔpH predisposed the post-ischemic myocardium to a more oxidized redox status. The above results were further evaluated and confirmed using tissue homogenates containing mitochondria and cytosol. As indicated in Fig. 8D, the redox activity of tissue homogenate from the post-ischemic myocardium was also significantly higher. Treatment of the non-ischemic tissue homogenates with nigericin enhanced their redox activity to the level of the post-ischemic tissue homogenates, whereas nigericin treatment did not significantly alter the redox activity of the tissue homogenates from the risk region.
The effect of nigericin on the redox status of mitochondria with SOD2 overexpression
To understand if the ·O2 − production by the mitochondria contributed to enhanced redox activity induced by nigericin or uncoupling ΔpH, the mouse model of SOD2-tg was employed. The mitochondria isolated from SOD2-tg exhibited no detectable NADH-linked ·O2 − generation and a higher NADH-linked H2O2 production under energized conditions [20]. The mouse model of SOD2-tg was thus an ideal system to evaluate whether or not mitochondrial ·O2 − generation can have an impact on enhanced redox activity mediated by uncoupling ΔpH (Fig. 9). Under state 2 and energized conditions, we found that the mitochondria from SOD2-tg murine hearts displayed a lower redox activity, which is consistent with the conclusion of a more reductive physiological setting in the myocardium of SOD2-tg mice [20]. Nigericin treatment significantly enhanced the redox activities of mitochondria from both wild-type and SOD2-tg mice. However, there was no statistically significant difference in nigericin-induced redox activity enhancement between wild-type and SOD2-tg mitochondria, supporting the conclusion that uncoupling of ΔpH predisposes mitochondria to a more oxidized physiological setting, independent of ·O2 − generation.

The effect of nigericin in the redox activity of CM-H oxidation mediated by the mitochondria isolated from the myocardium of wild-type and SOD2-tg mice. Mitochondria (0.4 mg/mL) isolated from the myocardium of wild-type and SOD2-tg mice (~27–33 g and 12 weeks old) in respiration buffer (as indicated in the legend of Fig. 6) containing 1 mM DTPA and 1 mM CM-H were subjected to EPR measurement at 298 oK. To assess the effect of nigericin (in gray bar), the mitochondrial preparation was pre-treated with nigericin (Nig., 5 µM) prior to CM-H incubation prior to EPR measurement. The instrumental EPR parameters were the same as described in the legend of Fig. 5 (n = 5, *p < 0.05)
Mitochondrial respiration, ΔpH, and Δψ in the 3 h post-ischemic rat heart
Multiple studies have shown substantial remodeling of mitochondria over the first 24 h of reperfusion, including fusion, fission, mitophagy, the inner membrane, and ETC itself. Additional studies were thus conducted in the model of rat heart subjected to 30 min of coronary ligation and an earlier time point of 3 h reperfusion in vivo. Similar respiratory impairments in the state 3 OCR, FCCP-induced uncoupling OCR, and respiratory control index were detected in the 3-h post-ischemic mitochondria (Supplementary Figure 1). We have further detected an enhanced NADH-linked ·O2 − generation at state 3 conditions as well as a less quenching effect of nigericin on the state 2 ·O2 − generation by the 3-h post-ischemic mitochondria as seen in the 24-h post-ischemic mitochondria (Supplementary Figure 1). However, the quenching effect of valinomycin on the state 2 ·O2 − generation in the 3-h post-ischemic mitochondria observed was more significant, suggesting a portion of Δψ was conserved in early point of reperfusion, which contributes to a moderately enhanced state 2 ·O2 − generation in part (Supplementary Figure 1).
Discussion
The main findings of this study elucidated (1) IR-induced ΔpH impairment prompted the post-ischemic myocardium to a more oxidized redox setting, which appears to be associated with enhanced uncoupled respiration, and (2) IR-induced impairment of both ΔpH and ΔΨ increased state 3 mitochondrial ·O2 − generation.
Uncoupled respiration in the post-ischemic heart
Current studies confirmed that the coupling of O2 consumption with ATP synthesis was impaired after in vivo myocardial ischemia and reperfusion injury, supporting a significant defect in respiration uncoupled in the post-ischemic myocardium. Current studies also supported the concept that mitochondrial dysfunction occurring in post-ischemic injury is caused by overproduction of mitochondria-derived oxygen free radicals (Fig. 1a, b vs. Fig. 2B under state 3 conditions). The results were in agreement with those studied by the Langendorff-perfused rat heart [11, 16]. These results further support the concept that increasing electrochemical gradients and proton backpressure drives a portion of the e− leakage for ·O2 − production under normal physiological conditions. Post-ischemic injury damaged the ETC activities and uncoupled the ΔpH across the inner membrane of the post-ischemic mitochondria. The ΔΨ, secondary to ΔpH, was the source of ·O2 − generation under normal physiological conditions, and uncoupled in the mitochondria of the post-ischemic heart. Together with the results from nigericin and valinomycin, it was concluded that neither ΔpH nor ΔΨ significantly contributes to ·O2 − generation by the mitochondria of 24-h post-ischemic heart.
It is worth noting that the mitochondria studied are predominately at 24-h reperfusion. Substantial impairment of ETC activity and oxidative phosphorylation and most important here of damage of inner membrane persist at this intermediate time of reperfusion. Therefore, mitochondrial remodeling has not resulted in tissue mitochondria with normal function.
It appears that direct damage to the ETC accounts for a significant component of the respiratory defect and increased ROS production in the post-ischemic myocardium of current study. However, the inner membrane damage during myocardial IR has been shown to involve significant damage or depletion of cardiolipin [30,31,32,33,34,35]. Cardiolipin of the inner membrane is postulated serving as the electrical “insulator” basis for the physiology of ΔpH and ΔΨ. IR-induced cardiolipin oxidation or cardiolipin depletion can further contribute to impairment of ΔpH and ΔΨ.
ΔpH in the post-ischemic heart
As indicated by EPR spin trapping analysis, nigericin treatment decreased NADH-linked ·O2 − generation in normal mitochondria (at state 2 and state 4 conditions in Fig. 5a, b), supporting the conclusion that ΔpH is a source of ·O2 − production under normal physiological conditions. The result was basically consistent with early studies with reverse electron flow-induced ROS through complex I [36]. Because the ΔpH of the mitochondrial inner membrane was high under conditions of low ADP (state 2) or inhibition of ATP synthase, external addition of ADP thus induced state 3 respiration by increased proton influx through the proton channel of ATP synthase, diminishing ΔpH and increasing ATP synthesis. The proton pumps of the ETC then responded with increased proton pumping and electron flow to maintain the electrochemical gradient, leading to the consequence of increasing O2 consumption (Fig. 1a, c). We have detected that the addition of ADP (state 3 conditions) significantly decreased ·O2 − production mediated by normal mitochondria (in Fig. 2) from the models of rat and mouse hearts [7, 8], further reinforcing the above conclusion.
In the post-ischemic myocardium, we have hypothesized that ischemia and reperfusion injury can mediate ΔpH impairment. The hypothesis was largely supported by the results of current studies such as an IR-mediated decreased state 3 OCR ex vivo (Fig. 1a, c), and in the Langendorff-perfused rat heart [11, 16]. The addition of ADP failed to reduce the ·O2 − production mediated by the mitochondria of the post-ischemic myocardium (Fig. 2). The result was corroborated by the respiratory analysis showing that post-ischemic mitochondria exhibited impaired OCRs for both ADP-enhanced (Fig. 1a, c) and nigericin-enhanced conditions (data not shown). Furthermore, the nigericin-mediated quenching effect of ·O2 − production was much less pronounced in the mitochondria of the post-ischemic myocardium due to the defect in ΔpH (Fig. 5a vs. Fig. 5b). The results generally supported that a decrease in ΔpH is correlated with decreasing ·O2 − generation by the mitochondria as predicted in previous report [1].
As the electrons pass through each complex of the ETC, there is an energy drop in reduction potential that provides the driving force required for proton pumping. Energy stored in the ΔpH and Δψ is the potential energy driving protons to return to the matrix for ATP synthesis. Therefore, damage of the ETC activity by the post-ischemic injury was likely directly involved in impaired proton pumping and ΔpH impairment, which has been reported in the complex I and complex IV of mouse model [3] as well as the complex I and complex III of rat model [11, 14, 16].
Our recent progress has indicated increased irreversible cysteine sulfonation in the complex I and complex III to be partially responsible for oxidative impairment of ETC in the post-ischemic heart. Specifically, several specific sites of irreversible cysteine S-sulfonation in the complex I of post-ischemic heart are involved in the ligands of iron–sulfur clusters, which control its electron transport activity [37]. Likely, IR-induced irreversible cysteine oxidation of ETC may directly or indirectly contributes to ΔpH impairment. Increased complex I S-glutathionylation has been detected in the post-ischemic heart [14]. It may not necessarily contribute to ΔpH impairment because reversal of glutathionylation by dithiothreitol (DTT) did not restore complex I activity [38].
ΔΨ in the post-ischemic heart
EPR analysis clearly revealed that valinomycin significantly diminished NADH-linked ·O2 − generation mediated by the isolated normal mitochondria, supporting mitochondrial membrane potential as one of components to regulate electron leakage for ·O2 − production under normal physiological conditions. The result was supported by early published work using vesicle-reconstituted complex III [6, 39]. Ischemia and reperfusion injury obviously impaired ΔΨ and subsequently disrupted the above regulation since ·O2 − production by the isolated post-ischemic mitochondria was less sensitive to the valinomycin treatment (Fig. 5a vs. Fig. 5b). This conclusion was upheld by respiratory analysis, which indicated that post-ischemic mitochondria displayed a declined FCCP-mediated (Fig. 1a, b) and valinomycin-induced uncoupling of OCRs (data not shown). Similar results have documented that reverse electron flow-induced ROS production can be mediated by the ΔΨ of the mouse heart [9] and brain mitochondria [40], but decreased due to the depolarized ΔΨ caused by ischemia of the mouse heart [9].
Redox dysfunction in the post-ischemic heart
The above results were further supported by the redox assays using the spin probe CM-H. In normal mitochondria, the ΔpH and ΔΨ build up when ΔpH is not being used for ATP synthesis. This “proton backpressure” derived from the ΔpH and ΔΨ can control and restrict electron transport and the OCR, predisposing the normal mitochondria to a more reduced physiological setting. Uncoupling of mitochondrial ΔpH and ΔΨ by nigericin (Fig. 4a) or FCCP (Figs. 1a, 4a) appreciably enhances the electron transfer and OCRs, prompting the redox status of mitochondria to become more oxidized. This hypothesis was substantiated by the results in which nigericin or FCCP was found to mediate enhancement of mitochondrial redox activity in converting CM-H to its stable nitroxide (Fig. 8).
We have further found that valinomycin treatment failed to enhance the redox activity of normal mitochondria under energetic conditions despite increasing the uncoupled OCR (Fig. 4a, b). It is likely that dissipation of the ΔΨ did not significantly promote the normal mitochondria to a more oxidized redox status. However, nigericin treatment of normal mitochondria increased its redox activity to the level of the mitochondria isolated from the risk region (Fig. 8A, B), suggesting ΔpH as the major component in regulating the redox status of the mitochondria and myocardium (Fig. 8), and impairment of ΔpH as one of the key factors in predisposing the post-ischemic myocardium to a more oxidized physiological setting.
In post-ischemic mitochondria, treatment with nigericin (Fig. 8) or FCCP (data not shown) exerted no significant impact on the redox activity of post-ischemic mitochondria or the post-ischemic myocardium, further confirming IR-mediated injury of the electrochemical gradient and the consequence of redox dysfunction caused by IR injury. Therefore, impairment of ΔpH and ΔΨ weakened the ability of the membrane instinct to restore “proton backpressure” under state 2 or state 4 conditions, provoking the more oxidized post-ischemic mitochondrial redox status.
There are other factors associated with the redox dysfunction during myocardial ischemia and reperfusion. These factors can include overproduction of ·O2 − and other ROS, which would increase the prooxidant activity of aconitase via producing hydroxyl radical by Fenton reaction and inactivating the [4Fe–4S] cluster (Fig. 3b) [27]. Aconitase in mitochondria was thus a redox sensor of ·O2 − (k ~ 107 M−1 s−1 for the reaction of aconitase with ·O2 −), and impairment of aconitase activity was thus correlated with redox dysfunction in the post-ischemic heart.
However, the results of evaluating the SOD2-tg mouse model has concluded that ·O2 − production may not necessarily correlate to a more oxidized redox status driven by ΔpH uncoupling, indicating the strength of SOD2-tg. As reported previously enhanced NADH-linked H2O2 generation as well as modest catalase upregulation was detected in the mitochondria of SOD2-tg heart [20]. It should not be ruled out that excess downstream H2O2 may exert a noticeable effect to increase redox activity of nigericin-treated SOD2-tg mitochondria. Investigation using the mouse model of mitochondrial catalase overexpression may compensate for the above weakness.
Post-ischemic injury of antioxidant enzymes associated with the mitochondrial GSH pool further contributed to the redox dysfunction of post-ischemic mitochondria, but might not necessarily enhance the redox activity of CM-H oxidation by the post-ischemic mitochondria. We further observed a defect in glutathione reductase (GR2) in enzymatic activity (a reduction of 69.5 ± 6.7%, n = 6) and protein expression (a reduction of 60.3 ± 14.5%, n = 3) in the mitochondria of the post-ischemic myocardium (Supplementary Figure 2A and 2C), which was correlated with the higher GSSG/GSH ratio and enhanced protein S-glutathionylation reported previously [14]. However, post-ischemic injury of the total glutathione reductase in the tissue homogenate was not as significant as the GR2 impairment detected in the post-ischemic mitochondria (Supplementary Figure 2B), reinforcing the critical role of mitochondrial dysfunction in the redox dysfunction of the post-ischemic myocardium.
A marginal decrease by 28.4 ± 8.6% (p < 0.05, n = 6) in the glutathione peroxidase (GPx2) activity in the post-ischemic mitochondria was further detected, supporting a decreased ability to scavenge H2O2, increased oxidant stress, and interrupted redox balance in mitochondria. In the tissue homogenate of the post-ischemic myocardium, we detected a similar decline in GPx activity, indicating no differential impairment between mitochondria and the cytosol during myocardial IR.
Relevance of cardioprotection
It has been well documented that mitochondria are a target of cardioprotection induced by humoral factors released during remote ischemic preconditioning [41], preconditioning effect of TNFα [42], and postconditioning activation of mitochondrial STAT3 [43]. However, the in-depth mechanism of cardioprotection remains to be elucidated. Current studies may provide insights into potential mechanisms involved in protecting ΔpH from dissipation, maintaining membrane potential, and stabilizing the redox status to preserve mitochondrial function and reduce infarction.
Conclusion
The present studies provide insights into the mechanisms of ΔpH and ΔΨ in controlling ·O2 − generation by mitochondria under conditions of normal physiology and myocardial infarction. As delineated in Fig. 10, the underlying mechanism has been characterized by impaired pH gradient and membrane potential, which enhance ·O2 − generation from ETC in the presence of ADP (state 3 conditions), and promote the post-ischemic mitochondria and myocardium to a more oxidized redox setting. Treatment with nigericin and valinomycin exerts a more pronounced effect on ΔpH- and ΔΨ-mediated ·O2 − generation by the normal mitochondria than by the post-ischemic mitochondria. Disruption of the ΔpH with nigericin predisposes the normal mitochondria to a more oxidized physiological setting as seen in the redox status of the post-ischemic mitochondria and myocardium. The mechanism addressed here provides a useful concept for understanding the mechanism of post-ischemic injury involved in ΔpH and ΔΨ. Recognition of this mechanism is valuable in understanding the fundamental basis of mitochondrial biology in the disease of myocardial infarction.

Diagram showing the mechanism by which pH gradient (ΔpH) and membrane potential (ΔΨ) mediates redox dysfunction-associated post-ischemic injury in the mitochondria of rat heart. a Under the basal conditions, electron flow mediated by ETC is accompanied by the transmembrane movement of protons to generate ΔpH and ΔΨ. The proton motive force (pmf) derived from electrochemical gradient (Δp) pushes the protons to re-enter the matrix through FiF0ATPase (ATPase, denoted by blue rectangle-oval object) for ATP synthesis. The presence of ADP (state 3 conditions) minimizes the electron leakage for ·O2 − generation by ETC. Oligomycin blocks the proton channel of ATPase, thus enhancing the ΔpH and ΔΨ of normal mitochondria (indicated by thick arrows). The higher ΔpH and ΔΨ of normal mitochondria as the source of electron leakage for ·O2 − generation are highly sensitive to nigericin and valinomycin. The higher pH and ΔΨ also predisposes the mitochondria to a less oxidized physiological setting (dashed arrow). b In comparison to basal conditions, myocardial ischemia and reperfusion (IR) induced oxidative damage of ETC and subsequently impaired ΔpH and ΔΨ, which further decreases the proton motive force for ATP synthesis. Oxidative injury of ETC also greatly enhances the electron leakage for ·O2 − generation in the post-ischemic mitochondria. Due to IR-induced impairment of pmf and ATPase, the electron leakage for ·O2 − generation driven by ΔpH and ΔΨ is less sensitive to oligomycin, nigericin, and valinomycin (indicated by fine arrows). The impairment of ΔpH by IR further predisposes post-ischemic mitochondria to a more oxidized physiological setting (dashed arrow)
Change history
27 June 2017
An erratum to this article has been published.
Abbreviations
- IR:
-
Myocardial ischemia and reperfusion
- Δp :
-
Electrochemical gradient
- ΔpH:
-
pH gradient or proton gradient
- ΔΨ:
-
Membrane potential
- ·O2 − :
-
Superoxide anion radical
- EPR:
-
Electron paramagnetic resonance
- OCR:
-
Oxygen consumption rate
- ETC:
-
Electron transport chain
- ETA:
-
Electron transfer activity
- ROS:
-
Reactive oxygen species
- RCR:
-
Respiratory control ratio
- TMRM:
-
Tetramethylrhodamine methyl ester CM-H, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- FCCP:
-
Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- Complex I:
-
NADH-ubiquinone reductase
- Complex II:
-
Succinate-ubiquinone reductase
- Complex III:
-
Ubiquinol-cytochrome c reductase
- Complex IV:
-
Cytochrome c oxidase
- DCPIP:
-
Dichlorophenyl indophenol
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- DMPO:
-
5,5-Dimethyl-1-pyrroline N-oxide
References
Chen YR, Zweier JL (2014) Cardiac mitochondria and reactive oxygen species generation. Circ Res 114:524–537. doi:10.1161/CIRCRESAHA.114.300559
Sack MN (2006) Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovas Res 72:210–219. doi:10.1016/j.cardiores.2006.07.010
Zhao X, He G, Chen YR, Pandian RP, Kuppusamy P, Zweier JL (2005) Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation 111:2966–2972. doi:10.1161/CIRCULATIONAHA.104.527226
Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M et al (1993) Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 268:18532–18541
Ferrari R, Ceconi C, Curello S, Cargnoni A, Pasini E, De Giuli F, Albertini A (1991) Role of oxygen free radicals in ischemic and reperfused myocardium. Am J Clin Nutr 53:215S–222S
Rottenberg H, Covian R, Trumpower BL (2009) Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem 284:19203–19210. doi:10.1074/jbc.M109.017376
Kang PT, Chen CL, Chen YR (2015) Increased mitochondrial prooxidant activity mediates up-regulation of Complex I S-glutathionylation via protein thiyl radical in the murine heart of eNOS(−/−). Free Radic Biol Med 79:56–68. doi:10.1016/j.freeradbiomed.2014.11.016
Kang PT, Chen CL, Ren P, Guarini G, Chen YR (2014) BCNU-induced gR2 defect mediates S-glutathionylation of Complex I and respiratory uncoupling in myocardium. Biochem Pharmacol 89:490–502. doi:10.1016/j.bcp.2014.03.012
Ross T, Szczepanek K, Bowler E, Hu Y, Larner A, Lesnefsky EJ, Chen Q (2013) Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition vs. depolarization of inner mitochondrial membrane. Biochim Biophys Acta 1830:4537–4542. doi:10.1016/j.bbagen.2013.05.035
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278:36027–36031. doi:10.1074/jbc.M304854200
Lee HL, Chen CL, Yeh ST, Zweier JL, Chen YR (2012) Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 302:H1410–H1422. doi:10.1152/ajpheart.00731.2011
Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G (2009) Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta 1794:476–485. doi:10.1016/j.bbapap.2008.12.008
Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PP, Zweier JL, Chen YR (2008) Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J Biol Chem 283:27991–28003. doi:10.1074/jbc.M802691200
Chen J, Chen CL, Rawale S, Chen CA, Zweier JL, Kaumaya PT, Chen YR (2010) Peptide-based antibodies against glutathione-binding domains suppress superoxide production mediated by mitochondrial complex I. J Biol Chem 285:3168–3180. doi:10.1074/jbc.M109.056846
Zhang L, Chen CL, Kang PT, Garg V, Hu K, Green-Church KB, Chen YR (2010) Peroxynitrite-mediated oxidative modifications of complex II: relevance in myocardial infarction. Biochemistry 49:2529–2539. doi:10.1021/bi9018237
Chen YR, Chen CL, Pfeiffer DR, Zweier JL (2007) Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem 282:32640–32654. doi:10.1074/jbc.M702294200
Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R (1998) Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol 275:H1375–H1387. doi:10.1016/j.bbabio.2004.09.004
Zhao X, Chen YR, He G, Zhang A, Druhan LJ, Strauch AR, Zweier JL (2007) Endothelial nitric oxide synthase (NOS3) knockout decreases NOS2 induction, limiting hyperoxygenation and conferring protection in the postischemic heart. Am J Physiol Heart Circ Physiol 292:H1541–H1550. doi:10.1152/ajpheart.00264.2006
Hinkle PC (2005) P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1706:1–11. doi:10.1016/j.bbabio.2004.09.004
Kang PT, Chen CL, Ohanyan V, Luther DJ, Meszaros JG, Chilian WM, Chen YR (2015) Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation. J Mol Cell Cardiol 88:14–28. doi:10.1016/j.yjmcc.2015.09.001
Duling DR (1994) Simulation of multiple isotropic spin-trap EPR spectra. J Magn Reson B 104:105–110
Doliba NM, Doliba NM, Chang Q, Babsky AM, Wroblewski K, Natelson BH, Osbakken MD (1999) Mitochondrial oxidative phosphorylation in heart from stressed cardiomyopathic hamsters. J Mol Cell Cardiol 31:543–553. doi:10.1006/jmcc.1998.0890
Ferrari R (1996) The role of mitochondria in ischemic heart disease. J Cardiovasc Pharmacol 28(Suppl 1):S1–10
Gardner PR, Fridovich I (1991) Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem 266:19328–19333
Gardner PR, Fridovich I (1992) Inactivation–reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem 267:8757–8763
Gardner PR, Nguyen DD, White CW (1994) Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci USA 91:12248–12252
Vasquez-Vivar J, Kalyanaraman B, Kennedy MC (2000) Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 275:14064–14069
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2008) Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294:C460–C466. doi:10.1152/ajpcell.00211.2007
Scaduto RC Jr, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76:469–477. doi:10.1016/S0006-3495(99)77214-0
Lesnefsky EJ, Hoppel CL (2008) Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta 1777:1020–1027. doi:10.1016/j.bbabio.2008.05.444
Lesnefsky EJ, Minkler P, Hoppel CL (2009) Enhanced modification of cardiolipin during ischemia in the aged heart. J Mol Cell Cardiol 46:1008–1015. doi:10.1016/j.yjmcc.2009.03.007
Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL (2001) Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 280:H2770–H2778
Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta 1837:408–417. doi:10.1016/j.bbabio.2013.10.006
Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM (2004) Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94:53–59. doi:10.1161/01.RES.0000109416.56608.64
Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G (2009) Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free Radic Biol Med 46:88–94. doi:10.1016/j.freeradbiomed.2008.09.031
Lambert AJ, Brand MD (2004) Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J 382:511–517. doi:10.1042/BJ20040485
Kang PT, Chen CL, Lin P, Zhang L, Chen YR (2017) Increased cysteine sulfonation of complex I, complex III, and aconitase is associated with mitochondrial dysfunction in the post-ischemic heart. FASEB J 31:680.1. http://www.fasebj.org/content/31/1_Supplement/680.1.abstract
Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP (2008) Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem 283:24801–24815. doi:10.1074/jbc.M803432200
Zhang L, Yu L, Yu CA (1998) Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J Biol Chem 273:33972–33976
Votyakova TV, Reynolds IJ (2001) DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem 79:266–277
Gedik N, Maciel L, Schulte C, Skyschally A, Heusch G, Kleinbongard P (2016) Cardiomyocyte mitochondria as targets of humoral factors released by remote ischemic preconditioning. Arch Med Sci 13:448–458. doi:10.5114/aoms.2016.61789
Lacerda L, McCarthy J, Mungly SF, Lynn EG, Sack MN, Opie LH, Lecour S (2010) TNFalpha protects cardiac mitochondria independently of its cell surface receptors. Basic Res Cardiol 105:751–762. doi:10.1007/s00395-010-0113-4
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. doi:10.1161/circresaha.111.255604
Acknowledgements
This work was supported by National Institutes of Health Grant HL83237 (to Y.-R C) and HL115114 (to WMC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
The original version of this article was revised: Figures 7 and 9 were interchanged.
On page 9 the word “oligomycin” was changed to “valinomycin” in the sentence “The effects of nigericin and valinomycin on the Δψ of normal mitochondria were highlighted in Fig. 7c, d.”
An erratum to this article is available at https://doi.org/10.1007/s00395-017-0632-3.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kang, P.T., Chen, CL., Lin, P. et al. Impairment of pH gradient and membrane potential mediates redox dysfunction in the mitochondria of the post-ischemic heart. Basic Res Cardiol 112, 36 (2017). https://doi.org/10.1007/s00395-017-0626-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-017-0626-1